DOI:
10.1039/C7SC01416C
(Edge Article)
Chem. Sci., 2017,
8, 5119-5125
Kinase-templated abiotic reaction†
Received
30th March 2017
, Accepted 20th May 2017
First published on 24th May 2017
Abstract
Protein kinases are quintessential regulators of cellular function. Numerous pathologies are intimately linked to the dysregulated activity of a particular protein kinase. Herein we report a technology based on a proximity-induced chemical transformation that enables the detection and imaging of specific kinases. Using two probes that target the nucleotide-binding site and substrate binding site of a target kinase respectively, the reagents appended on the probes are brought within reactive distance thereby enabling the chemical transformation. The reaction used for sensing is a ruthenium-photocatalyzed reduction of a pyridinium immolative linker, which uncages a fluorophore (rhodamine). We demonstrate that this technology can be used to discriminate between closely related kinases with a high signal to noise ratio. We further demonstrate that the technology operates within the complexity of a cellular context with a good correlation between the level of kinase activity and fluorescence output.
Introduction
Protein kinases (PKs) stand out as important regulators in post-translational control of protein activity.1 PKs exert their function by phosphorylating serine/threonine or tyrosine residues in target proteins which results in a change of activity in the phosphorylated protein. With 518 PKs encoded in the human genome, they account for one of the largest protein families.2 PKs are responsible for the phosphorylation of at least one third of the proteome, endowing them with a central role in regulating biochemical pathways. Not surprisingly, numerous pathologies ranging from cancers to chronic inflammation arise from an overexpression or constitutive activation of PKs and this protein family has been the subject of numerous drug discovery efforts.3–5 In parallel, technologies to sense and image kinase activity in cells for biomedical research have been developed. These technologies can be categorized into three groups: genetically encoded biosensors6,7 which are important for cellular biology but unsuitable for diagnostic applications; fluorogenic peptide substrates that respond to phosphorylation with a 2–5 fold change in fluorescence;8–12 and small molecule kinase inhibitors conjugated to a fluorophore; albeit it is generally challenging to achieve selectivity for a single kinase.13–15 Notwithstanding these important achievements, there remains a need for technologies that can deliver a kinase-specific response with high signal to noise ratio and good kinase-specific selectivity. Herein we report a novel strategy that combines the specificity of two ligands targeting the substrate-binding site and the nucleotide-binding site of a kinase, respectively, to yield a proximity-induced reaction (Fig. 1). Proximity-induced chemical reactions have proven to be a powerful technology for nucleic acid sensing.16–18 Nucleic acid probes conjugated to reagents are brought in close proximity following a sequence-specific hybridization; the proximity of the reagents enables a reaction that changes the fluorescent properties of a reporter. Such reactions have been shown to operate in living cells as well as in live vertebrate (zebra fish).19,20 More recently, this concept of proximity induced reaction has been used with ligands targeting oligomeric proteins,21 or peptides,22,23 but has not been applied to bi-substrate enzymes such as kinases. It is important to note that prior examples of templated chemistry made use of probes that bind the template with high affinity.
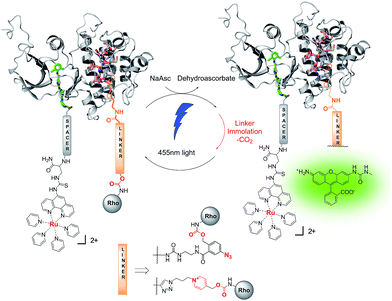 |
| | Fig. 1 Schematic representation of kinase-templated reaction. Using a nucleotide-binding site ligand derivatized with the ruthenium photocatalyst (Ru(bpy)2phen) and a substrate-binding site ligand conjugated to a caged rhodamine via a pyridinium linker, the close proximity of the reagents achieved upon binding accelerates the photo-reduction of the pyridinium resulting in the uncaging of rhodamine. | |
Results and discussion
