
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclohexa-1,4-dienes in transition-metal-free ionic transfer processes
Sebastian
Keess
 and
Martin
Oestreich
*
and
Martin
Oestreich
*
Institut für Chemie, Technische Universität Berlin, Strasse des 17. Juni 115, 10623 Berlin, Germany. E-mail: martin.oestreich@tu-berlin.de
First published on 24th May 2017
Abstract
Safe- and convenient-to-handle surrogates of hazardous chemicals are always in demand. Recently introduced cyclohexa-1,4-dienes with adequate substitution fulfil this role as El+/H− equivalents in B(C6F5)3-catalysed transfer reactions of El–H to π- and σ-donors (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C
C/C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C and CO/CN). Surrogates of Si–H/Ge–H, H–H and even C–H bonds have been designed and successfully applied to ionic transfer hydrosilylation/hydrogermylation, hydrogenation and hydro-tert-butylation, respectively. These processes and their basic principles are summarised in this Minireview. The similarities and differences between these transfer reactions as well as the challenges associated with these transformations are discussed.
C and CO/CN). Surrogates of Si–H/Ge–H, H–H and even C–H bonds have been designed and successfully applied to ionic transfer hydrosilylation/hydrogermylation, hydrogenation and hydro-tert-butylation, respectively. These processes and their basic principles are summarised in this Minireview. The similarities and differences between these transfer reactions as well as the challenges associated with these transformations are discussed.
 Sebastian Keess | Sebastian Keess (born in 1988 in Wuppertal/Germany) studied chemistry at the RWTH Aachen University (2008–2013), including a research internship at the University of York (2012). He obtained his bachelor's degree with Dieter Enders (2011) and his master's degree with Magnus Rueping (2013). His education was funded by a Deutschlandstipendium supported by the Bayer Science & Education Foundation (2012–2013). After a five-month internship at Bayer Pharma AG (Wuppertal/Germany), he moved to Berlin where he currently pursues graduate research in the group of Martin Oestreich at the Technische Universität Berlin. |
 Martin Oestreich | Martin Oestreich (born in 1971 in Pforzheim/Germany) is Professor of Organic Chemistry at the Technische Universität Berlin. He received his diploma degree with Paul Knochel (Marburg, 1996) and his doctoral degree with Dieter Hoppe (Münster, 1999). After a two-year postdoctoral stint with Larry E. Overman (Irvine, 1999–2001), he completed his habilitation with Reinhard Brückner (Freiburg, 2001–2005) and was appointed as Professor of Organic Chemistry at the Westfälische Wilhelms-Universität Münster (2006–2011). He also held visiting positions at Cardiff University in Wales (2005) and at The Australian National University in Canberra (2010). |
Concept
Transfer processes represent a practical strategy for performing challenging bond formations or avoiding handling hazardous reagents. Limited mainly to transfer hydrogenation1 for a long time, this technique has recently emerged as a powerful approach for the application of various toxic, flammable/explosive and/or gaseous chemicals that have otherwise only been rarely used in synthetic chemistry.2The aptitude of adequately substituted cyclohexa-1,4-dienes I to engage in ionic transfer reactions as synthetic equivalents of El+/H− (El = Si,3 Ge,4 H,5tBu6) was demonstrated by our laboratory during the last years (Scheme 1). The underlying concept relies on the ability of diene I to transiently form ion pair III+[HB(C6F5)3]− by B(C6F5)3-mediated hydride abstraction from the bisallylic methylene group (I → III+)7 and subsequently release electrofuge El+; aromatisation to furnish the respective arene is exploited as the driving force (Scheme 1, top). The fate of Wheland complex III+ was shown to be dependent on the nature of the attached El group, following divergent pathways: El–H release and subsequent activation by B(C6F5)3 or direct delivery of electrofuge El+ to substrate V (Scheme 1, bottom, grey pathways). Transfer hydrosilylation (El = Si)3 or hydrogermylation (El = Ge)4 were shown to pass through two interdependent catalytic cycles, liberating the hydrosilane and hydrogermane, respectively (III+ → El–H + RC6H5, left cycle), followed by B(C6F5)3-catalysed El–H bond activation. The thus-formed η1-adduct IV8 then participates in the reduction of C–C multiple bonds (right cycle).9,10 Conversely, transfer hydrogenation5 and hydro-tert-butylation6 proceed by direct transfer of the electrofuge El+ from Wheland intermediate III+ onto substrate V to eventually furnish adduct VII after hydride reduction by [HB(C6F5)3]− (III+ + V → VI+ → VII).11 Consistent with this dichotomy, liberation of the El–H functionality from I occurs even in the absence of a Lewis-basic substrate for hydrosilanes/hydrogermanes (El = Si and Ge)3,4 while degradation of the H–H and C–H surrogates (El = H and tBu) proceeds only slowly at room temperature.5,6
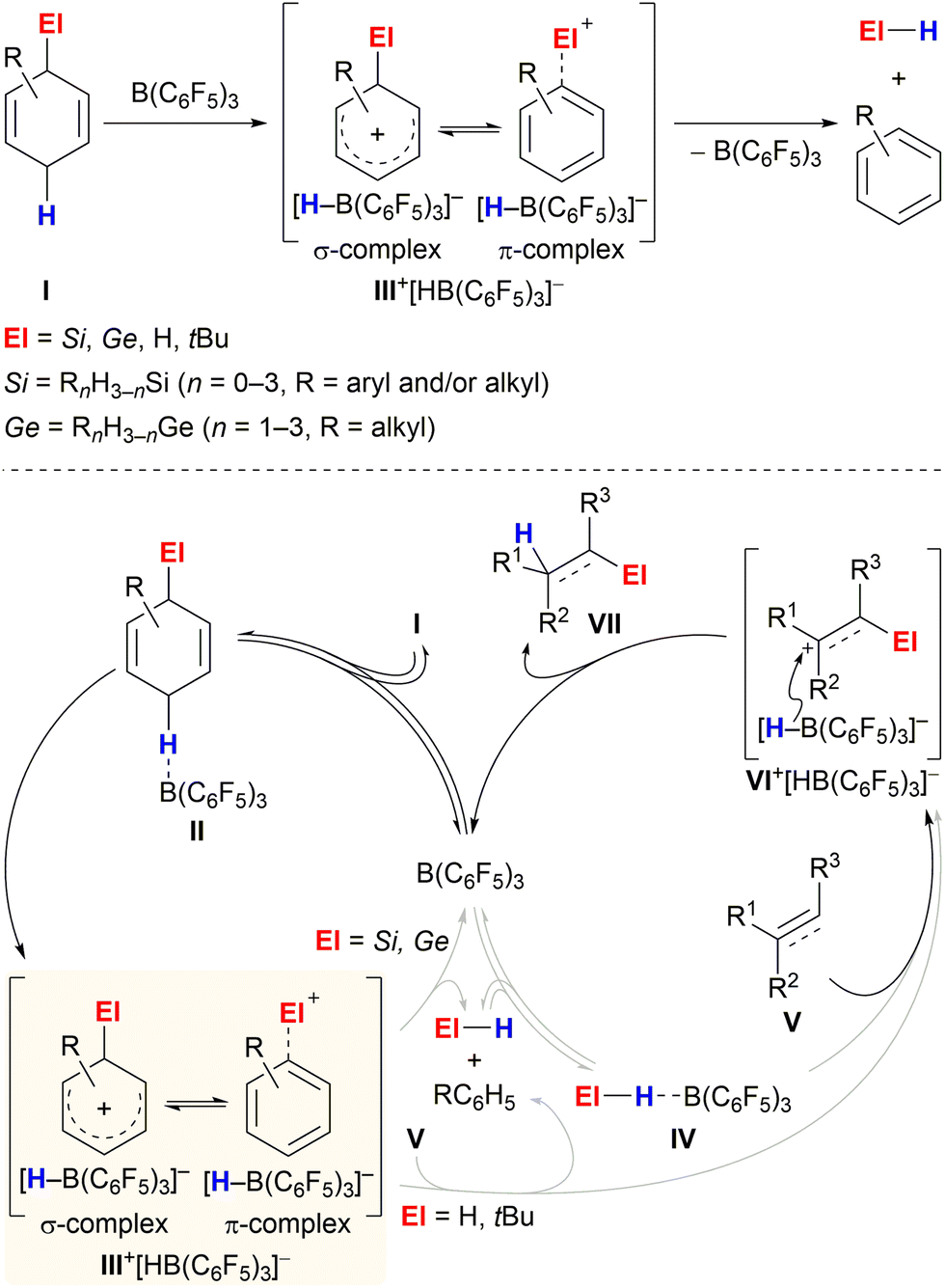 | ||
| Scheme 1 Reaction of cyclohexa-1,4-dienes with B(C6F5)3 in the absence (top) and presence (bottom) of π-basic substrates. | ||
The illustrated concept forms the foundation for all developed transition-metal-free ionic transfer processes using cyclohexa-1,4-dienes I as transfer reagents. This Minireview summarises the recent advances in these transformations. It outlines and discusses the challenges and limitations as well as the differences and similarities of the individual transfer processes.
