
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optical control of GPR40 signalling in pancreatic β-cells†
James Allen
Frank
 a,
Dmytro A.
Yushchenko
bc,
Nicholas H. F.
Fine
def,
Margherita
Duca
ag,
Mevlut
Citir
b,
Johannes
Broichhagen
ah,
David J.
Hodson
*def,
Carsten
Schultz
*bi and
Dirk
Trauner
*aj
a,
Dmytro A.
Yushchenko
bc,
Nicholas H. F.
Fine
def,
Margherita
Duca
ag,
Mevlut
Citir
b,
Johannes
Broichhagen
ah,
David J.
Hodson
*def,
Carsten
Schultz
*bi and
Dirk
Trauner
*aj
aDepartment of Chemistry, Center for Integrated Protein Science, Ludwig Maximilians University Munich, Butenandtstraße 5-13, 81377 Munich, Germany
bEuropean Molecular Biology Laboratory (EMBL), Cell Biology & Biophysics Unit, Meyerhofstraße 1, 69117 Heidelberg, Germany. E-mail: schultz@embl.de
cInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Flemingovo namesti 2, 16610 Prague 6, Czech Republic
dInstitute of Metabolism and Systems Research (IMSR), University of Birmingham, Birmingham, B15 2TT, UK. E-mail: d.hodson@bham.ac.uk
eCentre for Endocrinology, Diabetes and Metabolism, Birmingham Health Partners, Birmingham, B15 2TH, UK
fCOMPARE University of Birmingham and University of Nottingham Midlands, UK
gDepartment of Chemistry, University of Milan, Via Golgi 19, 20133, Milan, Italy
hMax-Planck Institute of Medical Research, Jahnstr. 29, 69120 Heidelberg, Germany
iDept. of Physiology and Pharmacology, Oregon Health and Science University, Portland, OR 97237, USA
jDepartment of Chemistry, New York University, 100 Washington Square East, New York, NY 10003-6699, USA. E-mail: dirktrauner@nyu.edu
First published on 30th August 2017
Abstract
Fatty acids activate GPR40 and K+ channels to modulate β-cell function. Herein, we describe the design and synthesis of FAAzo-10, a light-controllable GPR40 agonist based on Gw-9508. FAAzo-10 is a potent GPR40 agonist in the trans-configuration and can be inactivated on isomerization to cis with UV-A light. Irradiation with blue light reverses this effect, allowing FAAzo-10 activity to be cycled ON and OFF with a high degree of spatiotemporal precision. In dissociated primary mouse β-cells, FAAzo-10 also inactivates voltage-activated and ATP-sensitive K+ channels, and allows us to control glucose-stimulated Ca2+ oscillations in whole islets with light. As such, FAAzo-10 is a useful tool to study the complex effects, with high specificity, which FA-derivatives such as Gw-9508 exert at multiple targets in mouse β-cells.
Introduction
Although minimalistic in structure and often viewed as subunits of more complex lipids or simply an energy source, fatty acids can have profound effects on cell signalling.1–4 Free fatty acids most often consist of a long, unbranched carbon chain attached to a carboxyl headgroup, which is largely deprotonated and thus negatively charged at physiological pH.5 They are amphiphilic molecules with diverse structures that vary in the chain length and the level of unsaturation. A number of transmembrane signalling proteins, including G protein-coupled receptors (GPCRs) such as GPR40,6 are stimulated by free fatty acids,7 resulting in a rise in the intracellular Ca2+ concentration ([Ca2+]i) in insulin-secreting pancreatic β-cells through activation of phospholipase C.8–10 Given the role of GPR40 in glucose homeostasis, synthetic agonists for these receptors such as Gw-9508 (ref. 11 and 12) and TAK-875 (ref. 13 and 14) have received significant attention as potential treatments for type 2 diabetes mellitus.15,16 However, a phase III clinical trial for TAK-875 was recently terminated due to off-target effects and toxicity concerns.17,18Glucose-stimulated insulin secretion (GSIS) relies on transport of glucose into the β-cell, followed by its metabolism to ATP. The resulting increase in the ATP/ADP ratio leads to closure of ATP-sensitive K+ channels (KATP) and subsequent membrane depolarization. This causes the opening of voltage activated L-type Ca2+ channels (Cav) and an increase in [Ca2+]i, driving exocytosis of insulin secretory granules.19 Subsequent activation of delayed rectifier voltage-activated K+ (Kv) channels leads to repolarization of the membrane, reduced Ca2+ entry through Cav channels and termination of insulin secretion (Fig. 1).20 This is complemented by the action of other messengers, including those stemming from GPCRs (so-called “amplifying” signals). Notably, the amplifying effects of GPR40 activation on insulin secretion remain elusive due to conflicting results in different experimental conditions,12,21 which could be attributed to effects of FAs at different targets. For example, fatty acids are known to directly affect various K+ channels that are involved in modulation of the [Ca2+]i oscillation frequency,1,22,23 demonstrating their complex pharmacology and vital role in β-cell signalling. Therefore, a tool that could enable precise control over GPR40 signalling may be useful to better understand the effects of fatty acids, as well as specific agonists, on β- and other cell functions. This could lead to the development of novel therapeutics by delineating the receptor conformations required for biased signalling.18,24