We sought to develop kinase templated reactions that could selectively respond to Abl and Src. Abl and Src are two cytoplasmic tyrosine kinases which are involved in many cellular processes and signalling events by multiple protein complexes. Their aberrant activation results in oncogenic transformation and drives tumour growth. The fusion oncoprotein BCR-Abl, created by translocation of the Abl kinase from chromosome 9 with a breakpoint cluster region protein on chromosome 22, gives rise to the Philadelphia chromosome.24,25 Formation of the chimeric oncogenic kinase BCR-Abl results in constitutive Abl activity, which drives tumorigenesis26 and has been showed to be the main driver of chronic myelogenous leukemia (CML) and a subset of B-cell acute lymphoblastic leukemias. Importantly, BCR-Abl is the target of the first approved kinase inhibitor imatinib in 2001. Src and its eight human paralogue kinases are important proto-oncogenes. Increased Src family kinase activity is observed and involved in the development of various tumor types and in progression of metastasis in different human cancers.27,28 Due to the crucial role played by these kinases in cancer, they have attracted tremendous attention in inhibitor development. To date there are no small molecule ligands that uniquely binds to either kinase. For instance, imatinib, a type II inhibitor that binds the inactive conformation of Abl, is known to also bind KIT, PDGFR and a few other kinases;29 dasatinib,30,31 a type I inhibitor that binds to the active conformation of the kinase, is known to also bind to Src and many other kinases. Dasatinib has a low nM affinity for Abl and Src.32,33 While some level of PK promiscuity is pharmacologically acceptable or even therapeutically important, it does not allow for the discrimination of a single kinase for sensing purposes. Src and Abl share an identical fold with 62% sequence homology in the nucleotide-binding sites. While no inhibitor targeting the nucleotide-binding site of kinase is known to give selectivity for a unique kinase, we reasoned that the combined selectivity of nucleotide binding and substrate binding in a kinases-templated reaction would refine the selectivity of sensing. To this end, we targeted the nucleotide-binding site with a conjugate of dasatinib, which binds to the active conformation of the kinase and leaves the substrate-binding site open for interaction with a peptidic substrate.34 The inhibitor has been co-crystallized with the active form of both Src and Abl35,36 (Fig. 2A), the amino-thiazole moiety occupies the ATP binding site, while the piperazine moiety is solvent exposed and can be modified, extending in proximity of the N-terminus of a peptide substrate (Fig. 2B). Both peptide substrates were taken from optimal sequences identified from peptide libraries screens, measuring peptide phosphorylation for each kinase.34 The reported affinities are 4 μM for the Abl peptide and 30 μM for the Src peptide respectively.
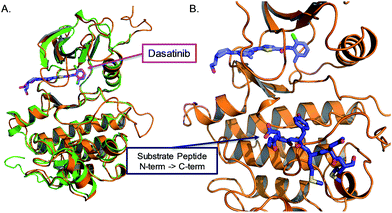 |
| | Fig. 2 Crystal structure of Abl and Src kinase domain. (A) Superposition of Abl and Src kinase domain (PDB ID: 2GQG and 3G5D) with dasatinib bound in the nucleotide-binding site. (B) Structure of Abl-kinase domain with dasatinib bound in the nucleotide-binding site and a peptide bound in the substrate-binding site (from the co-crystal structure of active insulin receptor kinase with a substrate peptide PDB ID: 1IR3). | |
We began our investigations using the ruthenium-photocatalyzed reduction of an azide-based immolative linker that had been applied to nucleic acid sensing both in human cells37 and in live vertebrate.20 This chemistry has two assets over other bioorthogonal reactions: first, the fact that light is required for the reaction provides temporal control in the sensor readout; second, the potential signal amplification by virtue of the ruthenium-catalysis (product yields in excess of the ruthenium-conjugate probe). Based on the higher affinity of dasatinib for the kinase (low nM) relatively to peptide substrate (low μM), we reasoned it would be preferable to conjugate the ruthenium catalyst to dasatinib and the immolative linker to the peptide substrate (Fig. 1) to maximize signal amplification through substrate turnover. However, no combination of probes afforded templated reaction (vide infra). More recently, we have shown that a pyridinium-based immolative linker was reduced 200-fold faster than the azide-based immolative linker.38 We speculated that the failure of the kinase-template reaction with the azide based immolative linker was due to the fast dissociation (koff) of the peptide substrate and that this reaction might benefit from the faster kinetics of the pyridinium-based linker. Fluorescence measurements of the pyridinium caged rhodamine prior and after photo-uncaging shows a 200-fold increase in fluorescence (Fig. 3A) which clearly surpasses the performance of fluorogenic substrate sensors.