Transfer reagents
The successful implementation of cyclohexa-1,4-dienes I in the different transfer reactions, i.e., transfer hydrosilylation/hydrogermylation, transfer hydrogenation and transfer hydro-tert-butylation, required deliberate modification of the substitution pattern of the cyclohexa-2,5-dien-1-yl unit (Fig. 1).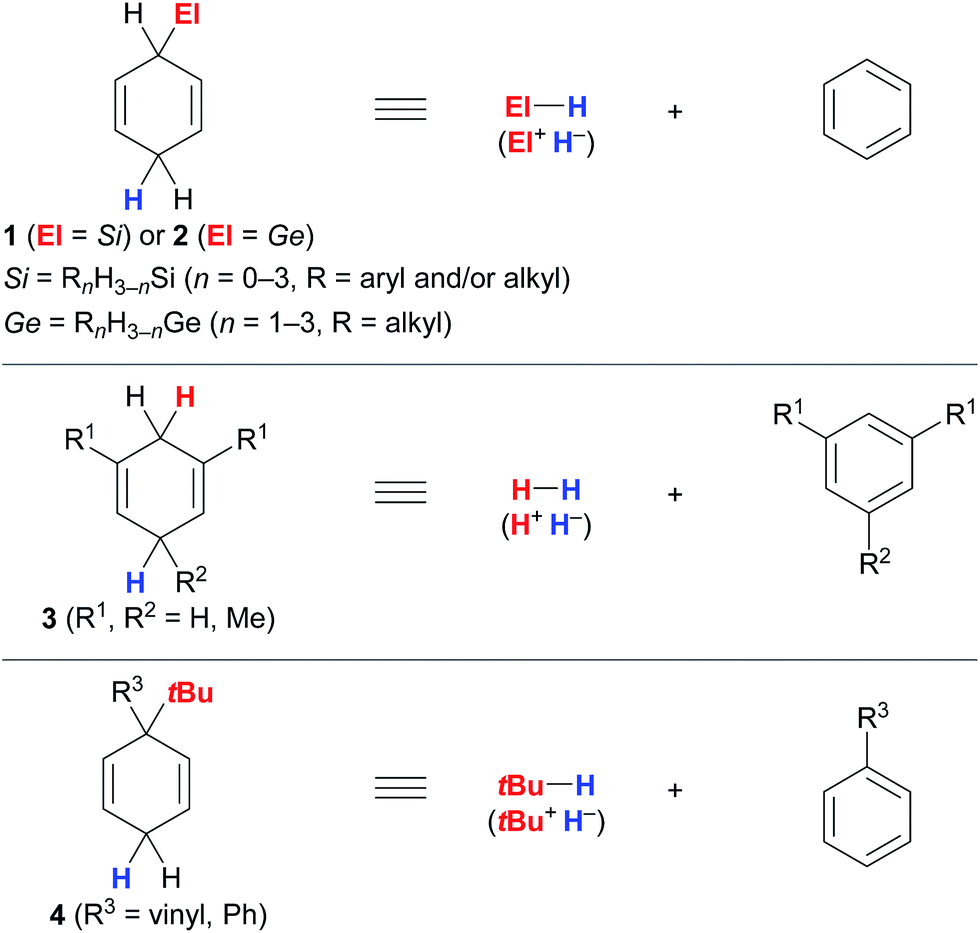 | ||
| Fig. 1 Substituted cyclohexa-1,4-dienes as synthetic equivalents of hydrosilanes/hydrogermanes (top), dihydrogen (middle) and isobutane (bottom). | ||
Unsubstituted cyclohexa-2,5-dien-1-ylsilanes 1 and -germanes 2 cleanly transform into the corresponding hydrosilane or hydrogermane and benzene at room temperature when treated with B(C6F5)3 (Fig. 1, top).3,4 Essential for this transformation to proceed is sufficient hydridic character of the bisallylic C(sp3)–H bond in 1 due to hyperconjugation with the C(sp3)–Si bond and the associated stabilisation of the resulting low-energy Wheland complex III+[HB(C6F5)3]− (cf.Scheme 1),12 as supported by computational studies by Sakata and Fujimoto.9 Comparable stabilisation from the C(sp3)–Ge bond is expected to facilitate the release of hydrogermanes from surrogates 2. Conversely, dihydrogen surrogates 3 are devoid of this stabilisation and require electron-donating substituents at C1/C5 (3a, R1 = Me, R2 = H)5a or C1/C3/C5 (3b, R1 = R2 = Me)5b to lend stabilisation to the resulting high-energy Wheland intermediates III+[HB(C6F5)3]− (middle), as well as to suppress undesired reaction pathways, e.g., dihydrogen release or cationic heterodimerisation of reactants. While unsubstituted cyclohexa-1,4-diene (3c) favoured side reactions in the transfer hydrogenation of alkenes catalysed by the Lewis acid B(C6F5)3,5b Brønsted acids such as Tf2NH were shown to selectively mediate transfer hydrogenation from this surrogate.5c,13 Likewise, adjustment of the substitution pattern at the cyclohexa-2,5-dien-1-yl core was necessary for the design of the transfer reagents 4 for the B(C6F5)3-catalysed transfer hydro-tert-butylation (bottom).6 Another substituent “ipso” to the tert-butyl group in 4 had to be introduced to avoid competing proton release from that position.
Transfer hydrosilylation/hydrogermylation
Simonneau and Oestreich introduced cyclohexa-1,4-dienes as reagents in ionic transfer processes14 and provided the proof-of-principle for the concept outlined above by employing surrogate 1a as an equivalent of gaseous Me3SiH (Fig. 2, left).3b Surrogates of various other hydrosilanes, e.g., functionalised (EtO)3SiH,3d were prepared and successfully tested in B(C6F5)3-catalysed ionic transfer hydrosilylations (not shown).3a The development of solid 1b as an easy-to-handle surrogate of pyrophoric and explosive SiH4 disclosed a rare strategy for the use of monosilane in organic synthesis.3e,15 Likewise, related 2a and 2b are surrogates of Et3GeH and gaseous MeGeH3 (Fig. 2, right) that enabled the first examples of transfer hydrogermylation.4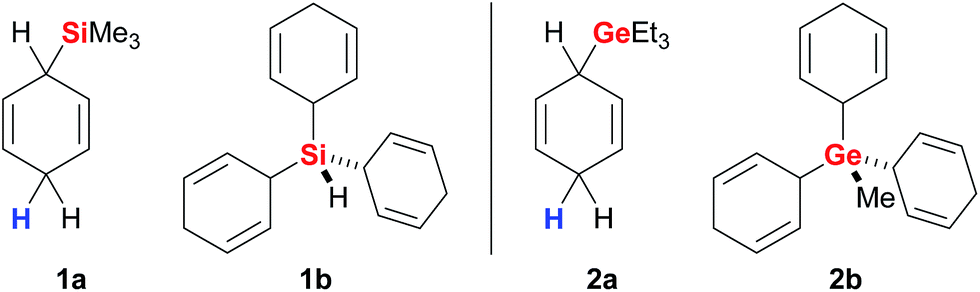 | ||
| Fig. 2 Representative cyclohexa-1,4-dienes as surrogates of hydrosilanes (left) and hydrogermanes (right). | ||
The ionic transfer hydrosilylation and hydrogermylation of π-basic substrates, i.e., alkenes and alkynes,16 proved to be applicable to a wide range of unfunctionalised derivatives (Scheme 2).3b,d,4 Both transfer processes proceeded at room temperature using catalytic amounts of the Lewis acid and a slight excess of surrogate 1a or 2a in CH2Cl2 or 1,2-F2C6H4, respectively. Terminal (→5–14), i.e., mono- and 1,1-disubstituted, as well as 1,2-disubstituted (→15–16) and trisubstituted (→17–18) alkenes were compatible with the transfer protocols and furnished tetraorganosilanes and -germanes in high yields. Reduction of an internal electronically unbiased alkyne selectively yielded the product of trans-addition (→(Z)-19–(Z)-20) whereas transfer hydrogermylation of electronically biased ethyl 3-phenylpropiolate proceeded selectively with cis addition, and the ester group was perfectly compatible (not shown).16b The exo selectivity in the reduction of norbornene (→15) and norborna-2,5-diene (→16) and predominant cis diastereoselectivity in the hydrosilylation of 1-methylcyclohexene (not shown) as well as the absence of products of radical cyclisation in the hydrogermylation of an acyclic 1,6-diene (not shown) confirmed the ionic nature of the mechanism for both transfer reactions.4,9 The regioselective formation of 17 and 18 emphasises the favoured formation of a benzylic (secondary) carbocation over a tertiary. A discrepancy in the performance of both surrogates 1a and 2a was observed in the reduction of functionalised substrates (grey box). Allyltriethylsilane reacted cleanly in the transfer hydrogermylation (→21) whereas only decomposition was observed when subjected to the setup of the transfer hydrosilylation (not shown). Acetophenone yielded alcohol 22 as product of hydrosilylation,17 but hydrogermylation of α,β-unsaturated esters and ketones furnished products with untouched carboxyl (→23) and carbonyl (→24) groups, respectively.