 | ||
| Fig. 1 Glucose-stimulated insulin secretion (GSIS) from pancreatic β-cells. Upon uptake into the pancreatic β-cell, glucose is metabolized into ATP. The rising ATP/ADP ratio inhibits KATP which causes membrane depolarization and the opening of Cav. The resulting increased [Ca2+]i triggers the fusion of secretory granules and the release of insulin. Kv channels work to repolarize the cell, generating oscillations in [Ca2+]i. GPR40 stimulation also leads to increased [Ca2+]i, further potentiating GSIS. | ||
Previous studies in our laboratories have focused on the development of photoswitchable sulfonylureas and incretins, with which we could place pancreatic β-cell function under the precise spatiotemporal control of light.25–29 We also showed that photoswitchable diacylglycerols30–32 affect β-cell [Ca2+]i and insulin secretion. These diacylglycerols were constructed from a photoswitchable fatty acid (FAAzo) chain, however the pharmacology of these FAAzos alone remains largely unexplored. Given the sensitivity of GPR40 to unsaturated, and sometimes aryl-containing free fatty acid-like molecules, we hypothesized that the FAAzos themselves could enable optical control of this GPCR. Herein, we describe a novel approach towards the optical control of fatty acid/GPR40 signalling in β-cells.
Results and discussion
Although GPR40 is activated by long-chain fatty acids such as arachidonic or linoleic acid,10 various aryl-containing carboxylic acids such as Gw-9508 are known to produce a similar effect (Fig. 2a).3 We recognized that the benzyl-aniline moiety of Gw-9508 could be easily substituted by a phenyl diazene, and would afford a photoswitchable ligand with little disturbance to the overall size and structure of the drug. Therefore, we synthesized the azologue33 of Gw-9508, FAAzo-10, using the Mills reaction after nitroso formation in two steps and 45% overall yield (Fig. 2b). Similar to the other members of the FAAzo family,30FAAzo-10 behaved as a regular azobenzene and could be isomerized between its thermally stable trans-form to the cis-form with UV-A light (Fig. 2c). The process could be reversed by irradiation with blue light, and photoswitching could be repeated over many cycles. | ||
| Fig. 2 Design and synthesis of photoswitchable GPR40 agonists. (a) The chemical structures of Gw-9508, AA and FAAzo-4. (b) Chemical synthesis of FAAzo-10, a photoswitchable derivative of Gw-9508. (c) The UV-Vis spectra of FAAzo-10 in its dark-adapted (black), UV-adapted (gray) and blue-adapted (blue) states (20 μM in PBS). | ||
We then characterized the effects of FAAzo-10 on GPR40 in HeLa cells using confocal fluorescence microscopy and the genetically encoded fluorescent [Ca2+]i reporter R-GECO.34 When transiently transfected with GPR40, a portion of cells displayed spontaneous [Ca2+]i oscillations without the addition of any external stimuli (Fig. 3a and S1a†). Gw-9508 induced a GPR40-dependent increase in the rate and intensity of [Ca2+]i oscillations, that was not affected by UV-A-irradiation (Fig. 3b and S1b†). In cells without GPR40, no response was observed (Fig. S1c and d†). Complementary to this result, the application of trans-FAAzo-10 (200 nM) stimulated a significant increase in [Ca2+]i in HeLa cells expressing GPR40 (Fig. 3c and d). On isomerization to cis with 375 nm irradiation, a sharp decrease in the [Ca2+]i was observed. The effect was reversed and [Ca2+]i increased on termination of the irradiation. In cells lacking GPR40, FAAzo-10 did not affect [Ca2+]i (Fig. 3e and S1e†). We also evaluated the effect of FAAzo-4, which possesses a similar structure to FAAzo-10, but was not active at this low concentration (Fig. 3f). Histamine35 (HIS, 10 μM) was used as a positive control and triggered a large increase in [Ca2+]i, independent of GPR40 expression (Fig. 3 and S1†).