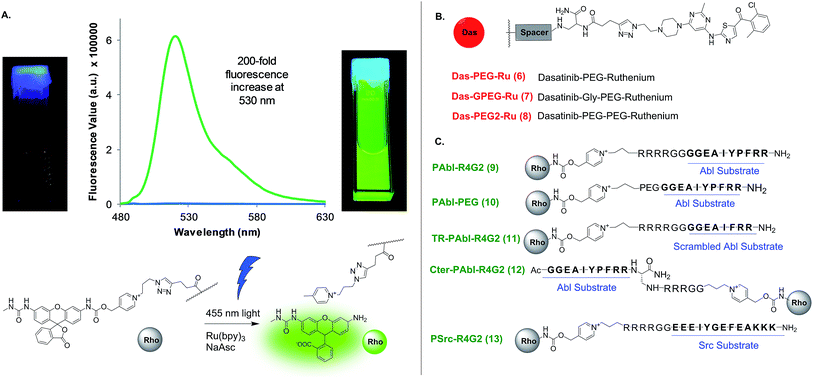 |
| | Fig. 3 Uncaging of rhodamine and structure of ligand conjugates used. (A) Emission of rhodamine-conjugate probe prior and after uncaging. (B) Structure of probes targeting the nucleotide-binding site. See Fig. S1† for explicit structures. (C) Structure of probes targeting the substrate-binding sites. | |
Optimization of Abl-templated photo-uncaging
In order to test and optimize the Abl-templated reaction and identify the optimal linker, we conjugated the ruthenium catalyst to dasatinib via three different linker lengths which included a single 9 atoms PEG linker (DAS-PEG-Ru 6, Fig. 2B), an additional glycine residue (DAS-GPEG-Ru 7) or an additional PEG linker (DAS-PEG2-Ru 8). The substrate for Abl, a 10-mer peptide was conjugated to the pyridinium linker via tetra-arginine bis-glycine linker (PAbl-R4G29) or a 9 atom PEG linker (PAbl-PEG 10). In addition, two control substrates were prepared; a scrambled peptide wherein the tyrosine residue is substituted with a phenylalanine (TR-PAbl-R4G211); the Abl substrate with the pyridinium linker at the C-terminus (Cter-PAbl-R4G212). Both controls should perform poorly in Abl-templated chemistry due to lower affinity for the scrambled peptide or misalignment of the reagent for the C-terminal conjugate. As initial conditions for the reaction, we used recombinant Abl kinase domain at 100 nM with 1 equivalent of the dasatinib conjugate with an excess of substrate conjugate (10 equivalents, 1 μM) to assess substrate turnover and signal amplification. Measuring the increase of fluorescence as a function of time reports on the progress of the reaction. Using a calibration curve of the rhodamine fluorophore, the reaction yield was calculated and shown in Fig. 4. Gratifyingly, the best combination of probes (PAbl-R4G29 with Das-GPEG-Ru 7) gave nearly 55% conversion in 40 min while the same reaction with carbonic anhydrase (CA) gave less than 3% yield (see Fig. S2† for prolonged measurement times). Comparing the initial rate of reaction, the Abl templated reaction was 18 times faster than the unspecific reaction with CA. Other linker combinations also provided Abl specific reactions albeit at lower rate suggesting that the PAbl-R4G29 with Das-GPEG-Ru 7 combination provides the best proximity of the reagents (Fig. 4B). The same reaction with the azide-based linker failed to deliver a fluorescence increase. It should be noted that under these conditions, a 5.5-fold excess of fluorophore is generated relative to the kinase using the pyridinium linker. As a control for the Abl-specific templated reaction, we next tested the optimal reagent combination (PAbl-R4G29 with Das-GPEG-Ru 7) in the presence of an excess (10 μM) of dasatinib (Das) resulting in a four-fold reduction in the initial rate of the reaction, reflecting the inhibition of the reaction due to competition for Abl-binding. The fact that a complete shutdown of the reaction is not observed likely results from the dynamic exchange of dasatinib 3 with Ru-conjugated dasatinib 7 in the nucleotide-binding site. Reaction of the optimal dasatinib ligand (Das-GPEG 7) with the scrambled peptide (TR-PAbl-R4G211) or the C-terminal conjugate (Cter-PAbl-R4G212) afforded reaction rates close to the one of the control with CA. Finally, reaction of the optimal dasatinib ligand (Das-GPEG 7) with Ru(bpy)31 showed the same low reactivity with Abl as with CA (Fig. 4C). Collectively, this data strongly supports the fact that the photocatalytic reduction of the immolative linker is accelerated by a favorable proximity of the conjugates that interact specifically with Abl. We next assessed the detection threshold of this Abl-templated reaction. To this end, we first used the same concentrations of reagents (Das-GPEG-Ru 7: 100 nM; PAbl-R4G29: 1 μM) and titrated down the Abl concentration from 100 nM to 0 nM. As shown in Fig. 4D, a statistically significant difference in signal was obtained down to 1 nM of Abl. We next measured the number of turnovers achieved (signal amplification) at different Abl concentrations keeping a stoichiometric quantity of the Das-GPEG-Ru 7 reagent with respect to the Abl concentration while keeping the PAbl-R4G29 substrate concentration at 1 μM (Fig. 4E). A 4–6-fold signal amplification was observed for concentrations ranging from 100 nM to 12 nM and began tapering off to 3-fold at the lowest tested protein concentration (6 nM).