 | ||
| Scheme 2 Transfer hydrosilylation and hydrogermylation of C–X multiple bonds using surrogates of Me3SiH and Et3GeH. aPerformed at 90 °C. | ||
A systematic study was recently reported by Oestreich and co-workers that provides comprehensive insight into the parameters that govern the transfer hydrosilylation.3d The analysis includes surrogates 1 with modified electronic and steric properties, fully or partially fluorinated triarylboranes as well as representative π- and σ-basic substrates.18 Selected data of this study are summarised in Fig. 3. Cyclohexa-1,4-diene 1a reacts readily with π-donor 25 at room temperature while elevated temperatures are required to split the Lewis acid/base adduct of σ-donor 26 and the borane catalyst (column 1). Derivative 1c with a methyl group ipso to the departing silicon group was less reactive than parent 1a (column 2), as was surrogate 1d bearing an extended π system (column 3). Introduction of +M substituents as in 1e significantly increased the reactivity due to an enhanced hydricity of the bisallylic methylene group, yielding quantitative conversion of σ-basic acetophenone (26) at room temperature within minutes (column 4). Conversely, the σ-donating methoxy substituents in resorcinol-derived 1e outcompete π-basic 1,1-diphenylethylene (25) for the transfer of the silicon electrophile, and only cleavage of the ether groups of 1e was observed (column 4).
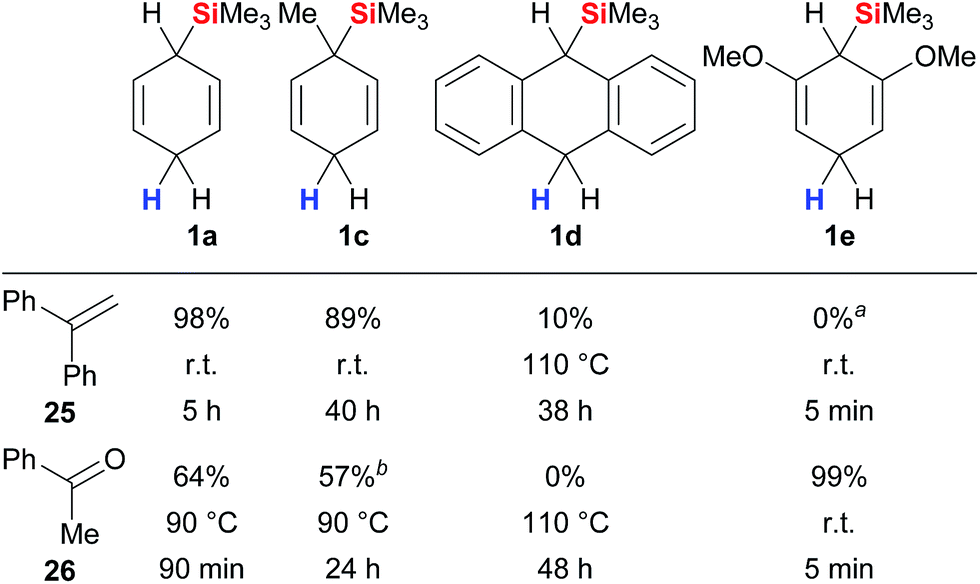 | ||
| Fig. 3 Interplay between surrogate structure and reactivity. aSurrogate 1e fully consumed. bPartial deoxygenation to styrene. | ||
Simonneau and Oestreich were able to further advance this strategy by introducing 1b as a surrogate of SiH4, a silane that is rarely used by synthetic chemists due to the associated safety issues.3e Later, this approach was unsuccessfully tested to access the related monogermane GeH4, and surrogate 2b as equivalent of MeGeH3 was prepared instead.4 Both surrogates 1b and 2b were shown to liberate SiH4 and MeGeH3, respectively, upon treatment with catalytic amounts of B(C6F5)3 followed by n-fold hydrosilylation or 3-fold hydrogermylation of typical alkenes (Scheme 3). Monohydro- (→27,30,31), dihydro- (→28,32) and tetraalkyl-substituted silanes (→29) became accessible dependent on the steric demand of the alkene; the degree of substitution at the silicon atom can usually not be controlled by the stoichiometry of the reagents. However, for 1,1-diphenylethylene (25), reversal of the chemoselectivity, that is the formation of 31 over 32, was achieved by adjustment of the stoichiometry and the use of di(cyclohexa-2,5-dien-1-yl)silane instead of 1b (grey box). Also, this method allowed for the mild preparation of tetraalkyl-substituted germanes (→33–37).