 | ||
| Fig. 3 FAAzos enable optical control of GPR40 in HeLa cells expressing GPR40, the diacylglycerol sensor C1-GFP and the genetically encoded [Ca2+]i sensor R-GECO. (a) Spontaneous oscillations of [Ca2+]i were observed before addition of any compound. (b) Gw-9508 (200 nM) caused an increase in [Ca2+]i that was not affected by 375 nm irradiation. HIS (10 nM) application caused an increase in [Ca2+]i (n = 179 cells from two experiments). (c, d) trans-FAAzo-10 (200 nM) increased [Ca2+]i, and isomerization to cis-FAAzo-10 with 375 nm light reversed this effect. Displayed as (c) individual [Ca2+]i traces from representative cells and (d) the average [Ca2+]i for many cells (n = 157 cells from two experiments). (e) In cells not expressing GPR40, FAAzo-10 (200 nM) did not affect [Ca2+]i. (f) At 200 nM, FAAzo-4 (n = 211 cells from two experiments) did not affect [Ca2+]i when compared to FAAzo-10 (n = 153 cells from two experiments). (g) C1-GFP translocated to the plasma membrane alongside an increase in [Ca2+]i when stimulated by trans-FAAzo-10 (20 μM, n = 10 cells from one representative experiment). Translocation (green) is displayed as the plasma membrane to cytoplasm (PM/CP) C1-GFP fluorescence intensity ratio. (h) Quantification of cell entry using fluorescence quenching of coumaryl-AA-loaded (100 nM) HeLa cells after application (100 nM, 2 experiments each) of FAAzo-4 (n = 29 cells, orange), FAAzo-10 (n = 23 cells, green) and PhoDAG-1 (n = 39 cells, grey), respectively. Error bars were calculated as ±s.e.m. | ||
To investigate the downstream effects of GPR40 activation, we expressed the fluorescent diacylglycerol reporter C1-GFP, which translocates to the plasma membrane in response to increased diacylglycerol levels following PLC activation.36 Gw-9508 (200 nM) triggered C1-GFP translocation towards the plasma membrane, indicating activation of the GPCR (Fig. S1f and g†). On application of trans-FAAzo-10 (20 μM), we observed a similar effect on C1-GFP translocation. This could be reversed following isomerization to cis-FAAzo-10 with 375 nm irradiation, and translocation could be repeated over many cycles (Fig. 3g). These results demonstrate that oscillations in GPR40 activity and its downstream effectors (i.e. PLC, [Ca2+]i and diacylglycerols) can be modulated with good temporal control.
Surprisingly, the effects induced by the FAAzos in HeLa cells did not diminish over time (Fig. 3), unlike those induced by the photoswitchable diacylglycerol PhoDAG-1, which decreased in magnitude over multiple UV-A pulses of the same length.31 To control for differences in cell loading, we applied the coumarinyl-ester of AA (cg-AA) to the HeLa cells.6 This fluorescent fatty acid-derivative localizes predominantly at the inner cellular membranes.31 By monitoring the quenching of coumarin fluorescence by the azobenzene of FAAzos, we demonstrated that this observed variance in activity was not due to variable FAAzo uptake by cells. Application of both FAAzos caused a rapid and large (>60%) decrease in coumarin fluorescence (Fig. 3h), especially when compared to the quenching effect of PhoDAG-1 (<20%), which is known to remain trapped on the outer plasma membrane.31 A cellular lipid analysis by thin layer chromatography (TLC) confirmed only minor FAAzo metabolism in cells incubated with FAAzo-4 and FAAzo-10 (100 μM) for up to 1 h (Fig. S2†). Together, these results demonstrate that the FAAzos are quickly taken up into cells, and only minimally metabolized over the timeframe of a typical imaging experiment.
A major advantage of FAAzo-10 when compared to conventional agonists is the ability to modulate GPR40 activity with increased spatial precision. By illuminating only cells of interest, we were able to selectively control GPR40 activity without affecting signalling in neighbouring unilluminated cells (Fig. 4). This allows GPR40 activity to be controlled in a spatially defined manner in large patches of cells or complex tissues.
 | ||
| Fig. 4 Spatial control of GPR40 signalling with FAAzo-10. (a) Confocal images of HeLa cells expressing GPR40 and R-GECO before and after treatment with FAAzo-10 (200 nM) and illumination with 375 nm light. The green rectangle indicates the area of illumination. After addition of FAAzo-10, all transfected cells showed increased [Ca2+]i. Following illumination, only cells within the green rectangle showed a sharp decrease in [Ca2+]i levels, which recovered after termination of illumination. Scale bar = 100 μm. (b) Normalized [Ca2+]i in illuminated cells (within the green rectangular in (a)) in blue (n = 52) and those in unilluminated cells (outside the green rectangular) in black (n = 82). Time points 1–4 correspond to the respective time frames in (a). Error bars were calculated as ±s.e.m. | ||
To evaluate the effects of FAAzo-10 on K+ channels, we used whole-cell electrophysiology in dissociated mouse β-cells, which express both Kv and KATP channels.37,38 Kv channel conductance is a major determinant of the [Ca2+]i oscillation frequency.20 Like AA31 and Gw-9508 (Fig. 5a), trans-FAAzo-10 reduced Kv channel conductance in the dark or under blue irradiation (Fig. 5b). On isomerization to cis-FAAzo-10, Kv channel activity was restored to a level comparable with the vehicle controls (Fig. 5a). FAAzo-10 could be switched ON and OFF repeatedly, effectively allowing us to quickly mimic the wash-in and wash-out of Gw-9508 using only a UV-A/blue irradiation (Fig. 5c). Furthermore, we could also fine-tune the effect of FAAzo-10 with greater precision by scanning through different irradiation wavelengths. The Kv conductance could be precisely controlled by gradually increasing the blocking effect of FAAzo-10 when scanning from UV-A to blue wavelengths. This was demonstrated by applying voltage ramps under 350–450 nm irradiation (Fig. 5d and e).
 | ||
| Fig. 5 Optical control of β-cell Kv channel activity. The whole-cell Kv channel current in dissociated wt mouse β-cells was measured using patch clamp electrophysiology. (a) An IV-plot showed that Gw-9508 (50 μM) (n = 8 cells from 2 animals) reduced the Kv conductance when compared to a vehicle control (n = 6 cells from 3 animals). (b) Under blue light, trans-FAAzo-10 (20 μM) reduced the whole-cell Kv current. Isomerization to cis-FAAzo-10 with UV-A light reversed this effect (n = 7 cells from 3 animals). (c) Similar to the wash-in and wash-out of Gw-9508, FAAzo-10 could be activated and inactivated over several cycles using irradiation. Shown are IV-steps from −70 to +80 mV from representative cells. (d, e) An action spectrum between 350–450 nm showed that Kv activity could be fine-tuned by changing the irradiation wavelength. Displayed as (d) overlaid sequential voltage ramps (−70 to +80 mV) from a representative cell and (e) the normalized current (to I350 nm) under each wavelength (n = 3 cells from 2 animals). Error bars were calculated as ±s.e.m. | ||
Gw-9508 has also been shown to potentiate KATP channels in mouse β-cells.11 We measured the whole-cell KATP current without extracellular glucose. IV-curves were measured between −110 and −50 mV to exclude any effect of the Kv channels. After dialysis of the cytoplasm with intracellular buffer to reduce the ATP/ADP ratio, the KATP current increased to a steady state (Fig. 6a and S3a†). In line with previous reports, Gw-9508 increased the KATP conductance further (Fig. 6a and c).
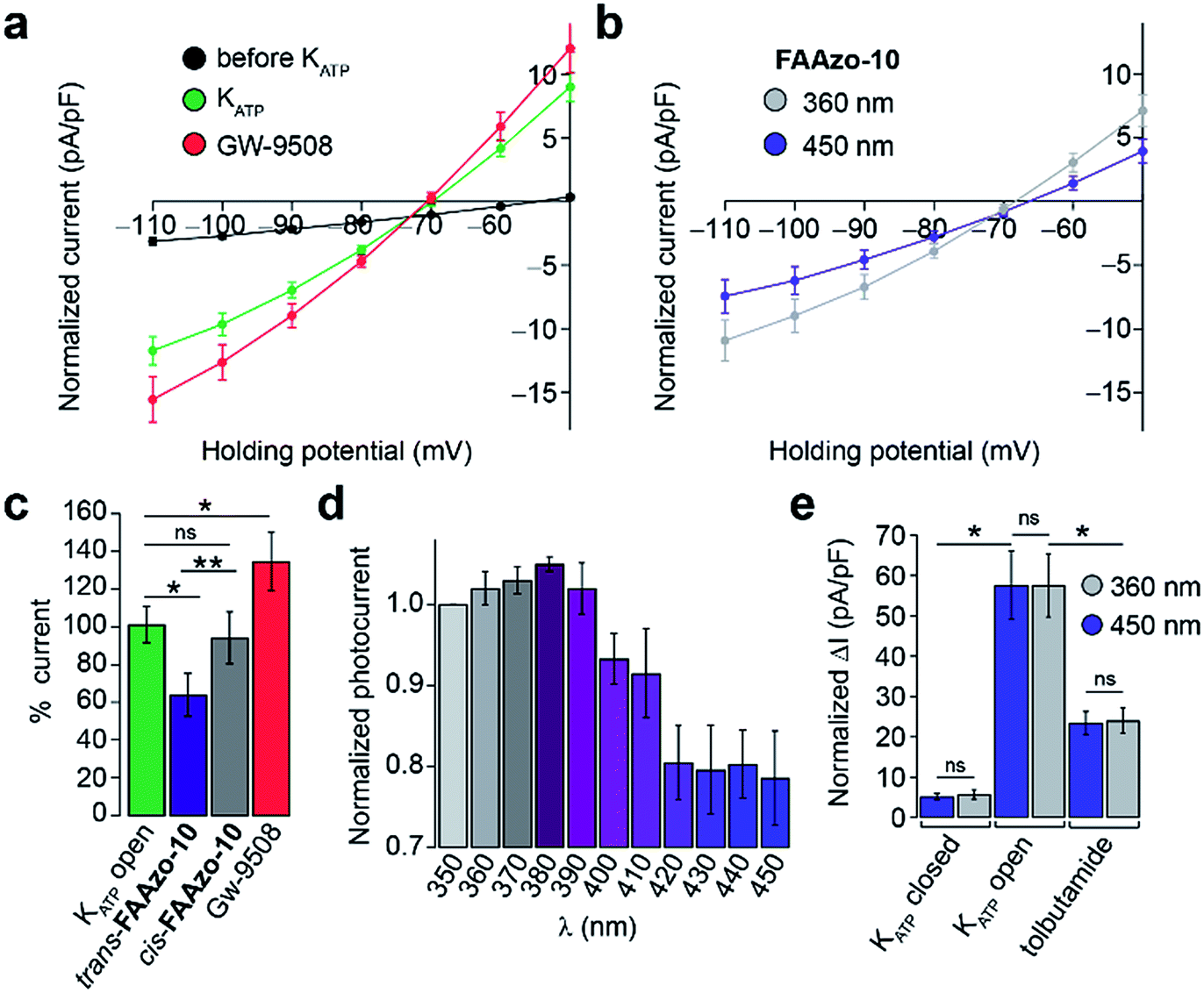 | ||
| Fig. 6 Optical control of β-cell KATP channels. The whole-cell KATP current from dissociated mouse β-cells was measured between −110 to −50 mV. (a–c) After dialysis of the cytoplasm with the pipette solution, the KATP current developed to a steady state (black = before, n = 21; green = after, n = 20 cells from 2 animals). Application of Gw-9508 (20 μM, red, n = 9 cells from 2 animals) increased KATP conductance. In contrast, the application of trans-FAAzo-10 (20 μM, blue) decreased the KATP current, while isomerization to cis-FAAzo-10 (gray) reversed this effect (n = 7 cells from 2 animals). Data is displayed as (a, b) the full IV relationship between −110 to −50 mV and (c) the % KATP current (at −110 mV) for multiple cells, normalized to the KATP open (green) state. (d) In the presence of FAAzo-10, an action spectrum between 350–450 nm revealed that KATP was inhibited the most under blue irradiation. Irradiation with UV-A light prevented FAAzo-10 from blocking the KATP current. Displayed as the normalized current (to I350 nm) under each wavelength (n = 3 cells from one animal). (e) UV-A or blue irradiation alone did not affect the KATP current, and tolbutamide (40 μM) significantly reduced the magnitude of the KATP current (ΔI from −110 to −50 mV, n = 3 cells from one animal). ns = P > 0.05, *P < 0.05, **P < 0.01. Error bars were calculated as ±s.e.m. | ||
Interestingly, trans-FAAzo-10 behaved differently, and reduced the KATP conductance, while isomerization to cis-FAAzo-10 reversed the effect (Fig. 6b). Similar to the effects observed on Kv channels, FAAzo-10 activity at KATP could be fine-tuned by altering the irradiation wavelength (Fig. 6d). Under blue irradiation, the KATP current was reduced, while the blockade was reversed towards UV-A wavelengths. In control experiments, application of the sulfonylurea tolbutamide reduced the KATP current significantly (Fig. 6e and S3a†), and neither UV-A nor blue irradiation alone affected the KATP conductance (Fig. 6e and S3†).
Finally, we evaluated our photoswitchable ligands for their effects on intact pancreatic islets using confocal fluorescence imaging. We employed the fluorescent small-molecule [Ca2+]i indicator Fluo-8 to monitor [Ca2+]i oscillations stimulated by a high glucose concentration (11 mM). Similar to the application of Gw-9508 (Fig. 7a and b), application of trans-FAAzo-10 (20 μM) caused a marked increase in the [Ca2+]i oscillation frequency (Fig. 7c and d). In line with the effects that would be expected from our results on GPR40, Kv, and KATP, isomerization to cis-FAAzo-10 with 365 nm irradiation reversed this effect entirely (Fig. 7c–f). Lower concentrations of FAAzo-10 (2.5 μM) did not affect oscillation frequency in either configuration (Fig. 7f). To exclude imaging artifacts, in particular fluorescence quenching, the cells were treated with a methyl ester FAAzo-derivative, FAAzo-5(OMe), which possesses an azobenzene photoswitch with similar spectral characteristics to FAAzo-10.30FAAzo-5(OMe) produced a small increase in the [Ca2+]i oscillation frequency in either configuration (Fig. S4a–c†), as methyl esterification of the acid group abolished cis-activity. Although FAAzo-10 effectively increased [Ca2+]i oscillations, we did not observe a significant increase in insulin secretion in either trans or cis at both low (3 mM) and high (11 mM) glucose concentrations (Fig. 7g). Similarly, benchmark Gw-9508 did not stimulate GSIS at 3 mM or 11 mM glucose (Fig. 7g). An effect of BSA on Gw-9508 and/or FAAzo-10 potency was unlikely, since assays with low (3 mM) glucose concentration but performed in the absence of the carrier were identical (data not shown). Experiments were also repeated at high (17 mM) glucose, but without BSA, showing a similar lack of stimulation with Gw-9508 or FAAzo-10 (Fig. S4d†). Neither FAAzo-10 nor Gw-9508 were able to suppress tolbutamide-stimulated insulin secretion, further supporting an effect on KATP channel conductance (Fig. S4e†).11 UV-A irradiation alone did not affect oscillatory behavior or insulin secretion levels, as expected from previous studies25,31 (Fig. S4f†).
 | ||
| Fig. 7 FAAzo-10 enables optical control of [Ca2+]i oscillations in pancreatic islets. [Ca2+]i oscillations were stimulated by a high glucose concentration (11 mM, G11) and monitored in intact mouse islets using the fluorescent [Ca2+]i indicator Fluo-8. (a, b) The application of Gw-9508 (50 μM) caused an increase in the [Ca2+]i oscillation frequency. Displayed as (a) a representative trace from a single islet and (b) the oscillation frequency averaged over multiple islets (n = 6 recordings). (c, d) The application of trans-FAAzo-10 (20 μM) also caused a marked increase in the oscillation frequency. Isomerization to cis-FAAzo-10 with 365 nm irradiation reversed this effect. Results are displayed as (c) a representative trace from a single islet and (d) the average oscillation frequency from multiple islets (n = 5 recordings). (e, f) FAAzo-10 enabled optical control of β-cell [Ca2+]i oscillations at 20 μM, but not at 2.5 μM (n = 4–5 recordings) (representative images cropped to show a single islet; scale bar = 25 μm). (g) FAAzo-10 (20 μM) did not afford a consistent effect on GSIS (3 mM glucose, G3). Gw-9508 (20 μM) also did not affect GSIS (n = 3–8 assays using islets from at least 3 animals) (* denotes significance between G3 and G11). Grey lines are raw traces (to show frequency effects), black lines are smoothed traces (to show amplitude effects). *P < 0.05 and **P < 0.01, ANOVA, with repeated measures as necessary. Error bars were calculated as ±s.e.m. | ||
Conclusions
In summary, we have demonstrated that FAAzo-10 is a potent photoswitchable agonist of GPR40, and reversibly inactivates K+ channels in dissociated mouse β-cells. Although our previous studies using the FAAzos conjugated to different headgroups afforded cis-active compounds,30,31 we found the opposite in this case. FAAzo-10 was more active in the trans-form at all targets, and can reversibly stimulate [Ca2+]i oscillations in pancreatic β-cells using light. Interestingly, stimulation of [Ca2+]i oscillations with FAAzo-10 did not translate to increased insulin secretion in primary mouse islets, in line with the effects of the benchmark drug, Gw-9508. This suggests that oscillations by themselves are potentially not a sufficient signal for effective granule fusion, and that an additional factor was not triggered under these conditions. Of note, previous studies using Gw-9508 have afforded either stimulatory, inhibitory or no effect on insulin secretion,11,21,39 with two conflicting reports in mouse islets.12,40 As previously alluded to using the PhoDAGs,31 the variation of the effects induced by Gw-9508 application may stem from different protein expression levels or membrane area between immortalized and primary cells, or conversely off-target effects on GPR120, which shares some homology with GPR40. Similarly, differential effects caused by plasma membrane vs. intracellular fatty acid-signalling, as was observed using caged AA-derivatives, may contribute to this effect.6 By contrast, long chain fatty acids such as linoleic and palmitic acid have been consistently shown to potently stimulate insulin secretion, and this can be abrogated by GPR40 knockdown/silencing.18 Our studies thus reinforce the notion that signals in addition to GPR40 activation may be required for fatty-acid-stimulated insulin release, highlighting the complexity of fatty acid signalling in the β-cell, and underscoring the importance of FAAzo-10 for studying the intricate relationship between [Ca2+]i oscillations and insulin secretion. More broadly, FAAzo-10 opens up the possibility to precisely interrogate the contribution of GPR40 signalling in different body compartments (e.g. brain and liver) to glucose homeostasis.Live subject statement
All studies were regulated by the Home Office per the Animals (Scientific Procedures) Act 1986 of the United Kingdom (PPL P2abc3a83), and study approval granted by the Animal Welfare and Ethical Review Body of the University of Birmingham.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We acknowledge financial support by the Deutsche Forschungsgemeinschaft (TRR 152 to D. T., J. A. F., J. B. and TRR 83 and TRR 186 to C. S.). We are grateful for the technical support of EMBL's Advanced Light Microscopy Facility. D. A. Y. was supported by an IOCB installation grant. D. J. H. was supported by Diabetes UK R.D. Lawrence (12/0004431), EFSD/Novo Nordisk Rising Star and Birmingham Fellowships, and Wellcome Trust Institutional Support and MRC Project (MR/N00275X/1) Awards. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Starting Grant 715884 to D. J. H. and Advanced Grant 268795 to D. T.). We thank Prof. Guy Rutter (Imperial College London) for allowing access to imaging equipment for pilot studies.References
- H. Meves, Br. J. Pharmacol., 2008, 155, 4–16 CrossRef CAS PubMed.
- N. Salem, B. Litman, H. Kim and K. Gawrisch, Lipids, 2001, 36, 945–959 CrossRef CAS PubMed.
- G. Milligan, B. Shimpukade, T. Ulven and B. D. Hudson, Chem. Rev., 2017, 117(1), 67–110 CrossRef CAS PubMed.
- L. M. Boland and M. M. Drzewiecki, Cell Biochem. Biophys., 2008, 52, 59–84 CrossRef CAS PubMed.
- IUPAC Gold Book – Fatty Acids, ed. A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997 Search PubMed.
- A. Nadler, D. A. Yushchenko, R. Müller, F. Stein, S. Feng, C. Mulle, M. Carta and C. Schultz, Nat. Commun., 2015, 6, 10056 CrossRef CAS PubMed.
- Y. Itoh, Y. Kawamata and M. Harada, Nature, 2003, 422, 173–176 CrossRef CAS PubMed.
- H. Shapiro, S. Shachar, I. Sekler, M. Hershfinkel and M. D. Walker, Biochem. Biophys. Res. Commun., 2005, 335, 97–104 CrossRef CAS PubMed.
- S. Schnell, M. Schaefer and C. Sch, Mol. Cell. Endocrinol., 2007, 263, 173–180 CrossRef CAS PubMed.
- C. P. Briscoe, M. Tadayyon, J. L. Andrews, W. G. Benson, J. K. Chambers, M. M. Eilert, C. Ellis, N. A. Elshourbagy, A. S. Goetz, D. T. Minnick, P. R. Murdock, H. R. Sauls, U. Shabon, L. D. Spinage, J. C. Strum, P. G. Szekeres, K. B. Tan, J. M. Way, D. M. Ignar, S. Wilson and A. I. Muir, J. Biol. Chem., 2003, 278, 11303–11311 CrossRef CAS PubMed.
- Y. Zhao, L. Wang, D. Zha, L. Qiao, L. Lu, J. Yu and P. Qu, J. Mol. Endocrinol., 2013, 51, 69–77 CrossRef CAS PubMed.
- C. P. Briscoe, A. J. Peat, S. C. Mckeown, D. F. Corbett, A. S. Goetz, T. R. Littleton, D. C. Mccoy, T. P. Kenakin, J. L. Andrews, C. Ammala, J. A. Fornwald, D. M. Ignar and S. Jenkinson, Br. J. Pharmacol., 2006, 148, 619–628 CrossRef CAS PubMed.
- C. Yabuki, H. Komatsu, Y. Tsujihata, R. Maeda, R. Ito, K. Matsuda, K. Sakuma, K. Miyawaki, N. Kikuchi, K. Takeuchi and Y. Habata, PLoS One, 2013, 8, 2–11 Search PubMed.
- N. Negoro, S. Sasaki, S. Mikami, M. Ito, M. Suzuki, Y. Tsujihata, R. Ito, A. Harada, K. Takeuchi, N. Suzuki, J. Miyazaki, T. Santou, T. Odani, N. Kanzaki, M. Funami, T. Tanaka, A. Kogame, S. Matsunaga, T. Yasuma and Y. Momose, ACS Med. Chem. Lett., 2010, 1, 290–294 CrossRef CAS PubMed.
- X. Feng, J. Leng, Z. Xie, S. Li, W. E. I. Zhao and Q. Tang, Int. J. Mol. Med., 2012, 30, 1261–1266 CrossRef CAS PubMed.
- T. Hara, A. Hirasawa, A. Ichimura, I. Kimura and G. Tsujimoto, J. Pharm. Sci., 2011, 100, 3594–3601 CrossRef CAS PubMed.
- K. Kaku, K. Enya, R. Nakaya, T. Ohira and R. Matsuno, Diabetes, Obes. Metab., 2015, 7, 675–681 CrossRef PubMed.
- A. D. Mancini and V. Poitout, Diabetes, Obes. Metab., 2015, 17, 622–629 CrossRef CAS PubMed.
- G. A. Rutter, T. J. Pullen, D. J. Hodson and A. Martinez-Sanchez, Biochem. J., 2015, 466, 203–218 CrossRef CAS PubMed.
- D. A. Jacobson, A. Kuznetsov, J. P. Lopez, S. Kash, C. E. Ammala and L. H. Philipson, Cell Metab., 2007, 6, 229–235 CrossRef CAS PubMed.
- H. Ou, H. Wu, H. Hung, Y. Yang, J. Wu and C. Chang, Am. J. Physiol. Endocrinol. Metabol., 2013, 67, 668–676 CrossRef PubMed.
- H. Meves, Prog. Neurobiol., 1994, 43, 175–186 CrossRef CAS PubMed.
- K. L. Hamilton, C. A. Syme and D. C. Devor, J. Biol. Chem., 2003, 278, 16690–16697 CrossRef CAS PubMed.
- M. Rendell, Drugs, 2004, 64, 1339–1358 CrossRef CAS PubMed.
- J. Broichhagen, J. a. Frank, N. R. Johnston, R. K. Mitchell, K. Šmid, P. Marchetti, M. Bugliani, G. a. Rutter, D. Trauner and D. J. Hodson, Chem. Commun., 2015, 51, 6018–6021 RSC.
- J. Broichhagen, S. C. Cork, J. A. Frank, P. Marchetti, J. Broichhagen, M. Scho, M. Bugliani, A. M. J. Shapiro, S. Trapp, G. A. Rutter, D. J. Hodson and D. Trauner, Nat. Commun., 2014, 5, 6116 Search PubMed.
- N. R. Johnston, R. K. Mitchell, E. Haythorne, M. P. Pessoa, F. Semplici, J. Ferrer, L. Piemonti, P. Marchetti, M. Bugliani, D. Bosco, E. Berishvili, P. Duncanson, M. Watkinson, D. Trauner, G. A. Rutter and D. J. Hodson, Cell Metab., 2016, 24, 389–401 CrossRef CAS PubMed.
- J. Broichhagen, N. R. Johnston, Y. Von Ohlen, H. Meyer-berg, B. J. Jones, S. R. Bloom, G. A. Rutter, D. Trauner and D. J. Hodson, Angew. Chem., Int. Ed., 2016, 55, 5865–5868 CrossRef CAS PubMed.
- J. Broichhagen, T. Podewin, H. Meyer-berg, Y. Von Ohlen, N. R. Johnston, B. J. Jones, S. R. Bloom, G. A. Rutter, A. Hoffmann-roeder, D. J. Hodson and D. Trauner, Angew. Chem., Int. Ed., 2015, 54, 15565–15569 CrossRef CAS PubMed.
- J. A. Frank, M. Moroni, R. Moshourab, M. Sumser, G. R. Lewin and D. Trauner, Nat. Commun., 2015, 6, 7118 CrossRef PubMed.
- J. A. Frank, D. A. Yuschenko, D. J. Hodson, N. Lipstein, J. Nagpal, G. A. Rutter, J.-S. Rhee, A. Gottschalk, N. Brose, C. Schultz and D. Trauner, Nat. Chem. Biol., 2016, 12, 755–762 CrossRef CAS PubMed.
- J. A. Frank, H. G. Franquelim, P. Schwille and D. Trauner, J. Am. Chem. Soc., 2016, 138, 12891–12896 Search PubMed.
- J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48, 1947–1960 CrossRef CAS PubMed.
- Y. Zhao, S. Araki, J. Wu, T. Teramoto, Y.-F. Chang, M. Nakano, A. S. Abdelfattah, M. Fujiwara, T. Ishihara, T. Nagai and R. E. Campbell, Science, 2011, 557, 1888–1891 CrossRef PubMed.
- M. Bootman, E. Niggli, M. Berridge and P. Lipp, J. Physiol., 1997, 499, 307–314 CrossRef CAS.
- E. Oancea, M. N. Teruel, A. F. G. Quest and T. Meyer, J. Cell Biol., 1998, 140, 485–498 CrossRef CAS PubMed.
- D. A. Jacobson, C. R. Weber, S. Bao, J. Turk and L. H. Philipson, J. Biol. Chem., 2007, 282, 7442–7449 CrossRef CAS PubMed.
- G. A. Rutter, Mol. Aspects Med., 2001, 22, 247–284 CrossRef CAS PubMed.
- N. Yaluri, S. Modi, M. L. Rodríguez and A. Stan, PLoS One, 2015, 10, e0142902 Search PubMed.
- B. M. Moran, Y. H. A. Abdel-Wahab, P. R. Flatt and A. M. Mckillop, Diabetes, Obes. Metab., 2014, 16, 1128–1139 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc01475a |
| This journal is © The Royal Society of Chemistry 2017 |