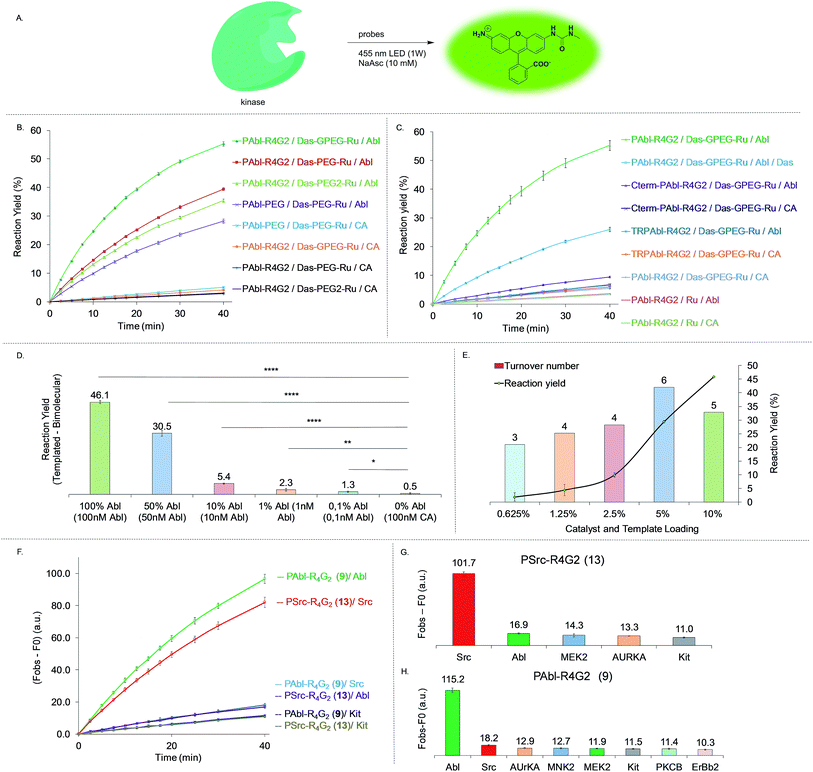 |
| | Fig. 4 Kinase-templated reaction. (A) Schematic representation of reactions performed in (B)–(H). (B) Abl-templated reaction using 100 nM of Abl, 100 nM of different dasatinib conjugates and 1 μM of different substrate conjugates. (C) Same conditions as in (B) with the addition of unconjugated dasatinib (Das) or different controls using C-terminal conjugated ligand, scrambled substrate ligand, control protein (carbonic anhydrase: CA), unconjugated photocatalyst Ru(bpy)3. (D) Titration of signal obtained with 100 to 0 nM of Abl. The experiment with 0 nM of Abl was performed with a control protein (CA) using 100 nM of dasatinib conjugate and 1 μM of substrate conjugate. (E) Signal amplification of the kinase-templated reaction through turnover of the substrate at varying kinase loading using stoichiometric dasatinib conjugate and 1 μM of substrate conjugate. (F) Selectivity of the kinase-templated reaction. Time curves of probes (dasatinib conjugate (50 nM) + PAbl substrate or PSrc substrate (1 μM)) reacting in the presence of Abl, Src and KIT (50 nM). (G) End-point measurement (40 min) for the reaction of dasatinib conjugate (50 nM) + PSrc substrate (1 μM) reacting in the presence of a panel of kinases. (H) End-point measurement (40 min) for the reaction of dasatinib conjugate (50 nM) + PAbl substrate (1 μM) reacting in the presence of a panel of kinases. (B)–(H): (1) = Ru(bpy)3Cl2; (3) = Das-N3; (6) = Das-PEG-Ru; (7) = Das-GPEG-Ru; (8) = Das-PEG2-Ru; (9) = PAbl-R4G2; (10) = PAbl-PEG; (11) = TR-PAbl-R4G2, (12) = Cterm-PAbl-R4G2. | |
Specificity of kinase-mediated photo-uncaging
Using the same design as for Abl, we prepared a substrate for Src conjugated at the N-terminus with the immolative linker-rhodamine adduct (PSrc-R4G213, Fig. 3C). We next tested the orthogonality of the reaction using the Abl specific substrate with Abl and Src, and likewise, the Src specific substrate for Abl and Src. As shown in Fig. 4F, we indeed observed a specific response in the reaction using the cognate kinase. Specifically, the Abl-templated reaction using the P-Abl substrate 9 was 7-fold faster than the same reaction with P-Src substrate 13. Conversely, the Src-templated reaction was 6-fold faster with P-Src substrate 13 than P-Abl substrate 9 (Fig. 4F). We next investigated the specificity of the reaction using a broader panel of PKs. To this end, we selected PKs with a range of affinities for dasatinib (Abl: 0.1 nM; Src: 0.21 nM; KIT: 0.81 nM; MEK2: 1.4 μM; ErBb2: 1.4 μM; AURKA: 9.8 μM; MNK2 and PKCB: >10 μM). As shown in Fig. 4G for the P-Src substrate and in Fig. 4H for the P-Abl substrate, the templated reaction was specific for the targeted kinase.