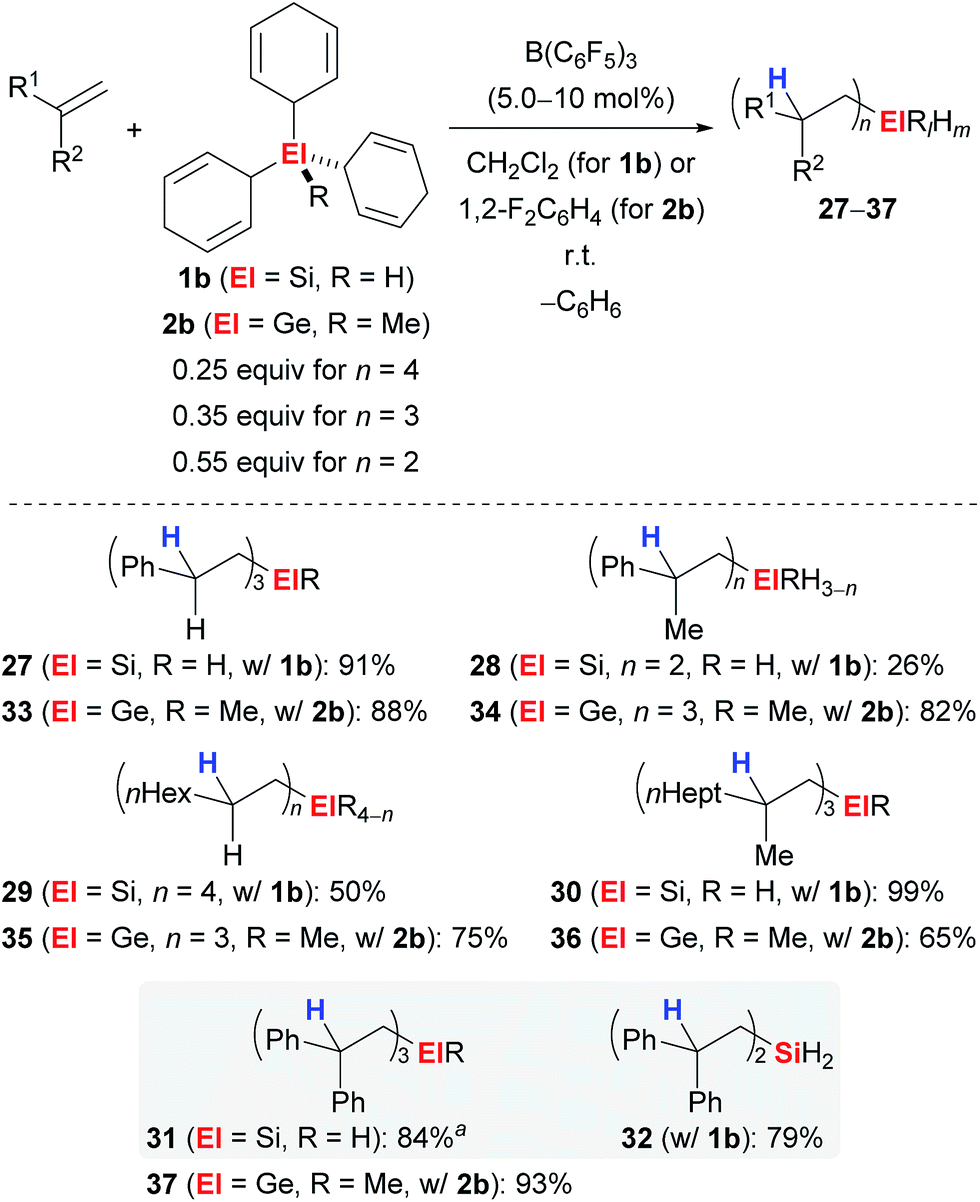 | ||
| Scheme 3 Transfer hydrosilylation/hydrogermylation of alkenes with surrogates of monosilane or methylgermane. aDicyclohexa-2,5-dien-1-ylsilane was used as the surrogate. | ||
Transfer hydrogenation
Kihara and co-workers introduced cyclohexa-1,4-diene (3c) as the dihydrogen source in the Lewis acid-catalysed reduction of dithioacetals to the corresponding sulfides (not shown).19,20 Later, the research group of Gandon reported a gallium(III)-assisted transfer hydrogenation of alkenes using the same hydrogen donor 3c (Scheme 4).13,21 Their protocol was applicable to 1,1-disubstituted (→38), 1,2-disubstituted (→39) as well as trisubstituted acyclic (→40–41) and cyclic (→42–46) alkenes and tolerated ketone (→39) or ester (→41) functionalities. Tetrasubstituted alkenes or those without an aryl substituent were unreactive (not shown).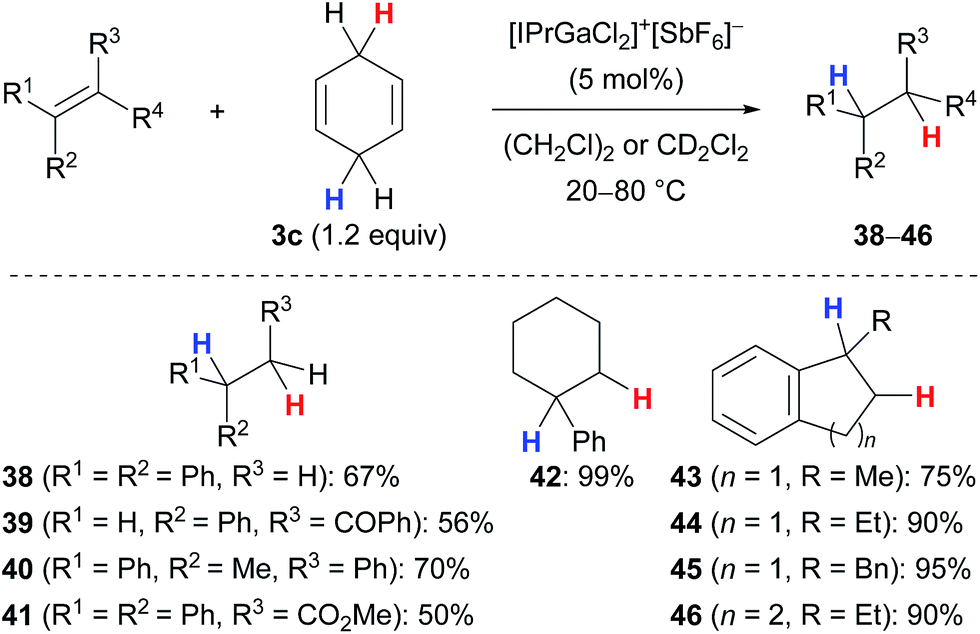 | ||
| Scheme 4 Gallium(III)-assisted transfer hydrogenation of alkenes. | ||
Aryl-substituted alkynes participated in a cascade hydroarylation/transfer hydrogenation sequence catalysed by the same gallium(III) complex with cyclohexa-1,4-diene (3c) as reductant to afford dicyclic (→47–49) as well as tricyclic (→50) products in high yields (Scheme 5).13 The formation of pentacyclic 51 gave a significantly lower yield. Although the mechanism of the gallium(III)-assisted transfer hydrogenation has not been studied in detail yet, an ionic process was proposed for the dihydrogen transfer (not shown).13a Later, it was demonstrated that the transformations depicted in Schemes 4 and 5 work equally well with an indium(III) complex (not shown).13b
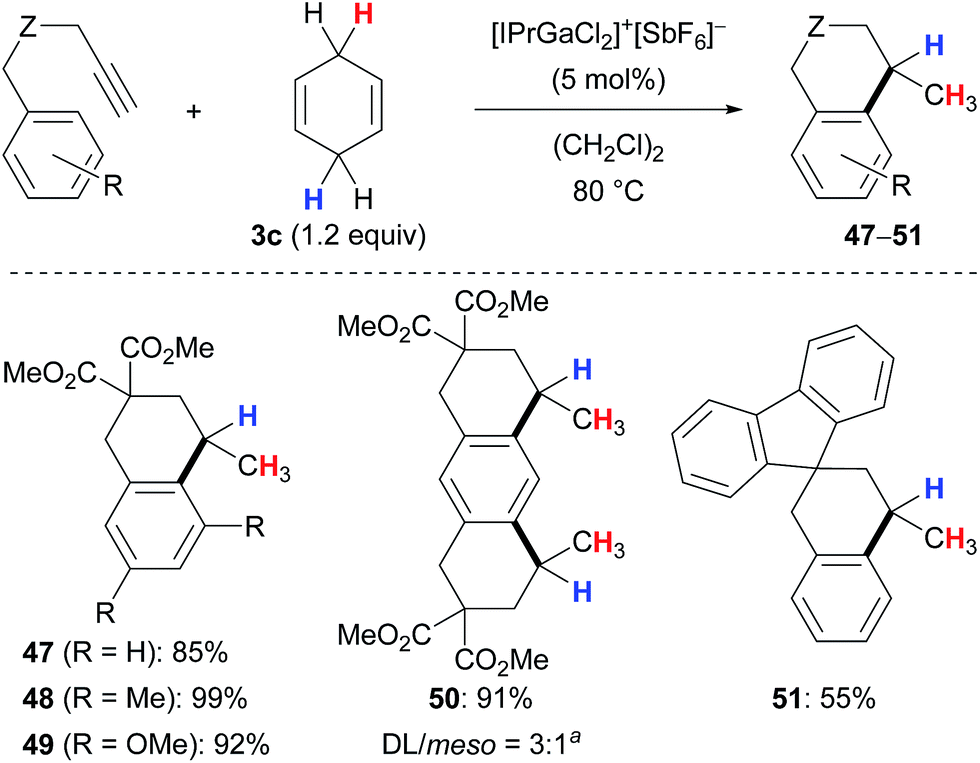 | ||
| Scheme 5 Gallium(III)-assisted hydrogenative cyclisation of alkynes. a2.4 equiv. of 3c used. | ||
Chatterjee and Oestreich disclosed the B(C6F5)3-catalysed ionic transfer hydrogenation of imines and related heteroarenes employing substituted cyclohexa-1,4-dienes 3a or 3b as the dihydrogen source.5a Later, Grimme and Oestreich showed that this transfer process also works with alkenes and confirmed the mechanism by quantum-chemical calculations.5b The catalytic cycle commences with rate-limiting Lewis acid-mediated hydride abstraction from surrogate 3a or 3b to give ion pair VIII+[HB(C6F5)3]− in low concentration (Scheme 6, left cycle). High-energy Wheland intermediate VIII+ acts as a strong Brønsted acid and protonates σ- or π-basic substrate IX to furnish ion pair XI+[HB(C6F5)3]− together with stoichiometric arene X. Dihydrogen release from VIII+ in the presence of a Lewis-basic substrate was excluded. Conversely, subsequent hydride transfer from [HB(C6F5)3]− to the carbenium ion in XI+ to regenerate B(C6F5)3 concomitant with the formation of product XII was proposed to compete with reversible dihydrogen liberation in the case of imines (grey pathway).22 The preference of either pathway depends on the electrophilicity of the carbon atom of the iminium ion intermediate as well as the basicity of the imine nitrogen atom. The formation of highly Brønsted-acidic Wheland intermediate VIII+ in the course of the Lewis acid-mediated transfer hydrogenation inspired Chatterjee and Oestreich to investigate potentially competing Brønsted-acid catalysis. As part of these studies, these authors successfully showed that reasonably strong Brønsted acids such as Tf2NH are equally able to initiate transfer hydrogenation23 from cyclohexa-1,4-dienes 3a or 3b by catalytically generating the same Wheland intermediate VIII+ (right cycle).5c It seems plausible that protonation of substrate IX occurs from either Brønsted acids VIII+ or Tf2NH to furnish intermediate XIII+[Tf2N]−. In the absence of the borohydride [HB(C6F5)3]−, cyclohexa-1,4-diene 3a or 3b steps in as the hydride donor for the reduction of XIII+[HB(C6F5)3]−, thereby closing the catalytic cycle.20g
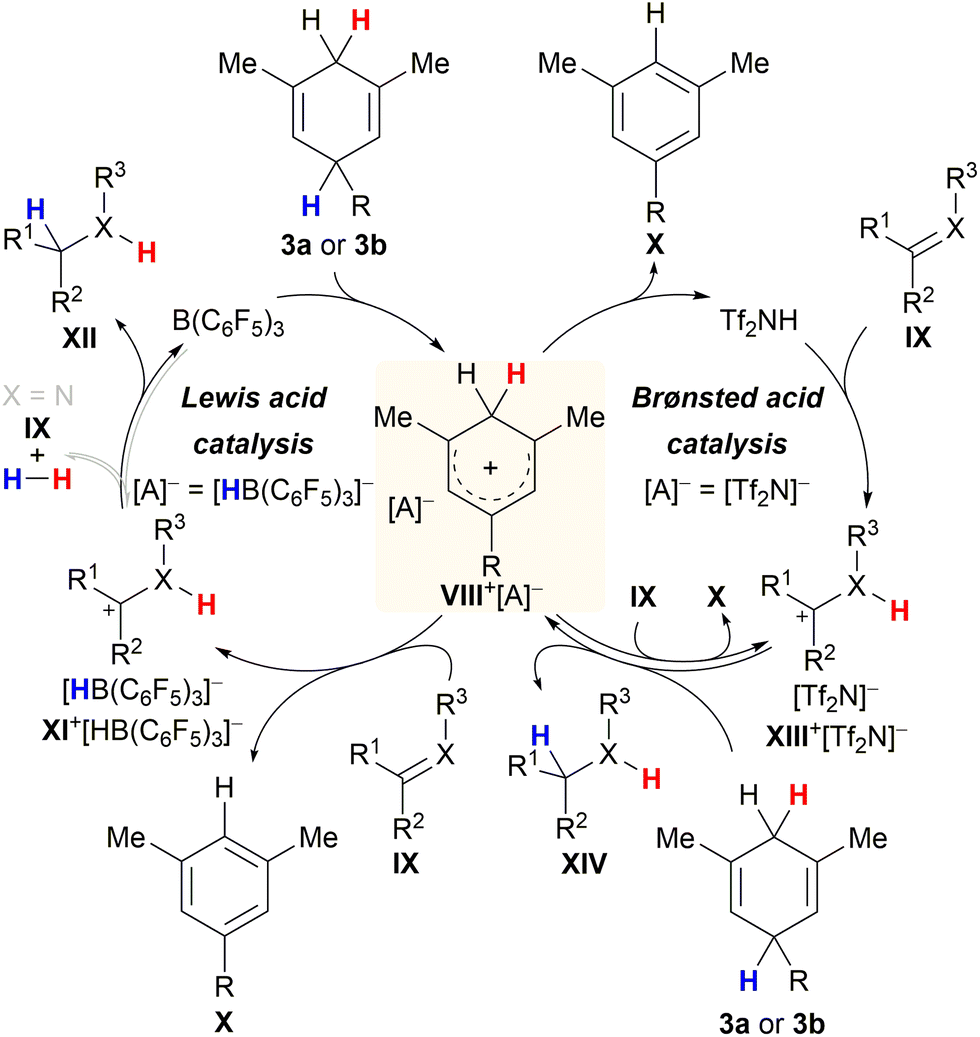 | ||
| Scheme 6 Catalytic cycles for Lewis and Brønsted acid-catalysed transfer hydrogenation of imines (X = NPG) and alkenes (X = CH). | ||
The transfer hydrogenation of imines requires forcing reaction conditions, i.e., 125 °C and 10 to 15 mol% of the catalyst, and is limited to certain protecting groups at the nitrogen atom to secure optimal steric shielding, sufficient Lewis basicity and stability (Scheme 7).5a,c The protocol is compatible with differently functionalised ketimines (→52–55) and aldimines (→57–61) and tolerated electron-withdrawing substituents (→54,55,59–61) and even ortho substitution (→59). A cyclohexanone-derived imine was completely unreactive (not shown), and a 4-anisyl-substituted ketimine showed only moderate reactivity in the presence of the Brønsted acid Tf2NH and no reactivity when subjected to catalysis with B(C6F5)3 (→56), likely due to lower hydride affinity of the respective iminium ion intermediate. Nitrogen-containing heterocycles participated well in the B(C6F5)3-catalysed transfer hydrogenation affording 62–64 in high yields.