Imaging BCR-Abl in a cellular context (K562 cell line)
Having established the dynamic range and selectivity of Abl-templated reaction biochemically, we turned our attention to whole cell imaging. We initiated our study with K562 cells, a commonly used chronic myelogenous leukemia cell line, expressing the constitutively active fusion protein BCR-Abl. The cells were treated at two different concentrations of reagents after PFA fixation, namely 5 μM of peptide 9 and 1 μM of dasatinib 7 (Fig. 5A) or 1 μM of peptide 9 and 0.2 μM of dasatinib probe 7 (Fig. 4B). The cells were imaged after 30 min irradiation under 455 nm LED (1 W). We repeated the same controls as the ones performed for the biochemical characterization, namely using the scrambled peptide 11, unconjugated ruthenium photocatalyst (Ru(bpy)31) and in addition, the ruthenium catalyst conjugated to 2-aminoglucose 2. For both sets of concentrations, a notably stronger fluorescence signal was observed with the functional probes than with the controls (4- to 18-fold higher signal relative to the different controls at the higher reagent concentration, Fig. 5A; 3- to 6-fold higher signal relative to the different controls at the lower reagent concentration, Fig. 5B).
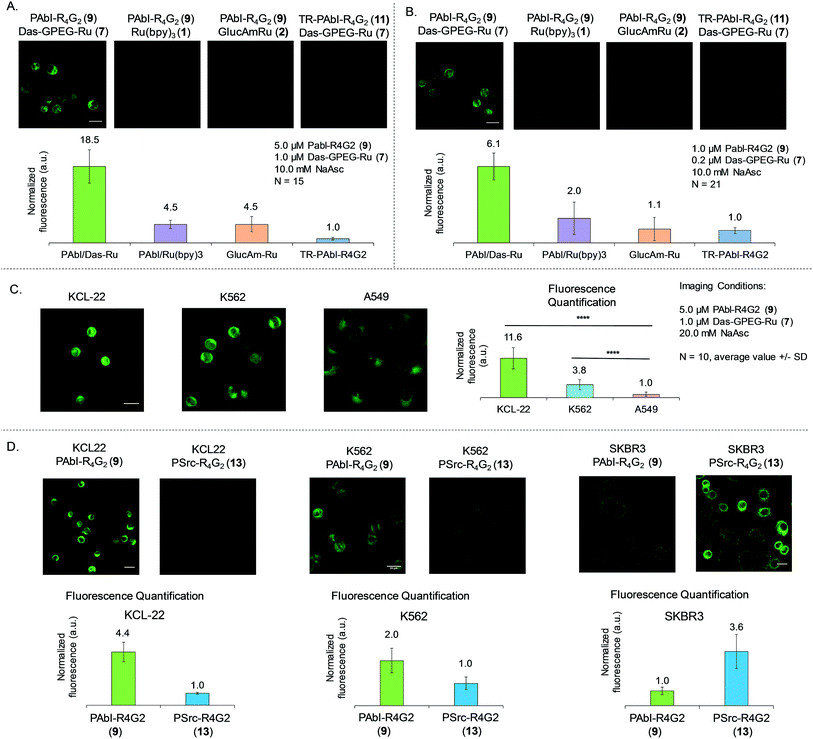 |
| | Fig. 5 Whole cell imaging using kinase-templated reaction (A) fixed K562 Cell Abl imaging with probes and negative controls. Reaction conditions displayed on figure. Quantification displayed with average values and standard deviation with N = 15. Scalebar = 20 μm. (B) Same experiment as in (A) using lower concentrations of probes. Quantification displayed with average values and standard deviation with N = 21. Scalebar = 20 μm. (C) BCR-Abl expression quantification in cancer cell lines. All images taken with same microscope settings for comparison purposes. Images displayed and quantification done by sum projection of Z-stack. Scalebar = 20 μm. (D) Specificity of templated reaction in cellulo. Reaction conditions used: peptide probe 5 μM, dasatinib probe 1 μM and sodium ascorbate 20 mM. Each pair of images were taken using the same microscope settings. Images and quantification realized on average projection of Z-stack, average values with standard deviation displayed with N = 10. Scalebar = 20 μm. | |
BCR-Abl expression quantification in different cancer cell lines (KCL-22, K562 and A549 cells)
We next asked if the templated reaction could quantitatively report on the level of BCR-Abl across different cell lines using K562, KCL-22 (another CML cell line expressing c.a. twice as much BCR-Abl as K562),39 A549 (a lung cancer cell line with WT Abl-1). We envisioned that using our probes and the same settings to image each cell line, we should be able to quantify the level of Abl expression by fluorescence microscopy. Indeed, comparing the images obtained for these 3 cell lines (Fig. 5C), we could observe a 12-fold difference between KCL-22 and A-549. In the case of the two different CML cell lines, quantification of the fluorescence shows a 3-fold difference between the two cell lines. Our system allows the quantification of Abl by fluorescence readout with results similar to the ones obtained by western blot39 or miRNA quantification.40
Specificity of Abl and Src templated reaction in cellulo (KCL-22, K562 and SKBR3 cells)
We next investigated the ability of our system to discriminate between Abl and Src in a cellular context. For that purpose, we used 3 different cell lines, KCL-22 and K562 expressing high and moderate levels of BCR-Abl and low levels of Src and SKBR3 a Src-driven breast cancer cell line with no BCR-Abl and low levels WT-Abl-1. For the CML cancer cell lines, KCL-22 and K562, the reaction with Abl probes (7 and 9) shows respectively 4.4 and 2-fold more fluorescence than the reaction with Src probes (7 and 13). On the contrary, in the Src driven SKBR3 line, the reaction using Src probes (7 and 13) gave a 3.6-fold difference with the reaction using the Abl probes (7 and 9). Collectively, the results demonstrate that the designed templated reactions discriminate between Src and Abl in different cell lines with fluorescence intensities correlating to the kinase function.
Experimental section
Synthetic procedures for all compounds are described in the ESI.†
Kinase-templated reactions in vitro
Template reactions were carried out in opaque 96 well plates in 100 mM TBS (Tris Buffer Saline 150 mM NaCl) + 0.05% Tween 20 buffer and 20 mM of sodium ascorbate. Stock solutions of probes were made in 10% DMSO in mQ water at 1 mM and stored either at −20 °C or in aliquots at −80 °C. A stock solution of sodium ascorbate was made in water at 1 M. Stock solutions were diluted with reaction buffer to the desired concentration and used in the experiment. Each experiment was performed in triplicate. After measurement of initial fluorescence, the plate was irradiated using a 455 nm collimated LED lamp (http://www.thorlabs.com, 1 W, 30 cm distance) and the fluorescence of rhodamine (exc = 490 nm; em = 530 nm) was measured at different time points. The reactions were carried out in 200 μL volume at two different reagent concentrations: at higher concentration with 1 μM of peptide-rhodamine probe and 100 nM of dasatinib–ruthenium probe and 100 nM of protein; at lower concentration using 500 nM of peptide-rhodamine probe and 100 nM of dasatinib–ruthenium probe and 50 nM of template protein. The raw data was treated by subtracting the initial fluorescence value for each conditions and averaging the values of triplicate experiment while calculating a standard deviation. A picture of the LED setup is available in the ESI† in the template reaction section.
Cellular imaging of kinase-templated reaction
Adherent cell lines, A549 and SKBR3, were seeded in 35 mm glass bottom dishes and grown for 24 h. Cells were then washed with DPBS and fixed for 20 min with 4% PFA. For K562, the cells were seeded in 35 mm glass bottom dishes, coated with poly-L-lysine and grown for 24 h. Cells were then washed with DPBS and fixed with 4% PFA for 20 min. For KCL-22, cells were seeded in 35 mm glass bottom dishes coated with poly-L-lysine and gently centrifuged (300 rpm, 3 min, rt). The cells were then fixed with 4% PFA for 20 min.
Peptide and dasatinib conjugates were added to the cells in DPBS at the concentrations indicated and incubated at 37 °C for 2 h. Sodium ascorbate (20 mM) was added and the cells were further incubated at 37 °C for 1 h. The media was aspirated and replaced with 2 mL of DPBS containing 20 mM sodium ascorbate. The cells were then irradiated for 30 min with a 455 nm collimated LED lamp. After irradiation the cells were imaged using confocal microscopy. Images were acquired using a Zeiss LSM780 inverted confocal microscope, or a Leica SP8 inverted confocal microscope with 488 nm laser lines. Acquired images were analyzed and quantified using ImageJ software. Information on cell culture can be found in the ESI section.†
Conclusion
Our experiments demonstrate that kinases can be used to template an abiotic reaction, a ruthenium photocatalyzed reduction of pyridinium immolative linker. The fast kinetics of the pyridinium photoreduction were found to be critical for the success of this reaction. The combined selectivity of a nucleotide binding site pharmacophore and a ligand binding the substrate site delivers a kinase-specific reaction with 4- to 6-fold amplification relatively to the kinase. In light of the breadth of small molecules targeting the nucleotide binding site of kinase with some level of selectivity and the substrate requirement of protein kinases, this design is generalizable to other protein kinases. Furthermore, the reaction is amenable to unmask other effectors than a fluorophore. Given the central role of kinases in regulatory pathways, this technology offers an inroad to redirect messaging cascades in synthetic biology and for smart therapeutics.
Acknowledgements
The authors thank the SNSF (grant: 200020_157106; 200020_169141) and the NCCR Chemical Biology for financial support.
Notes and references
- P. Cohen, Eur. J. Biochem., 2001, 268, 5001–5010 CrossRef CAS PubMed.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS PubMed.
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309–315 CrossRef CAS PubMed.
- L. K. Chico, L. J. Van Eldik and D. M. Watterson, Nat. Rev. Drug Discovery, 2009, 8, 892–909 CrossRef CAS PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- A. Y. Ting, K. H. Kain, R. L. Klemke and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 15003–15008 CrossRef CAS PubMed.
- J. Zhang and M. D. Allen, Mol. BioSyst., 2007, 3, 759–765 RSC.
- R. H. Yeh, X. W. Yan, M. Cammer, A. R. Bresnick and D. S. Lawrence, J. Biol. Chem., 2002, 277, 11527–11532 CrossRef CAS PubMed.
- M. D. Shults and B. Imperiali, J. Am. Chem. Soc., 2003, 125, 14248–14249 CrossRef CAS PubMed.
- M. D. Shults, K. A. Janes, D. A. Lauffenburger and B. Imperiali, Nat. Methods, 2005, 2, 277–283 CrossRef CAS PubMed.
- E. Lukovic, E. Vogel Taylor and B. Imperiali, Angew. Chem., Int. Ed., 2009, 48, 6828–6831 CrossRef CAS PubMed.
- Q. Wang, E. I. Zimmerman, A. Toutchkine, T. D. Martin, L. M. Graves and D. S. Lawrence, ACS Chem. Biol., 2010, 5, 887–895 CrossRef CAS PubMed.
- Z. J. Zhang, N. Kwiatkowski, H. Zeng, S. M. Lim, N. S. Gray, W. Zhang and P. L. Yang, Mol. BioSyst., 2012, 8, 2523–2526 RSC.
- C. Zambaldo, K. K. Sadhu, G. Karthikeyan, S. Barluenga, J. P. Daguer and N. Winssinger, Chem. Sci., 2013, 4, 2088–2092 RSC.
- M. L. Vetter, Z. J. Zhang, S. Liu, J. H. Wang, H. Cho, J. M. Zhang, W. Zhang, N. S. Gray and P. L. Yang, ChemBioChem, 2014, 15, 1317–1324 CrossRef CAS PubMed.
- A. P. Silverman and E. T. Kool, Chem. Rev., 2006, 106, 3775–3789 CrossRef CAS PubMed.
- A. Shibata, H. Abe and Y. Ito, Molecules, 2012, 17, 2446–2463 CrossRef CAS PubMed.
- K. Gorska and N. Winssinger, Angew. Chem., Int. Ed., 2013, 52, 6820–6843 CrossRef CAS PubMed.
- Z. Pianowski, K. Gorska, L. Oswald, C. A. Merten and N. Winssinger, J. Am. Chem. Soc., 2009, 131, 6492–6497 CrossRef CAS PubMed.
- L. Holtzer, I. Oleinich, M. Anzola, E. Lindberg, K. K. Sadhu, M. Gonzalez-Gaitan and N. Winssinger, ACS Cent. Sci., 2016, 2, 394–400 CrossRef CAS PubMed.
- K. K. Sadhu, T. Eierhoff, W. Römer and N. Winssinger, J. Am. Chem. Soc., 2012, 134, 20013–20016 CrossRef CAS PubMed.
- N. Brauckhoff, G. Hahne, J. T. H. Yeh and T. N. Grossmann, Angew. Chem., Int. Ed., 2014, 53, 4337–4340 CrossRef CAS PubMed.
- U. Reinhardt, J. Lotze, S. Zernia, K. Morl, A. G. Beck-Sickinger and O. Seitz, Angew. Chem., Int. Ed., 2014, 53, 10237–10241 CrossRef CAS PubMed.
- D. Cilloni and G. Saglio, Clin. Cancer Res., 2012, 18, 930–937 CrossRef CAS PubMed.
- O. Hantschel, Genes Cancer, 2012, 3, 436–446 CrossRef PubMed.
-
M. Pérez-Caro and I. Sánchez-Garcia, in Apoptosis, Cell Signaling, and Human Diseases: Molecular Mechanisms, ed. R. Srivastava, Humana Press, Totowa, NJ, 2007, pp. 3–34, DOI:10.1007/978-1-59745-200-7_1.
- M. C. Frame, Biochim. Biophys. Acta, 2002, 1602, 114–130 CAS.
- L. C. Kim, L. Song and E. B. Haura, Nat. Rev. Clin. Oncol., 2009, 6, 587–595 CrossRef CAS PubMed.
- O. Hantschel, U. Rix, T. Buerckstuemmer, U. Schmidt, M. Kneidinger, K. L. Bennett, I. Kaupe, W. Ellmeier, P. Valent and G. Superti-Furga, Blood, 2007, 110, 207b Search PubMed.
- L. J. Lombardo, F. Y. Lee, P. Chen, D. Norris, J. C. Barrish, K. Behnia, S. Castaneda, L. A. M. Cornelius, J. Das, A. M. Doweyko, C. Fairchild, J. T. Hunt, I. Inigo, K. Johnston, A. Kamath, D. Kan, H. Klei, P. Marathe, S. Pang, R. Peterson, S. Pitt, G. L. Schieven, R. J. Schmidt, J. Tokarski, M.-L. Wen, J. Wityak and R. M. Borzilleri, J. Med. Chem., 2004, 47, 6658–6661 CrossRef CAS PubMed.
- N. P. Shah, C. Tran, F. Y. Lee, P. Chen, D. Norris and C. L. Sawyers, Science, 2004, 305, 399–401 CrossRef CAS PubMed.
- U. Rix, O. Hantschel, G. Dürnberger, L. L. Remsing Rix, M. Planyavsky, N. V. Fernbach, I. Kaupe, K. L. Bennett, P. Valent, J. Colinge, T. Köcher and G. Superti-Furga, Blood, 2007, 110, 4055–4063 CrossRef CAS PubMed.
- O. Hantschel, U. Rix and G. Superti-Furga, Leuk. Lymphoma, 2008, 49, 615–619 CrossRef CAS PubMed.
- Z. Songyang, K. L. Carraway 3rd, M. J. Eck, S. C. Harrison, R. A. Feldman, M. Mohammadi, J. Schlessinger, S. R. Hubbard, D. P. Smith and C. Eng,
et al.
, Nature, 1995, 373, 536–539 CrossRef CAS PubMed.
- J. S. Tokarski, J. A. Newitt, C. Y. Chang, J. D. Cheng, M. Wittekind, S. E. Kiefer, K. Kish, F. Y. Lee, R. Borzillerri, L. J. Lombardo, D. Xie, Y. Zhang and H. E. Klei, Cancer Res., 2006, 66, 5790–5797 CrossRef CAS PubMed.
- M. Getlik, C. Grutter, J. R. Simard, S. Kluter, M. Rabiller, H. B. Rode, A. Robubi and D. Rauh, J. Med. Chem., 2009, 52, 3915–3926 CrossRef CAS PubMed.
- K. Gorska, I. Keklikoglou, U. Tschulena and N. Winssinger, Chem. Sci., 2011, 2, 1969–1975 RSC.
- D. Chang, E. Lindberg and N. Winssinger, J. Am. Chem. Soc., 2017, 139, 1444–1447 CrossRef CAS PubMed.
- W. Haass, H. Kleiner, C. Weiss, C. Haferlach, B. Schlegelberger, M. C. Muller, R. Hehlmann, W. K. Hofmann, A. Fabarius, W. Seifarth, Schweizerische Arbeitsgemeinschaft fur Klinische Krebsforschung and German CML Study Group, PLoS One, 2015, 10, e0129648 Search PubMed.
- M. Uhlen, P. Oksvold, L. Fagerberg, E. Lundberg, K. Jonasson, M. Forsberg, M. Zwahlen, C. Kampf, K. Wester, S. Hober, H. Wernerus, L. Bjorling and F. Ponten, Nat. Biotechnol., 2010, 28, 1248–1250 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc01416c |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
N.
Winssinger
b and
N.
Winssinger