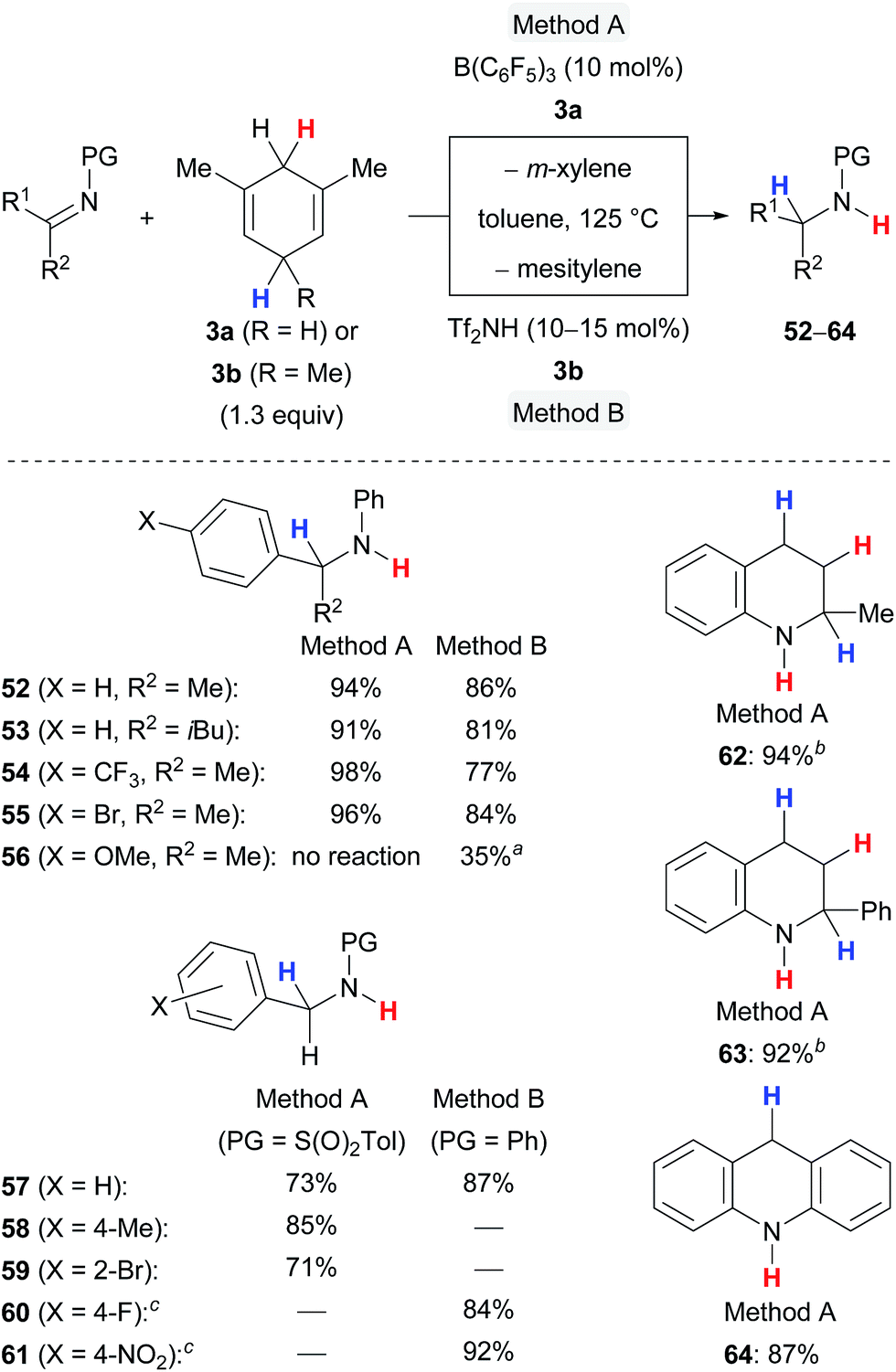 | ||
| Scheme 7 Lewis and Brønsted acid-catalysed transfer hydrogenation of imines and nitrogen-containing heteroarenes. aMessy reaction. b2.6 equiv. of surrogate 3a used. cPrepared by reductive amination. | ||
The transfer hydrogenation of π-basic alkenes proceeds equally well with B(C6F5)3 or Tf2NH under mild reaction conditions using 5.0 mol% of the catalyst at room temperature (Scheme 8).5b,c The transfer process, however, requires an additional methyl group in the bisallylic position of the cyclohexadienyl group where the hydride abstraction occurs to prevent undesired side reactions, that is liberation of dihydrogen and heterodimerisation of cationic intermediates. The method can be applied to a wide range of 1,1-disubstituted alkenes (→38,65–71) and works also with trisubstituted derivatives (→42). 1,1-Diarylalkenes furnished the corresponding alkanes 38, 65 and 66 in high yields, irrespective of the electronic properties of the arene. α-Alkyl-substituted styrenes as well as 1,1-dialkylalkenes required sterically demanding substituents, e.g., an isopropyl (→67) or a cyclohexyl group (→68–69), to prevent thermoneutral cationic dimerisation as observed for 70 and 71.
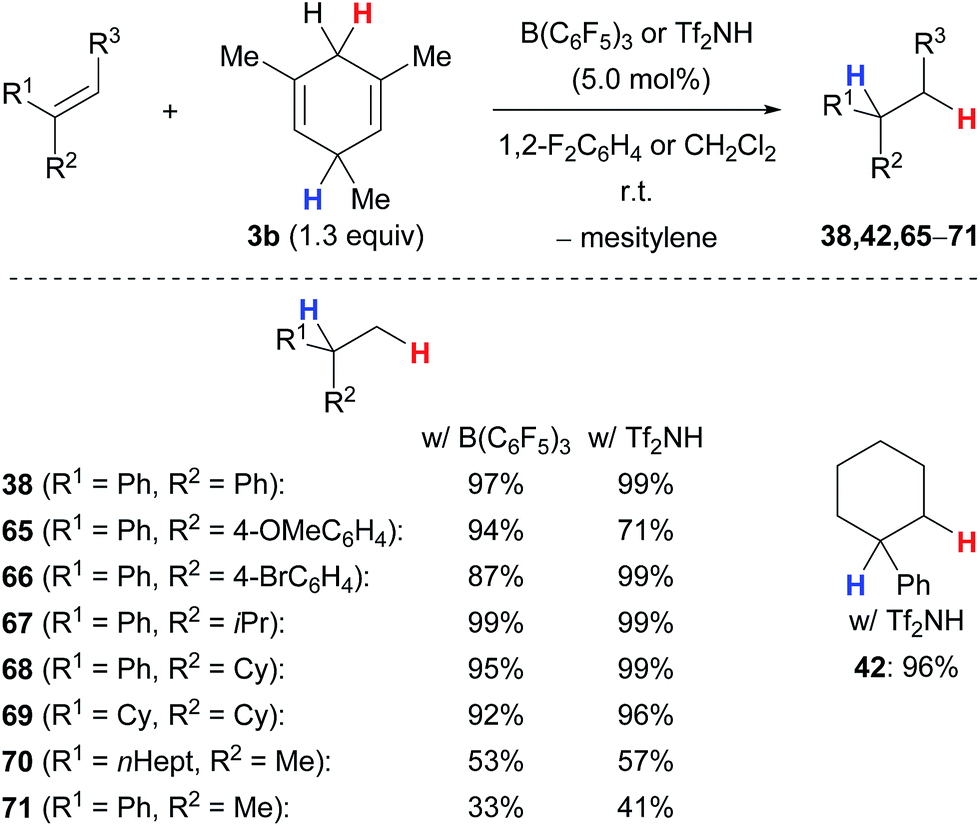 | ||
| Scheme 8 Lewis acid- and Brønsted acid-catalysed transfer hydrogenation of alkenes. | ||
Transfer hydro-tert-butylation
Keess and Oestreich introduced 3-tert-butyl-substituted cyclohexa-1,4-dienes 4 as transfer reagents in the transfer hydro-tert-butylation of alkenes.6 This methodology represents an unprecedented approach to install tertiary alkyl groups at carbon frameworks24 but competing reaction channels that could not be completely suppressed still limit its synthetic utility.The transfer of the tert-butyl group proceeds smoothly at room temperature with only little excess of transfer reagent 4, yielding quantitative conversion of the alkene (Scheme 9).6 Extensive optimization of the reaction conditions using 1,1-diphenylethylene (25) as model substrate could not fully prevent the formation of byproducts 75a and 76a (column 1). Electronically modified 1,1-diarylalkenes 72 and 73 were also tested but favoured the formation of the byproducts 75 and 76 to an even greater extent (columns 2 and 3). The influence of the surrogate structure, namely the substituent “ipso” to the tert-butyl group, is profound, resulting in superior selectivities for surrogate 4a (R = Ph) compared to 4b (R = vinyl). In the latter case, separation of the stoichiometrically formed arene byproduct is conveniently achieved as styrene polymerises under the reaction conditions.
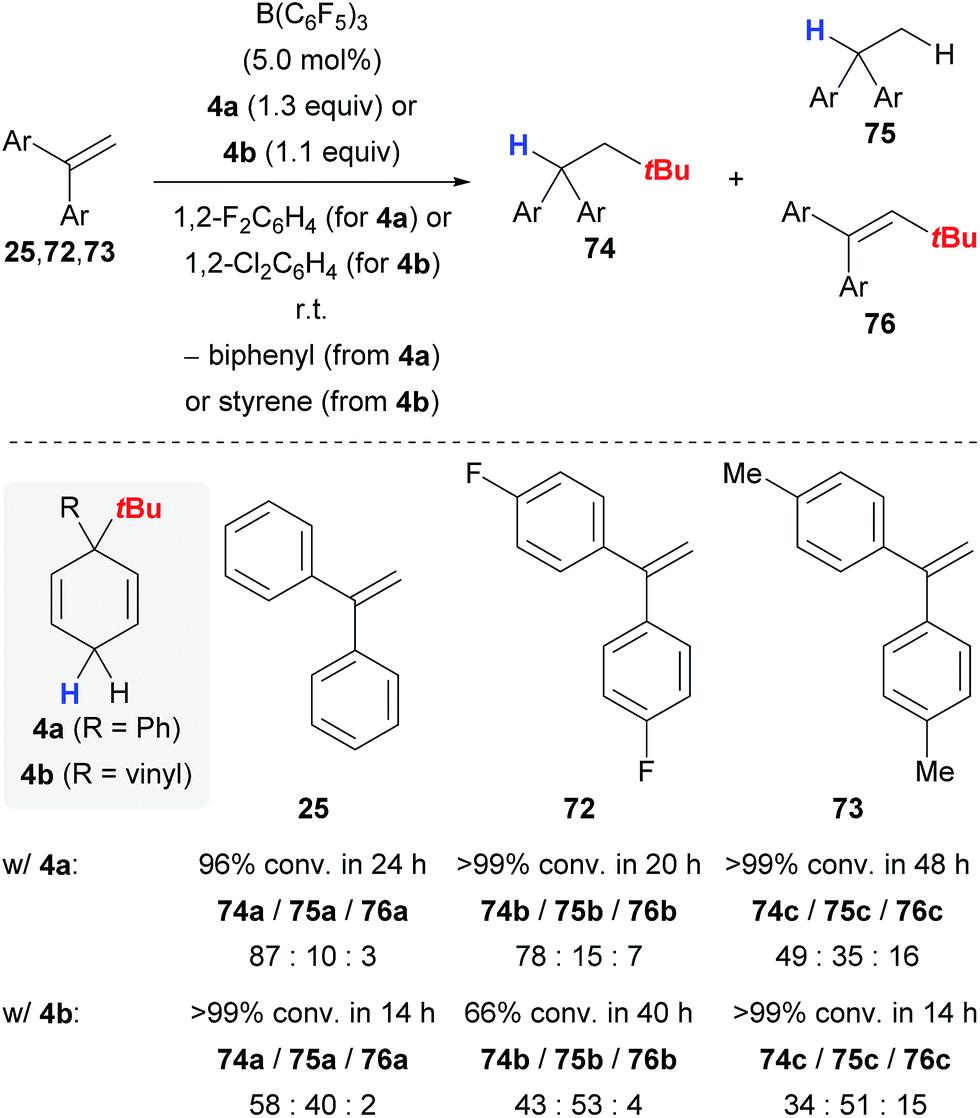 | ||
| Scheme 9 Transfer hydro-tert-butylation of 1,1-diarylalkenes. | ||
The proposed catalytic cycle that rationalises the pathways for byproduct formation commences with the B(C6F5)3-triggered abstraction of a bisallylic hydride from surrogate 4 to furnish Wheland complex XV+[HB(C6F5)3]− (Scheme 10), followed by transfer of either the tert-butyl cation (XV+ → XVII+, left cycle) or a distal proton (XV+ → XVIII+, right cycle) to alkene XVI; stoichiometric liberation of gaseous isobutene likely accounts for the latter pathway. The carbenium ion in XVIII+ is eventually reduced by borohydride [HB(C6F5)3]− to afford byproduct 75, thereby closing the catalytic cycle. Likewise, intermediate XVII+ can either directly collapse and form the desired alkane 74 or first transfer a proton from the β position in XVII+ to another molecule of alkene XVI and form byproducts 75 and 76 after hydride transfer from [HB(C6F5)3]− (grey pathway).
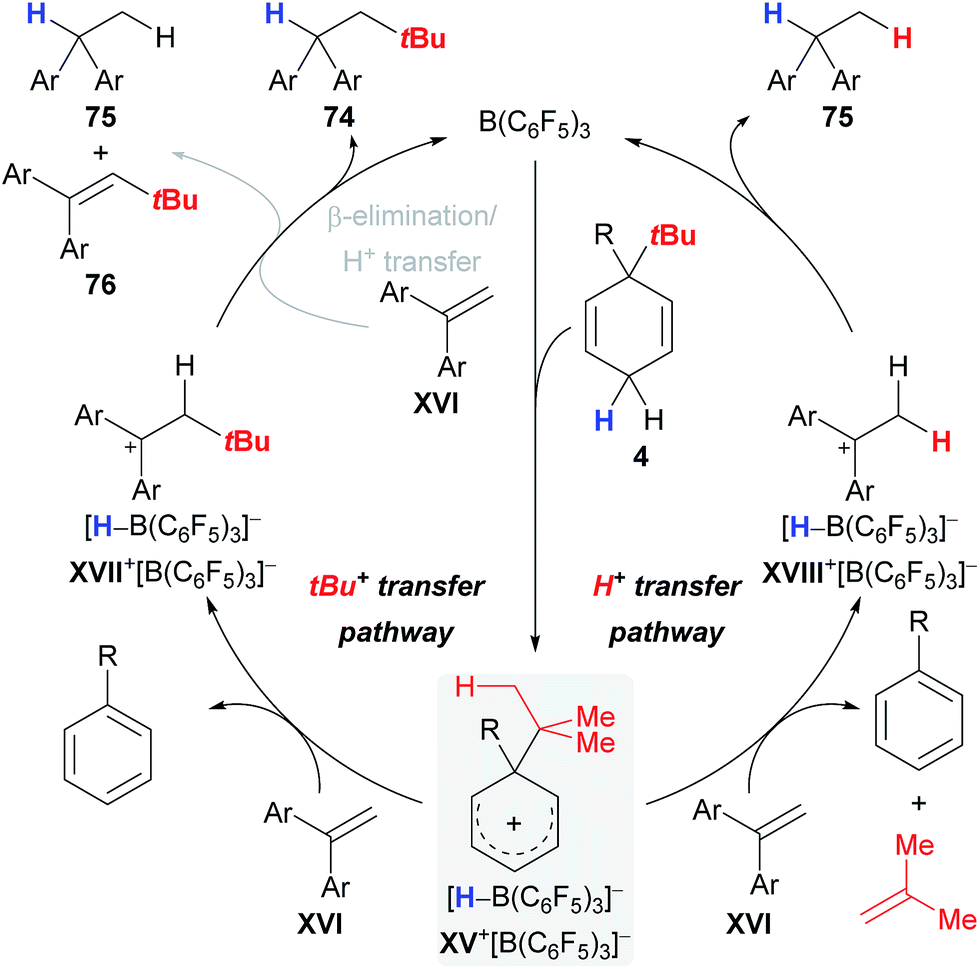 | ||
| Scheme 10 Proposed catalytic cycle for the transfer hydro-tert-butylation. | ||
Outlook
The recent advances in transition-metal-free ionic transfer processes using substituted cyclohexa-1,4-dienes as transfer reagents hint its great promise. While still at the early stages of development, we believe that these transformations are about to emerge as useful synthetic tools. Particularly, unleashing small reactive molecules such as SiH4 from cyclohexa-1,4-dienes by straightforward treatment with a Lewis-acid catalyst could also prove valuable for inorganic chemists.Transfer hydrosilylation is feasible for several π- and σ-donors with a variety of hydrosilane surrogates, particularly of SiH4 and (EtO)3SiH. Lack of chemoselectivity and, hence, functional-group tolerance is the obvious limitation of this method. That problem is less pronounced in the related transfer hydrogermylation. Issues in the transfer hydrogenation such as hetero- and homodimerisation of the reactants have been successfully addressed by judicious choice of the substituents at cyclohexa-1,4-diene core. The substrate scope for both CN and CC bonds is however still relatively narrow. The transfer of a tert-butyl group is currently the biggest challenge. While it represents promising precedence for the transfer of carbon electrofuges, the surrogate synthesis still remains unsolved. The design of new (short) synthetic routes and extension to other stabilised carbenium ions as departing groups will hopefully allow for more efficient transfer hydroalkylation reactions in the future.
On the basis of the knowledge gained from these efforts, we will continue improving the existing procedures and devise new El+/H− equivalents. We also hope that our findings serve as an inspiration for others in the field.
Acknowledgements
This research was supported by the Deutsche Forschungsgemeinschaft (Oe 249/11-1). Parts of the findings summarised herein were funded by the Humboldt Foundation and the Cluster of Excellence “Unifying Concepts in Catalysis” of the Deutsche Forschungsgemeinschaft (EXC 314/2). M. O. is indebted to the Einstein Foundation (Berlin) for an endowed professorship. M. O. thanks Dr Antoine Simonneau and Dr Indranil Chatterjee for their enthusiasm and commitment.References
- For a review, see: D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- For recent overviews of transition-metal-catalysed transfer processes, see: (a) B. N. Bhawal and B. Morandi, Chem.–Eur. J., 2017, 23 DOI:10.1002/chem.201605325; (b) B. N. Bhawal and B. Morandi, ACS Catal., 2016, 6, 7528–7535 CrossRef CAS.
- For a review of transfer hydrosilylation, see: (a) M. Oestreich, Angew. Chem., Int. Ed., 2016, 55, 494–499 CrossRef CAS PubMed; (b) A. Simonneau and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 11905–11907 CrossRef CAS PubMed; (c) A. Simonneau, J. Friebel and M. Oestreich, Eur. J. Org. Chem., 2014, 2077–2083 CrossRef CAS; (d) S. Keess, A. Simonneau and M. Oestreich, Organometallics, 2015, 34, 790–799 CrossRef CAS; (e) A. Simonneau and M. Oestreich, Nat. Chem., 2015, 7, 816–822 CrossRef CAS PubMed.
- S. Keess and M. Oestreich, Org. Lett., 2017, 19, 1898–1901 CrossRef CAS PubMed.
- (a) I. Chatterjee and M. Oestreich, Angew. Chem., Int. Ed., 2015, 54, 1965–1968 CrossRef CAS PubMed; (b) I. Chatterjee, Z.-W. Qu, S. Grimme and M. Oestreich, Angew. Chem., Int. Ed., 2015, 54, 12158–12162 CrossRef CAS PubMed; (c) I. Chatterjee and M. Oestreich, Org. Lett., 2016, 18, 2463–2466 CrossRef CAS PubMed.
- S. Keess and M. Oestreich, Chem.–Eur. J., 2017, 23, 5925–5928 CrossRef CAS PubMed.
- For the related borane-mediated hydride abstraction from 1,4-dihydropyridines, see: (a) J. D. Webb, V. S. Laberge, S. J. Geier, D. W. Stephan and C. M. Crudden, Chem.–Eur. J., 2010, 16, 4895–4902 CrossRef CAS PubMed; (b) D. V. Gutsulyak, A. van der Est and G. I. Nikonov, Angew. Chem., Int. Ed., 2011, 50, 1384–1387 CrossRef CAS PubMed; (c) G. Hamasaka, H. Tsuji and Y. Uozumi, Synlett, 2015, 26, 2037–2041 CrossRef CAS.
- The molecular structure of a hydrosilane–borane adduct was reported by Piers, Tuononen and co-workers. See: A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- For the quantum-chemical analysis of the transfer hydrosilylation, see: K. Sakata and H. Fujimoto, Organometallics, 2015, 34, 236–241 CrossRef CAS.
- For a review of Si–H and H–H bond activation catalysed by B(C6F5)3, see: M. Oestreich, J. Hermeke and J. Mohr, Chem. Soc. Rev., 2015, 44, 2202–2220 RSC.
- For the hydride donor abilities of various main-group hydrides, see: Z. M. Heiden and A. P. Lathem, Organometallics, 2015, 34, 1818–1827 CrossRef CAS.
- For the preparation of donor-stabilised silylium ions from cyclohexa-2,5-dien-1-yl-substituted silanes, see: A. Simonneau, T. Biberger and M. Oestreich, Organometallics, 2015, 34, 3927–3929 CrossRef CAS.
- (a) B. Michelet, C. Bour and V. Gandon, Chem.–Eur. J., 2014, 20, 14488–14492 CrossRef CAS PubMed; (b) B. Michelet, J.-R. Colard-Itté, G. Thiery, R. Guillot, C. Bour and V. Gandon, Chem. Commun., 2015, 51, 7401–7404 RSC; (c) B. Michelet, S. Tang, G. Thiery, J. Monot, H. Li, R. Guillot, C. Bour and V. Gandon, Org. Chem. Front., 2016, 3, 1603–1613 RSC.
- The concept of transfer hydrosilylation from silicon-substituted cyclohexa-1,4-dienes based on a radical mechanism was introduced by Studer and co-workers. See: (a) S. Amrein, A. Timmermann and A. Studer, Org. Lett., 2001, 3, 2357–2360 CrossRef CAS PubMed; (b) S. Amrein and A. Studer, Chem. Commun., 2002, 1592–1593 RSC; (c) S. Amrein and A. Studer, Helv. Chim. Acta, 2002, 85, 3559–3574 CrossRef CAS.
- I. Buslov, S. C. Keller and X. Hu, Org. Lett., 2016, 18, 1928–1931 CrossRef CAS PubMed.
- For seminal reports on the B(C6F5)3-catalysed direct hydrosilylation/hydrogermylation of π-basic substrates, see: (a) M. Rubin, T. Schwier and V. Gevorgyan, J. Org. Chem., 2002, 67, 1936–1940 CrossRef CAS PubMed (hydrosilylation); (b) T. Schwier and V. Gevorgyan, Org. Lett., 2005, 7, 5191–5194 CrossRef CAS PubMed (hydrogermylation).
- (a) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS; (b) D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed; (c) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed; (d) K. Sakata and H. Fujimoto, J. Org. Chem., 2013, 78, 12505–12512 CrossRef CAS PubMed.
- For the related transfer silylation of alcohols, see: ref. 3c.
- K.-i. Ikeshita, N. Kihara, M. Sonoda and A. Ogawa, Tetrahedron Lett., 2007, 48, 3025–3028 CrossRef CAS.
- For transition-metal-catalysed transfer hydrogenations with cyclohexa-1,4-dienes, see: (a) R. P. Linstead, E. A. Braude, P. W. D. Mitchell, K. R. H. Wooldridge and L. M. Jackman, Nature, 1952, 169, 100–103 CrossRef CAS; (b) A. M. Felix, E. P. Heimer, T. J. Lambros, C. Tzougraki and J. Meienhofer, J. Org. Chem., 1978, 43, 4194–4196 CrossRef CAS; (c) P. J. McDermott and R. A. Stockman, Org. Lett., 2005, 7, 27–29 CrossRef CAS PubMed; (d) O. Verho, A. Nagendiran, E. V. Johnston, C.-w. Tai and J.-E. Bäckvall, ChemCatChem, 2013, 5, 612–618 CrossRef CAS; (e) O. Verho, A. Nagendiran, C.-w. Tai, E. V. Johnston and J. E. Bäckvall, ChemCatChem, 2014, 6, 205–211 CrossRef CAS; (f) S. M. King, X. Ma and S. B. Herzon, J. Am. Chem. Soc., 2014, 136, 6884–6887 CrossRef CAS PubMed; (g) A. Lefranc, Z.-W. Qu, S. Grimme and M. Oestreich, Chem.–Eur. J., 2016, 22, 10009–10016 CrossRef CAS PubMed.
- For a radical-based transfer hydrogenation of alkenes using cyclohexa-1,4-diene, see: M. K. Eberhardt, Tetrahedron, 1967, 23, 3029–3031 CrossRef CAS.
- For the cooperative activation of dihydrogen by B(C6F5)3 and imines, see: (a) P. A. Chase, T. Jurca and D. W. Stephan, Chem. Commun., 2008, 1701–1703 RSC; (b) D. Chen and J. Klankermayer, Chem. Commun., 2008, 2130–2131 RSC; (c) T. A. Rokob, A. Hamza, A. Stirling and I. Pápai, J. Am. Chem. Soc., 2009, 131, 2029–2036 CrossRef CAS PubMed; (d) Z. M. Heiden and D. W. Stephan, Chem. Commun., 2011, 47, 5729–5731 RSC.
- For leading reviews on Brønsted acid-catalysed transfer hydrogenations, see: (a) C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC; (b) S. G. Ouellet, A. M. Walji and D. W. C. MacMillan, Acc. Chem. Res., 2007, 40, 1327–1339 CrossRef CAS PubMed.
- Examples of the related hydro-tert-alkylation of alkenes are exceedingly rare. See: (a) D. W. K. Yeung and J. Warkentin, Can. J. Chem., 1976, 54, 1345–1348 CrossRef CAS; (b) U. Biermann and J. O. Metzger, J. Am. Chem. Soc., 2004, 126, 10319–10330 CrossRef CAS PubMed . For a review of Friedel-Crafts-type alkene alkylation, see: ; (c) H. Mayr, Angew. Chem., Int. Ed. Engl., 1990, 29, 1371–1384 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |