
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible C–C bond formation at a triply cyclometallated platinum(IV) centre†
Paul A.
Shaw
,
Guy J.
Clarkson
and
Jonathan P.
Rourke
 *
*
Department of Chemistry, Warwick University, Coventry, UK CV4 7AL. E-mail: j.rourke@warwick.ac.uk
First published on 5th June 2017
Abstract
The oxidation of the tribenzylphosphine derivative of the doubly cylcometallated platinum(II) complex of diphenylpyridine, 1, with PhICl2 led, as a first step, to the formation of a highly electrophilic metal centre which attacked the benzyl phosphine to give a triply cyclometallated species as the arenium ion. The highly acidic arenium ion protonated unreacted starting 1, a reaction that could be supressed by the addition of water, and gave the neutral species 2(t). Octahedral complex 2(t) was induced to reductively couple, with two five-membered rings coupling to give square planar complex 5 containing a nine-membered ring. The crystal structure of 5 showed the nine-membered ring to span trans across the square planar metal accompanied by considerable distortion: the P–Pt–N bond angle is 155.48(5)°. Oxidation of 5 with PhICl2 resulted in the addition of two chlorides and a change of the nine-membered ring ligand coordination to cis at an octahedral centre, still with considerable distortions: the P–Pt–N bond angle in the crystal structure of 6 is 99.46(5)°. Treatment of 2(t) with AgBF4 also induced a coupling to give a nine-membered ring, and the fluxional three coordinate complex 7. A mono-methylated version of 1, Me-1, was prepared and similar reactions were observed. The presence of the methyl group allowed us to observe selectivity in the coupling reaction to give the nine-membered ring, with two products (a-Me-7 and b-Me7) being initially formed in the ratio 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The concentrations of two products changed with time giving a final ratio of 1:8 at room temperature (half-life 48 hours), the equilibration being made possible by a reversible C–C bond forming reaction. Reaction of complexes 7 with CO or hydrogen left the nine-membered ring intact, though oxidative degradation resulted in decomplexation of the phosphine donor, accompanied by formation of a P
:1. The concentrations of two products changed with time giving a final ratio of 1:8 at room temperature (half-life 48 hours), the equilibration being made possible by a reversible C–C bond forming reaction. Reaction of complexes 7 with CO or hydrogen left the nine-membered ring intact, though oxidative degradation resulted in decomplexation of the phosphine donor, accompanied by formation of a P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group.
O group.
Introduction
The making and breaking of carbon–carbon bonds is key to the field of organic chemistry. Transition metals have a considerable role to play in these processes, and two of this century's Nobel Prizes in Chemistry (20051 and 20102) were awarded for developments in the formation of, respectively, double and single carbon to carbon bonds, mediated by transition metal catalysts. Even today, control over these processes is far from complete and fundamental mechanistic studies on C–C bond formation and cleavage at a metal centre are still necessary, with both the oxidative addition of a C–C bond, and its microscopic reverse, the reductive elimination of a C–C bond, being studied. Under some circumstances, typically where a strained or otherwise sterically constrained molecule is used, oxidative addition of the C–C bond is studied.3 However, normally the energetics are such that the formation of a single carbon–carbon bond from two metal–carbon bonds is strongly favoured, as one strong bond replaces two much weaker ones. This energetic preference, often combined with the difficulty of getting a carbon–carbon bond in close proximity to a metal centre, results in an irreversible elimination reaction, and thus it is the formation of the C–C bond that is studied.4 In some circumstances, a reversible C–C bond formation/cleavage process is observed, allowing study of both the forward and the reverse reactions.5
Organometallic platinum complexes are used in the cyclisation of enynes,6 but are more commonly the subject of fundamental, mechanistic study.7 In part this is due to their amenability to study, but it is also due to their relevance to actual processes and their ability to activate methane.8 Our own work in the area has been to study the oxidation reactions9 and reductive couplings10 at cyclometallated platinum. In particular, we have been studying the oxidation of square planar platinum(II) complexes with PhICl2. Oxidation with PhICl2 (which can be thought of as a convenient and easy to handle source of Cl2) is a two step process11 with initial delivery of a Cl+ generating a very electrophilic metal centre, which would normally then react with a Cl− to complete the oxidation. Thus, for many complexes, the end result is simply the addition of two chloride ligands to the metal centre giving an octahedral Pt(IV) centre;9a,c,10a,b isomerisation may take place, and this may be assisted by the intermediacy of agostic interactions.9a,12 However, this electrophilic metal can also react before combining with chloride. Thus, interesting reactions result when the organic groups on the ligand systems can interact with, and intercept, the metal centre. We have observed agostic intermediates at low temperatures (e.g. −60 °C) prior to transcyclometallation13 reactions that lead to cyclometallated alkyl phosphines.14 Even though the size of the ring containing the agostic interaction could be of different sizes, the cyclometallation selectively gave five-membered rings. Fast intra-molecular C–H activation of aromatic groups (which proceed via an electrophilic attack of the metal on an aryl ring), also at low temperature, has also been observed.9c,15 Crucial to the efficacy of intramolecular electrophilic attack on aryl groups is the size of the ring that is formed. Thus, when the co-ligand is triphenyl phosphine an unfavoured four-membered ring is the only possibility and C–H activation is not observed.14b However, a benzyl group on a phosphine ligand would lead to the formation of a favourable five-membered ring and here we report on our study of such complexes. In addition to the expected electrophilic attack on the benzyl groups, we see a reductive coupling reaction from the newly formed metallacycle, a reaction that is ultimately identified as being reversible.
Results and discussion
Tricyclometallation via Wheland intermediates
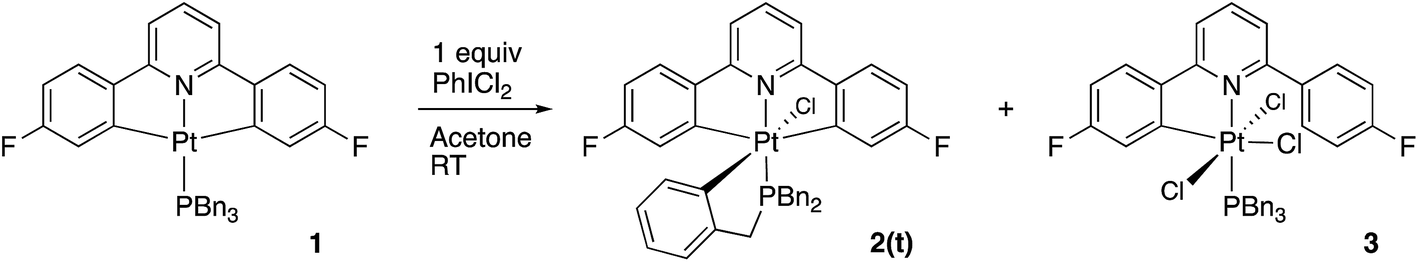
Using our established methodology,16 doubly cyclometallated C^N^C tribenzylphosphine complex 1 was synthesised and characterised, including by X-ray crystallography, Fig. 1. Oxidation of 1, with one equivalent of PhICl2 proceeds with initial delivery of a Cl+ generating a very electrophilic metal centre, which would normally then react with a Cl− to complete the oxidation. However, when 1 was oxidised, in anhydrous chloroform, two products were formed in a 1:1 ratio, neither of which was simply the addition of two chlorides to the original complex. The two complexes were separable and were identified and fully characterised, Scheme 1.
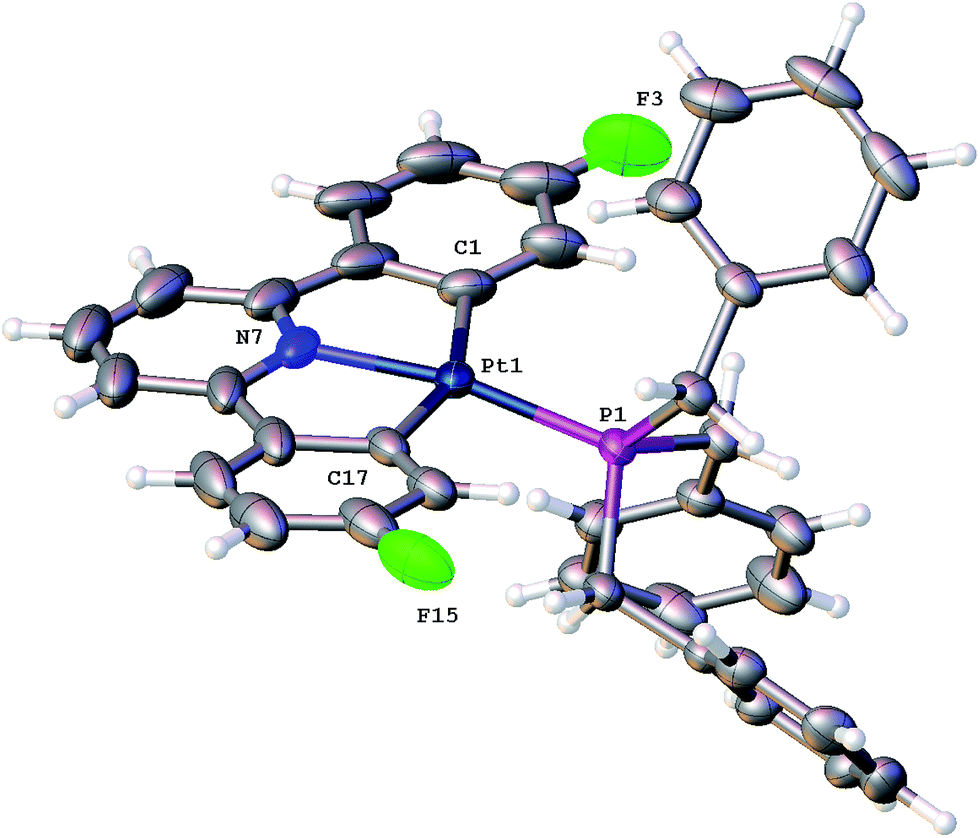 | ||
| Fig. 1 The solid-state structure of 1, thermal ellipsoids drawn at 50% probability level. Selected bond lengths (Å) and angles (°): Pt1–P1 2.2200(8); Pt1–N7 2.033(3); Pt1–C1 2.075(4); Pt1–C17 2.085(4); N7–Pt1–P1 168.72(10); N7–Pt1–C1 80.05(17); N7–Pt1–C17 79.84(16); C1–Pt1–P1 103.82(12); C1–Pt1–C17 159.19(17); C17–Pt1–P1 96.93(11). | ||
 | ||
| Scheme 1 | ||
The identity of the first product, 2(t), became clear from an analysis of the NMR spectra: only one 19F resonance (with 20 Hz 195Pt satellites), a single 31P resonance (satellites at 2649 Hz suggesting Pt(IV)17), a 195Pt chemical shift of −2911 ppm (again suggesting Pt(IV)18), two different aryl protons (integral ratio 2:1) with strong coupling to Pt, three different benzyl protons (ratio 2:2:2) and NOE measurements that showed four of the benzyl protons to be close to the protons on the cyclometallated ring with integral two. All this data pointed towards an intact C^N^C Pt system with an additional cyclometalation to one ring of the phosphine; the structure was definitively confirmed by the solving of the single crystal X-ray structure, Fig. 2. The structure of 2(t) shows a six-coordinate geometry at platinum, with some distortion away from perfect octahedral: the C–Pt–P angle of the new five-membered metallacycle is 80.68(10)°, the N–Pt–C angles in the cyclometallated diphenyl pyridine are 79.61(14)° and 79.78(13)°, but otherwise there is nothing to indicate major stresses or strains within the molecule.
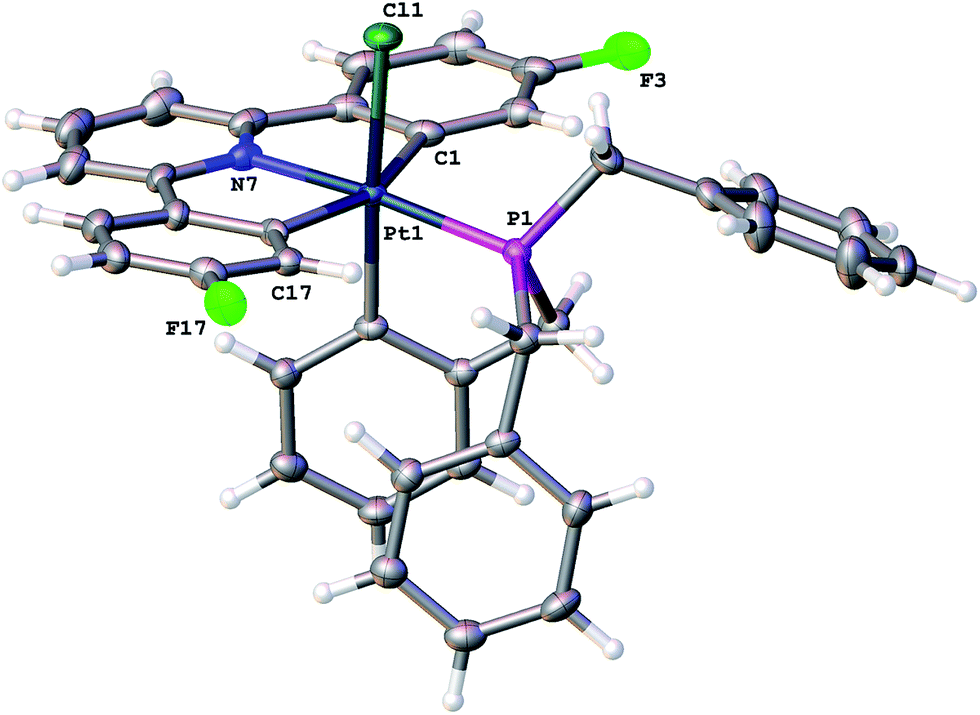 | ||
| Fig. 2 The solid-state structure of 2(t), thermal ellipsoids drawn at 50% probability level. Selected bond lengths (Å) and angles (°): Pt1–Cl1 2.4425(8); Pt1–P1 2.2609(9); Pt1–C1 2.106(3); Pt1–N7 2.039(3); Pt1–C17 2.097(3); Pt1–C20 2.057(3); P1–Pt1–Cl1 96.45(3); C1–Pt1–Cl1 91.42(9); C1–Pt1–P1 96.30(11); N7–Pt1–Cl1 91.88(8); N7–Pt1–P1 170.83(8); N7–Pt1–C1 79.61(14); N7–Pt1–C17 79.78(13); N7–Pt1–C20 90.93(13); C17–Pt1–Cl1 86.02(9); C17–Pt1–P1 104.57(10); C17–Pt1–C1 159.13(15); C20–Pt1–Cl1 176.97(10); C20–Pt1–P1 80.68(10); C20–Pt1–C1 87.96(12); C20–Pt1–C17 95.61(12). | ||
The identity of the second product, 3, is clear too: it has a much simpler set of NMR spectra than 2(t). Once again the Pt shift (−1719 ppm) indicates Pt(IV), as does the 31P signal (satellites at 2434 Hz). Two 19F signals (in the ratio 1:1, one with 195Pt satellites, one without), a set of 1H resonances indicating a monocylcometallated diphenyl pyridine (only one H with significant coupling to Pt, six signals of relative integral 1, two with relative integral 2), and a simple set of signals for the tribenzylphosphine (one benzyl resonance, relative integral 6, three aryl signals relative integrals 6:6:3) suggest structure 3.
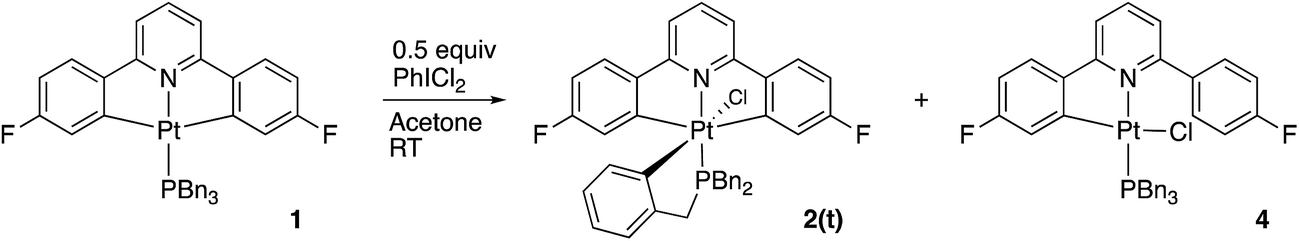
Reasoning that the obvious route to this second product, 3, was oxidation of a mono-cyclometallated platinum(II) species, we repeated the reaction with only half an equivalent of PhICl2, whereupon the products were 2(t), as before, and a new platinum(II) species, 4, with the same pattern of resonances as 3 in the 1H NMR spectrum, Scheme 2. For this material, we were able to grow crystals and solve the X-ray structure, confirming our assignment, Fig. 3.
 | ||
| Scheme 2 | ||
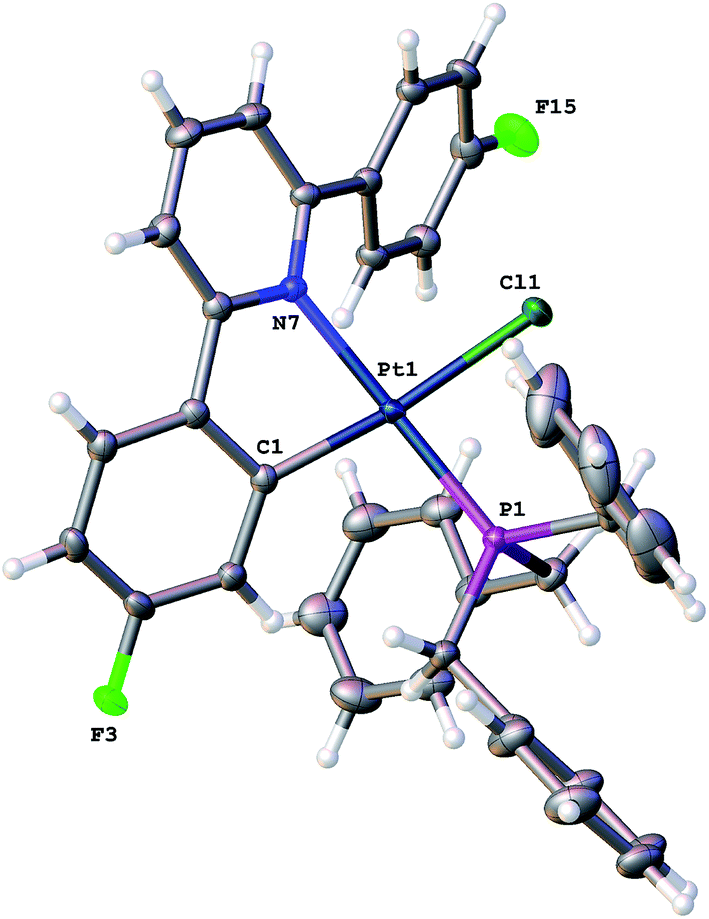 | ||
| Fig. 3 The solid-state structure of 4, thermal ellipsoids drawn at 50% probability level. Selected bond lengths (Å) and angles (°): Pt1–Cl1 2.3884(4); Pt1–C1 1.9947(17); Pt1–P1 2.2384(4); Pt1–N7 2.1274(13); C1–Pt1–Cl1 172.43(5); C1–Pt1–P1 99.70(5); C1–Pt1–N7 79.14(6); P1–Pt1–Cl1 86.424(16); N7–Pt1–Cl1 94.32(4); N7–Pt1–P1 174.23(4). | ||
To rationalise the formation of the products, we need to look at the role of the oxidant. As an electrophilic reagent we would expect PhICl2 to initially deliver a Cl+ to one face of the starting platinum(II) complex 1.11 This generates an extremely electrophilic cationic metal centre that can attack the adjacent aryl ring of the benzyl phosphine. This in turn generates a highly acidic arenium ion19 (Wheland intermediate), which protonates further starting material, it itself rearomatising in the process and forming neutral 2(t). Protonation of the platinum(II) complex 1 would result in cleavage of a metal carbon bond, with a final combination with the chloride generated in the initial oxidation giving 4, Scheme 3. Control experiments show that hydrochloric acid itself, under equivalent conditions, is insufficiently acidic to protonate 1 to give 4.
 | ||
| Scheme 3 | ||
Thus, under the conditions we used, the arenium ion protonated unreacted 1 faster than the PhICl2 could oxidise 1, so fast, in fact, that we were unable to observe the arenium ion spectroscopically. So, in order to try and maximise the yield of 2(t), we took the simple expedient of adding water, which we expected to be preferentially protonated, allowing all the starting 1 to be oxidised. This strategy worked well, and it proved possible, with an acetone (80%)/water (20%) solvent mixture, and one equivalent of PhICl2, to generate essentially quantitative yields of 2(t).
Reductive coupling of two five-membered metallacycles to give nine-membered rings
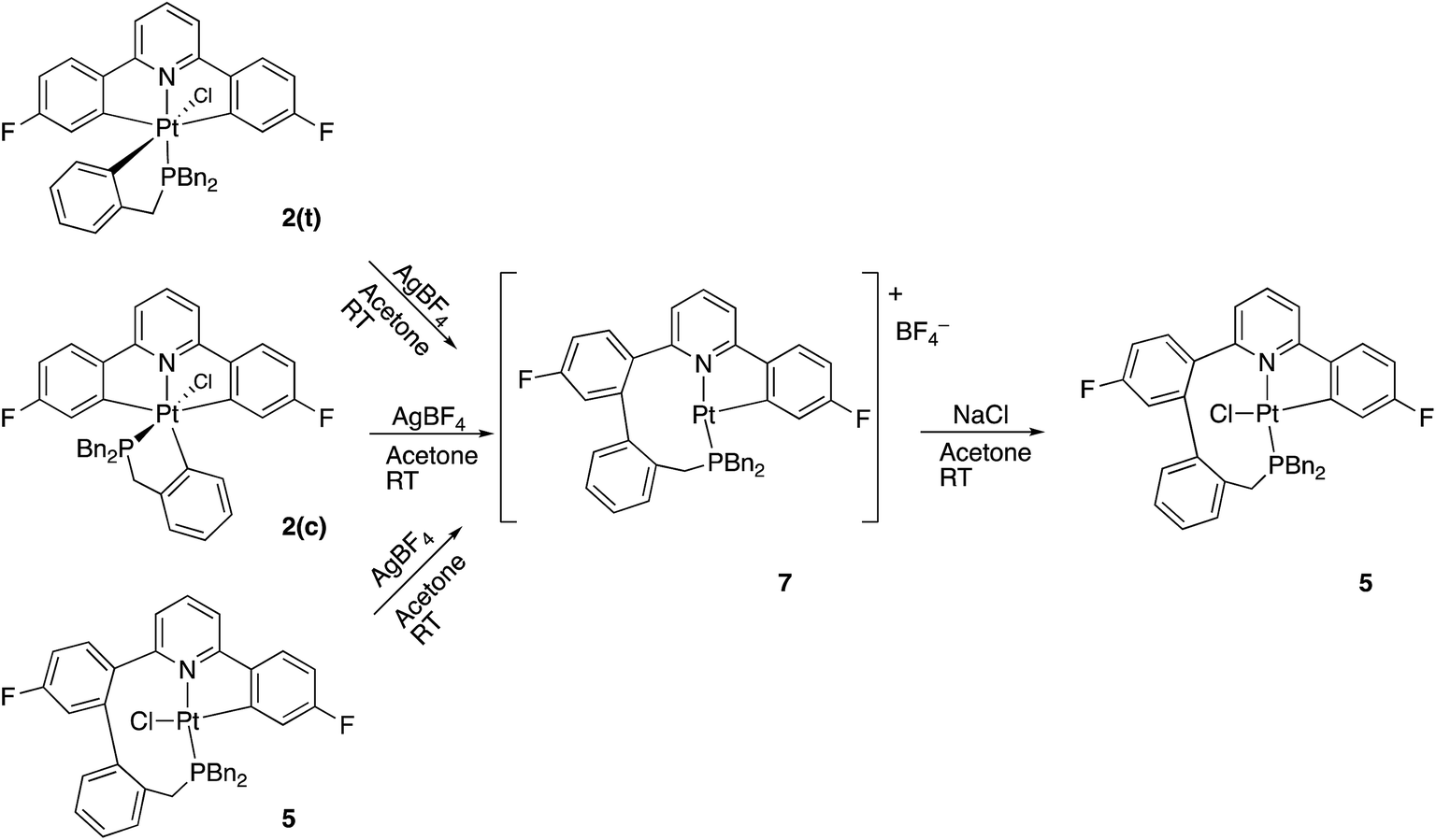
Having maximised the yield of 2(t), we set about investigating its reactivity. Simply attempting further purification on a silica column caused isomerisation and the formation of two new complexes, which could be separated by chromatography. One of these new complexes, 2(c), is simply the geometric isomer of 2(t), where the P and C of the cyclometallated phosphine have exchanged positions. This isomer has the P and N cis to each other, and can be rationalised on the basis of bringing the bulky phosphine away from the plane of the cyclometallated diphenylpyridine to a less crowded site above that plane.Solution NMR data on the other product clearly indicated a platinum(II) species (Pt shift of −3985 ppm, 1JPt–P = 4552 Hz), only a singly cyclometallated diphenyl pyridine (two different 19F resonances, one with Pt satellites, one without) and the joining of the one of the rings of the diphenyl pyridine to one of those of the tribenzyl phosphine (clear from the HMBC 13C spectrum). A crystal structure, Fig. 4, provided unambiguous evidence for the formation of 5, the complex that forms from the reductive coupling of two of the metal–carbon bonds such that one side of the doubly cyclometallated diphenyl pyridine has coupled to the cyclometallated benzyl group, giving a nine-membered ring, Scheme 4. Typically these two new compounds were separated out at a ratio of 4:1, 2(c):5.
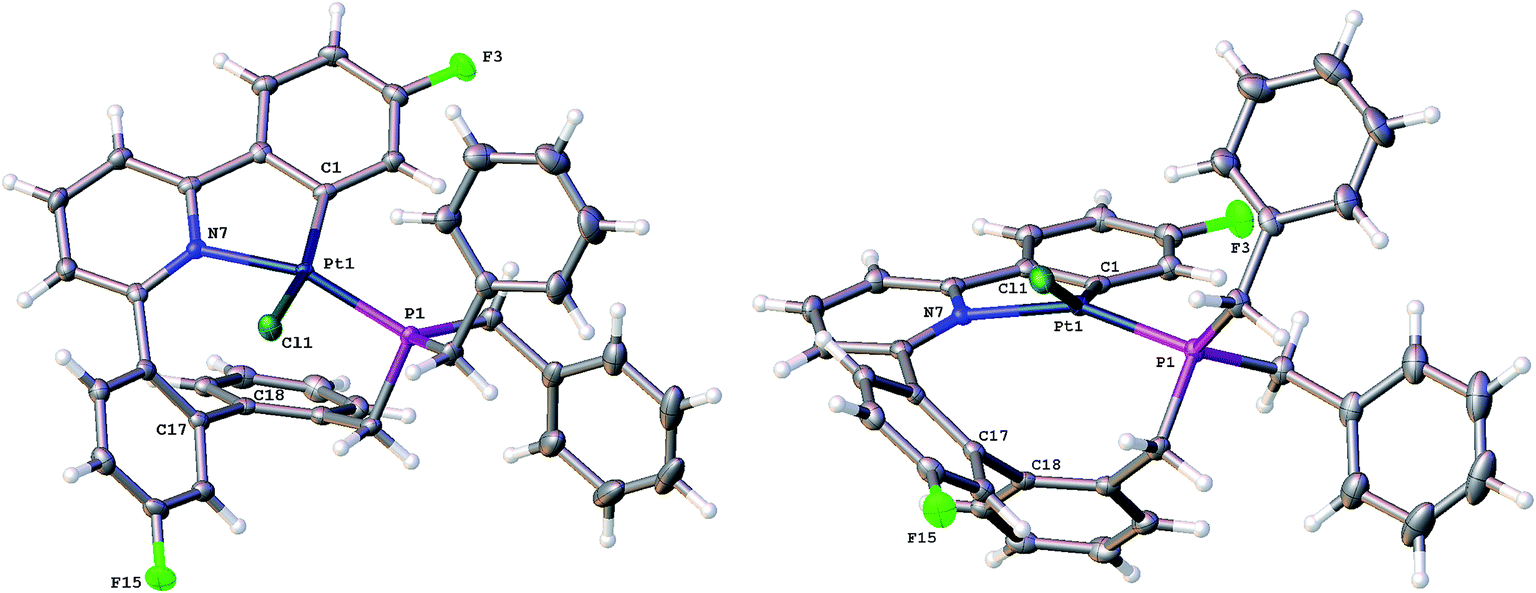 | ||
| Fig. 4 Two views of the solid-state structure of 5, thermal ellipsoids drawn at 50% probability level. Selected bond lengths (Å) and angles (°): C1–Pt1 1.987(2); Cl1–Pt1 2.4001(6); P1–Pt1 2.2346(6); P1–C24 1.857(2); Pt1–N7 2.095(2); C24–P1–Pt1 96.81(8); C1–Pt1–Cl1 158.48(6); C1–Pt1–P1 104.44(7); C1–Pt1–N7 80.56(9); P1–Pt1–Cl1 87.97(2); N7–Pt1–Cl1 95.35(6); N7–Pt1–P1 155.83(5). | ||
 | ||
| Scheme 4 | ||
The nine-membered ring in 5 spans two trans coordination sites at the platinum and induces significant distortions away from perfect square planar geometry (the N–Pt–P and C–Pt–Cl bond angles are 155.83(5) and 158.48(6)° respectively). The nine-membered ring itself mostly has unexceptional bond lengths and angles, but it is pertinent to note that in addition to the N–Pt–P angle of 155.83(5)°, the Pt–P–C angle is only 96.81(8)°, suggesting the ring is rather tight. The phenyl ring that was derived from the phosphine is located over one side of the square planar platinum, effectively blocking approach to the Pt from that side. A close analysis of the 1H NMR spectrum shows all six of the benzyl protons to be different from each other and variable temperature (−60 to +60 °C) studies shows no broadening of these signals and no hint of any exchange processes.
Quite what it is about the silica column that causes the isomerisation is unclear to us (potentially the Lewis acidic silica assists in the removal of the chloride20) and, in any event, it is a rather inefficient method of inducing the reductive coupling that gives 5. Simply heating 2(t) also gives 2(c) and 5, even more inefficiently, as this process is accompanied by significant decomposition. Pure 2(c) does not transform to 5 either on a column, or upon heating. Once formed 5 is indefinitely stable at room temperature, in solution and in air. It is possible to oxidise it with PhICl2 and generate quantitative yields of 6, simply adding the oxidant at room temperature, Scheme 5. A cis relationship for the P and N in the oxidised product is suggested (but cannot be definitively assigned) on the basis of a change in the pattern of the six inequivalent benzyl protons, when compared with 5.
 | ||
| Scheme 5 | ||
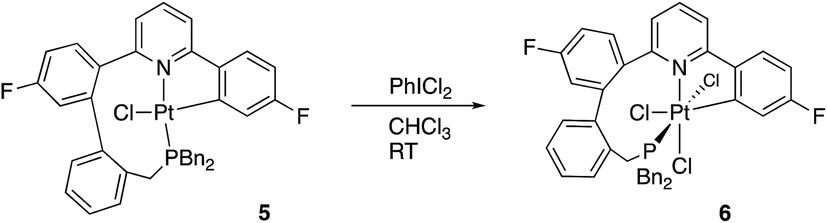
Crystallisation of 6 and solving the X-ray structure, Fig. 5, confirmed the suggested cis P–N geometry. In this arrangement the nine-membered ring seems as if it is on the large side for a cis arrangement at the metal with the N–Pt–P and P–C–C bond angles being 99.46(5) and 122.04(15)° respectively. However, given the strains in the nine-membered ring when it was forced into a trans arrangement in the square planar 5, and the positioning of a phenyl ring across one face of the platinum in that complex, it seems unlikely that such a trans arrangement could be possible in an octahedral complex as an additional two chloride ligands need to be accommodated.
 | ||
| Fig. 5 Two views of the solid-state structure of 6, thermal ellipsoids drawn at 50% probability level. Selected bond lengths (Å) and angles (°): Pt1–Cl1 2.3060(5); Pt1–P1 2.3162(5); Pt1–Cl3 2.3870(5); Pt1–Cl2 2.4175(5); Pt1–N7 2.1070(16); Pt1–C1 2.031(2); Cl1–Pt1–P1 87.370(19); Cl1–Pt1–Cl3 90.032(19); Cl1–Pt1–Cl2 85.42(2); P1–Pt1–Cl3 175.622(19); P1–Pt1–Cl2 84.619(19); Cl3–Pt1–Cl2 91.66(2); N7–Pt1–Cl1 169.78(5); N7–Pt1–P1 99.46(5); N7–Pt1–Cl3 83.59(5); N7–Pt1–Cl2 102.69(5); C1–Pt1–Cl1 90.83(6); C1–Pt1–P1 95.26(6); C1–Pt1–Cl3 88.30(6); C1–Pt1–Cl2 176.26(6); C1–Pt1–N7 81.03(7); C24–P1–Pt1 115.49(7); C23–C24–P1 122.04(15). | ||
A much more efficient, and more readily understandable, method of inducing the reductive coupling of the phenyl pyridine with the benzyl phosphine is the addition of one equivalent of silver tetrafluoroborate to 2(t). The silver salt removes the chloride, generating a reactive five-coordinate intermediate,10d,21 and a reductive coupling reaction between two of the carbon bonded groups takes place. The rapid reductive elimination from an unsaturated intermediate has been rationalised theoretically,22 with the argument hinging on the fact that the coupling process results in the population of a metal orbital that is only non-bonding in the five-coordinate complex, but anti-bonding in a six-coordinate complex. When the reagents are mixed at −40 °C and warmed up slowly, the reaction starts at around −10 °C and exclusively gives what is presumably the three-coordinate 7 as the tetrafluoroborate salt. Not only does the initial trans2(t) give this new compound, but so does the cis2(c) and so does the coupled 5; subsequent treatment of 7 with NaCl gives 5 in quantitative yield, Scheme 6.
 | ||
| Scheme 6 | ||
Careful analysis of the variable temperature NMR spectra does not reveal any loosely bound ligands, such as water or BF4−, coordinated to 7, even at −60 °C. It does, however, reveal fluxionality in the compound. At low temperature (−60 °C) all six of the benzyl protons have unique resonances, similar to the situation for 5. However as the temperature is increased, broadening and merging of signals is observed and the high temperature limit appears to be one in which there are only three signals for the benzyl protons, each of integral two. We can envisage a process whereby the coupled phenyl ring moves from its position above one side of the approximately T-shaped platinum coordination plane to above the other, bringing the newly formed C–C bond past the vacant coordination site, interconverting the appropriate hydrogens as it does so. From the NMR data, we can estimate (see ESI†) the barrier for this interconversion to be of the order of 62 ± 8 kJ mol−1. The presence of a chloride in 5 will provide an addition steric barrier to this process that is sufficient to render it unobservable by NMR; we noted above that all six benzyl proton signals in 5 were inequivalent, with no signs of broadening or an hint of an exchange process over the temperature range −60 to +60 °C. Looking at the X-ray structure of 5, Fig. 4, it is easy to believe the barrier to interconversion in 5 to be so high that it is insurmountable under normal conditions.
However, an alternative process for the equilibration of the benzyl protons in 7 can be envisaged, one that involves the cleavage of a C–C bond to generate a five-coordinate triply cyclometallated species (i.e.2(t) without the chloride). If this species were then to reductively couple the benzyl ring with the fluorophenyl ring to which it was not previous coupled (the two fluorophenyl rings are chemically identical in the triply cyclometallated species), the effect would be to exchange the benzyl protons. At first sight this process seems rather unlikely but, as we demonstrate below, it is not impossible, though the energy barrier is too high for it to significantly contribute to the effects we see in the NMR spectra.
Distinguishing between the two fluoro-phenyl rings: reversible C–C bond formation
Using our previously published methodology10d we were able to cleanly synthesise a non-symmetric mono-methylated version of 1, i.e.Me-1, Scheme 7. Me-1 was fully characterised, including solving the X-ray crystal structure, Fig. 6. The presence of a methyl group on one of the fluoro-phenyl rings significantly affects the coupling pattern in the 1H NMR of the protons on that ring, and has a minor effect on the 19F signals; taken together, these differences are sufficient for us to be able to distinguish between reactions on one ring rather than the other. We therefore set about repeating the reactions we had observed with 1 to see how the presence of a methyl group would affect the reactivity, using its presence to distinguish reactions at the two fluoro-phenyl rings, and to elucidate the dynamic behaviour of the intermediates.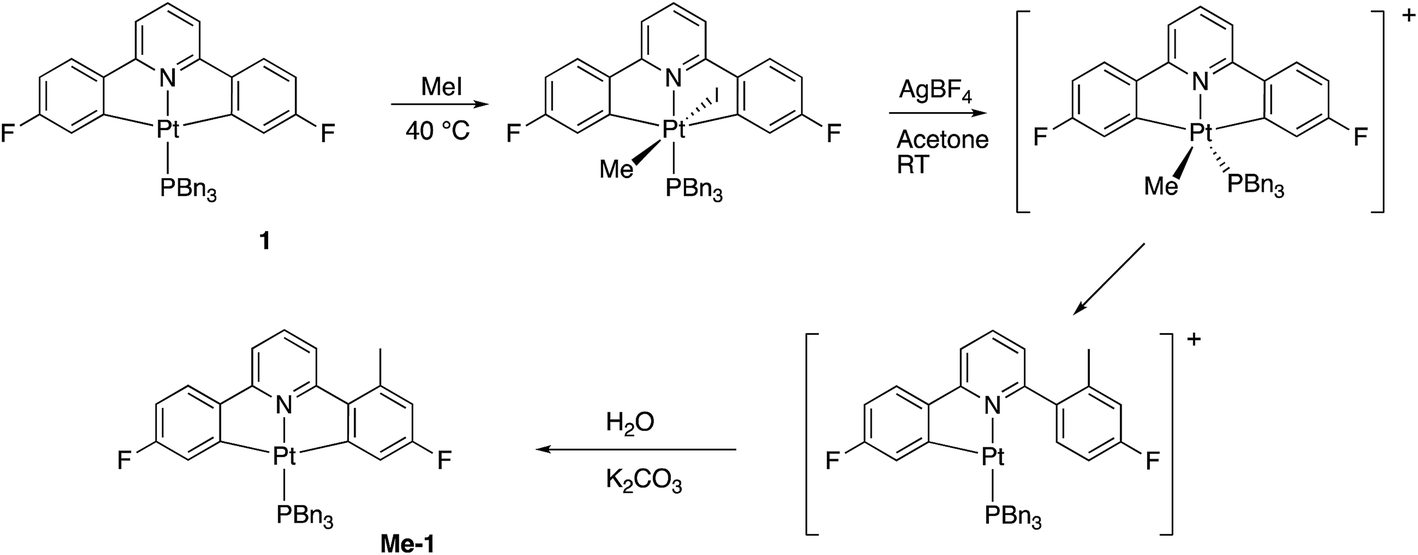 | ||
| Scheme 7 | ||
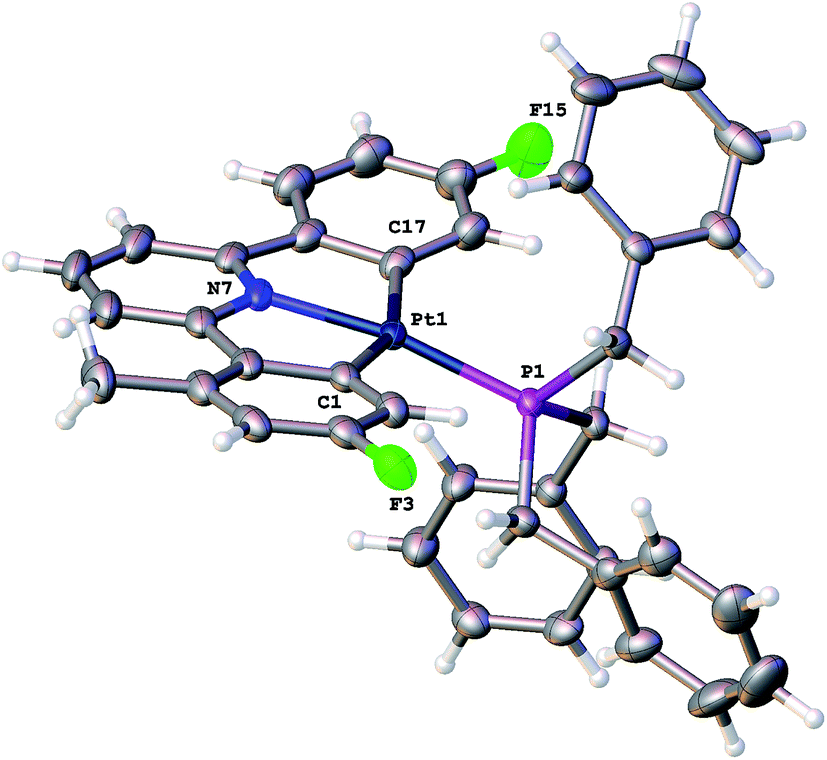 | ||
| Fig. 6 The solid-state structure of Me-1, thermal ellipsoids drawn at 50% probability level. Selected bond lengths (Å) and angles (°): Pt1–P1 2.2409(9); Pt1–C1 2.062(4); Pt1–N7 2.029(3); Pt1–C17 2.071(4); C1–Pt1–P1 98.67(11); C1–Pt1–C17 158.79(15); N7–Pt1–P1 169.79(8); N7–Pt1–C1 79.69(14); N7–Pt1–C17 80.22(14); C17–Pt1–P1 102.30(10). | ||
The initial reaction with half an equivalent of PhICl2 in dry solvent gave the expected Me-2(t) and only one of the two possible isomers of Me-4, Scheme 8. Protonation of the platinum centre, will be followed by aryl-Pt cleavage and could take place with either aryl ring, however we would expect b-Me-4 to be favoured thermodynamically over a-Me-4, as cleavage of the methylated metallacycle would allow a release of a steric clash associated with the methyl group and the pyridine. That it is indeed the only product formed is, however, due to a lower energy pathway to that isomer, rather than the thermodynamic preference, as we would not expect the two compounds to be in equilibrium with each other under the reaction conditions used. As the methyl group will be electron releasing, in comparison to a hydrogen, perhaps it is the more electron rich nature of this ring that means it preferentially takes place in what is a reductive coupling reaction.
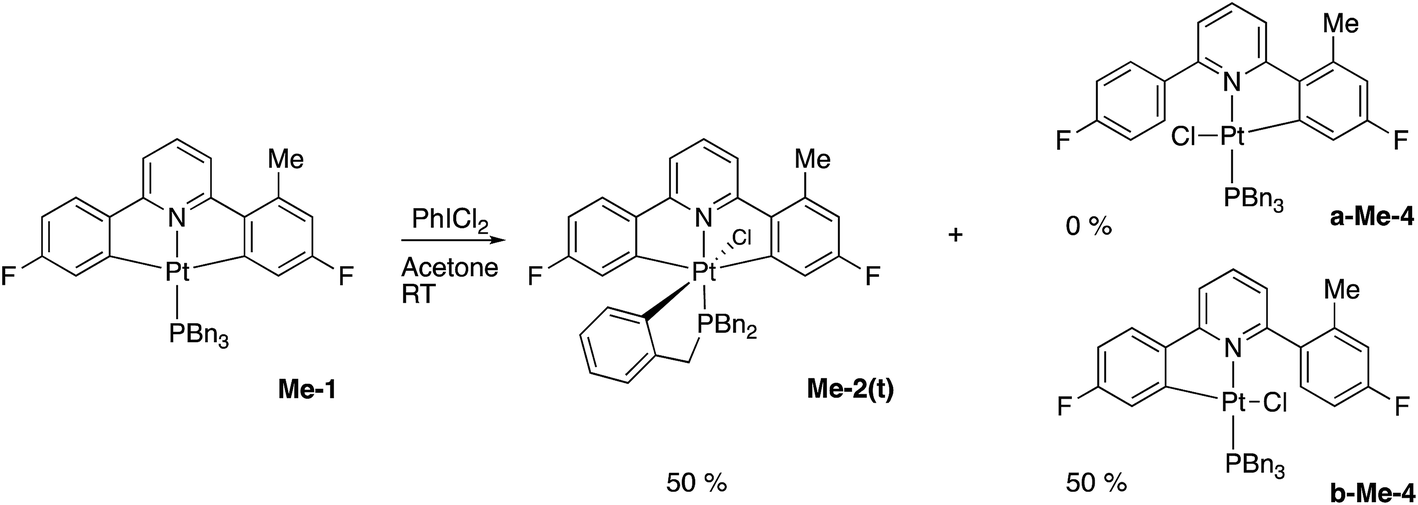 | ||
| Scheme 8 | ||
In a similar fashion to the chemistry reported earlier, column chromatography induces an isomerisation reaction with Me-2(t) giving three compounds, the methyl analogues of 2(c) and two coupled products, a-Me-5 and b-Me-5, Scheme 9, approximate ratio 6:1:1, respectively. Though it proved impossible to completely separate a-Me-5 and b-Me-5 from each other, samples enriched in one isomer over the other allowed complete sets of solution data on each to be acquired. Given that these two Me-5 compounds were formed by this method in only low yields via an indeterminate mechanism, we should not read too much into the fact that they formed in roughly equal quantities. What we can do, though, is note that the relative proportions of these two isomers in solution does not change, whatever that proportion is, even after 1 month at 50 °C; this clearly implies an irreversible coupling reaction in their formation.
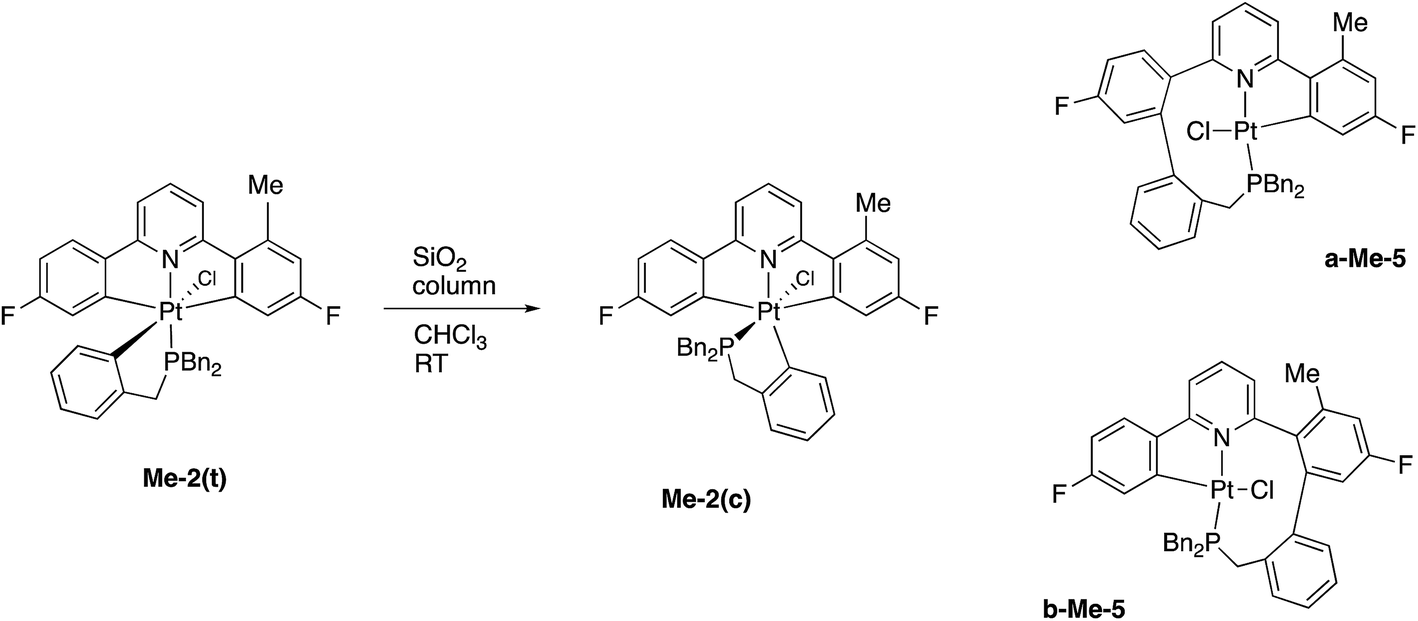 | ||
| Scheme 9 | ||
In contrast, and more interesting, is the chemistry we observe when we attempt the reaction of the triply-cyclometallated Me-2(t) with AgBF4. The first step of the reaction will be to generate a five-coordinate intermediate from which one of the aryl rings will couple with the cyclometallated phosphine, Scheme 10. Unlike the earlier situation, Scheme 6, we can now distinguish exactly which fluoro-phenyl ring the benzyl group couples with.
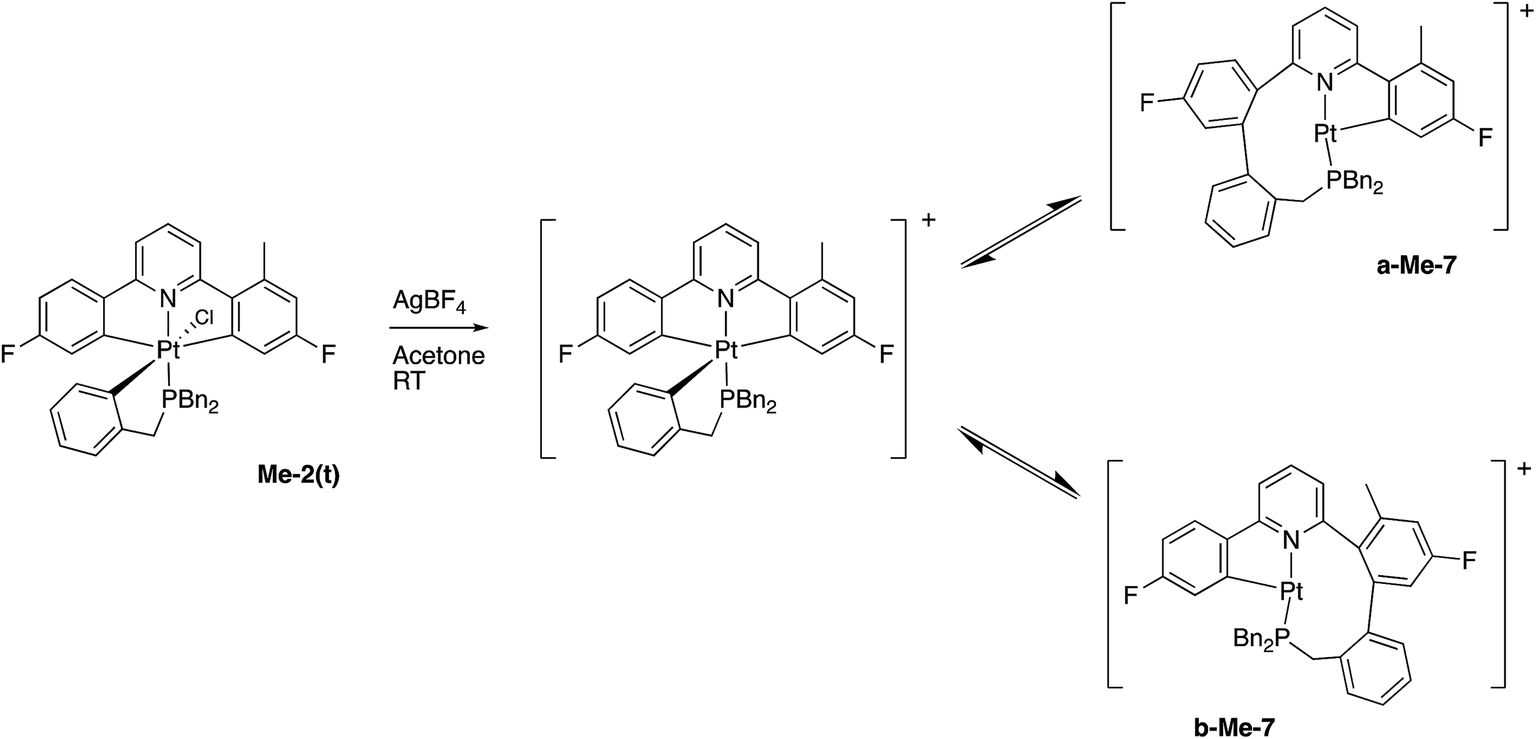 | ||
| Scheme 10 | ||
Once again we can see that b-Me-7 will be thermodynamically favoured, but it is in fact a-Me-7 that is formed preferentially (rough ratio a:b = 7:1) when the reaction is carried out at room temperature. Quite what causes this selectivity is unclear, but it is reproducible. Presumably the presence of the methyl group has a tangible influence of the orientation of the three metallacycles and this subtly affects the activation energies of the two competing coupling reactions, with the result that we get the product distribution observed.
It is also clear that, in solution, the proportions of the two isomers changes with time: the concentration of a-Me-7 decreases with a corresponding increase in the concentration of b-Me-7 and an equilibrium exists between the two of them, Fig. 7. A full analysis is given in the ESI,† and we only need to note here that the reaction exhibits first order kinetics with a half-life of around 48 hours at room temperature and a final position of equilibrium of around 1:7.5, a:b.
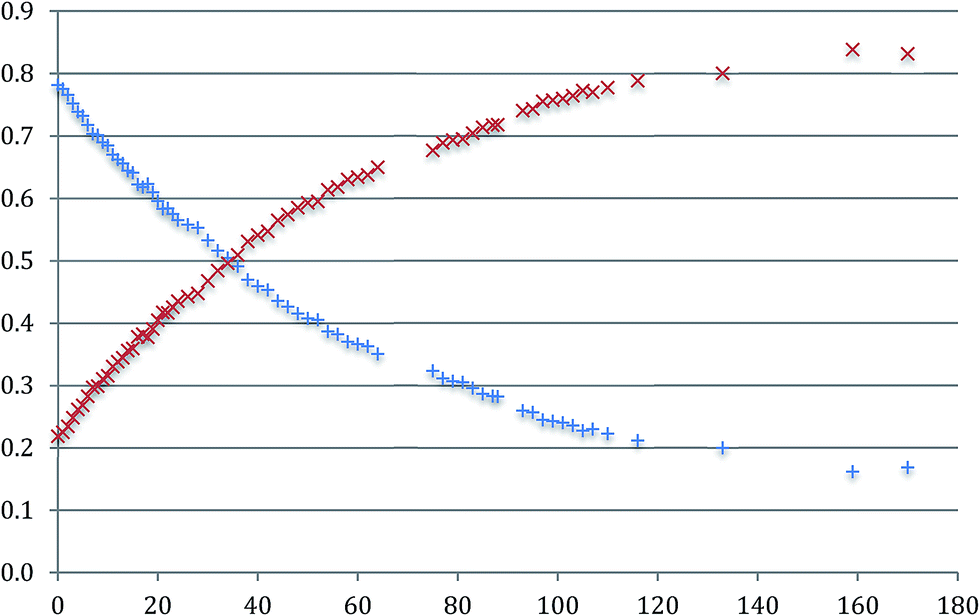 | ||
Fig. 7 The change in the normalised concentrations of a-Me-7 ( ) and b-Me-7 ( ) and b-Me-7 ( ) with respect to time (hours). ) with respect to time (hours). | ||
The only sensible intermediate between the two isomers is the triply cyclometallated cation that forms when chloride is abstracted from Me-2(t), i.e. the coupling reaction is reversible. The three coordinate complexes 7 have a vacant site at platinum, and we know from the variable temperature 1H NMR data recorded on the non-methylated complexes that the nine-membered ring system is flipping in conformation on an NMR timescale, and that this process must bring the appropriate C–C bond right across the vacant site, so such a reaction makes sense. In addition, we know that the nine-membered ring is strained, so that the balance of energies does not so clearly lie on the side of the reductively coupled product (a C–C bond, part of a strained nine-membered ring), compared with two five-membered rings with two Pt–C bonds. Earlier, Scheme 9, we had noted that there is no interchange between the a and b isomers of Me-5, reinforcing the fact that the vacant site at platinum is crucial for the reversible reaction.
Nine-membered rings have considerable precedence in the literature, though there are only seven crystal structures of transition metal complexes with one P and one N donor reported; of these, six are of chiral spiro iridium phosphine complexes used for enantioselective transformations23 and the last one is of a dirhenium phosphino-fullerene.24 There are rather more nine-membered rings where both donors to the metal are nitrogen. In some of the crystal structures of these κ2N donors the donors span trans25 coordination sites, some cis26 and there is one example where a change from trans to cis occurs upon addition of CO.27 The formation of the eight-membered linkage that becomes the nine-membered metallacycle from two equal sized rings does not appear to have been seen before with two different cyclometallated ligands (as in the phosphine and pyridine reported here), but has been seen with two identical ligands. Thus, reported examples include the coupling of two cyclometallated phenylpyridines to give a nine-membered metal containing ring,25a,27 and a similar coupling but where the coupled product detaches from the metal.28 One of these examples also illustrates the fine balance that is present: the coupling of two five-membered cyclometallated phenylpyridines to give a nine-membered ring is favourable when the metal is palladium, but not when it is platinum.25a In our case, the fine balance shows up as a reversible reaction: though the coupling of the two five membered rings is thermodynamically favoured, it is not so favoured as to present an insurmountable barrier to its reverse.
We had also earlier raised the possibility of the reversible C–C bond forming reaction in 7 being responsible for the exchange processes seen in the 1H NMR of the benzyl protons. We can now discount such a process as it is obvious that the timescale of the chemical exchange (48 hour half-life) is far too slow to be responsible for the interconversion of NMR signals.
Other reactions of the nine-membered ring species
We noted above, Scheme 6, that complex 7 reacted with chloride ions to give 5, and this reaction is general: bromide or iodide gave equivalent complexes. The reaction of 7 with other potential ligands was not fully explored, but we did note that reaction did occur with CO and H2. Carbon monoxide reacted rapidly (<1 min reaction time) and cleanly giving 8, Scheme 11.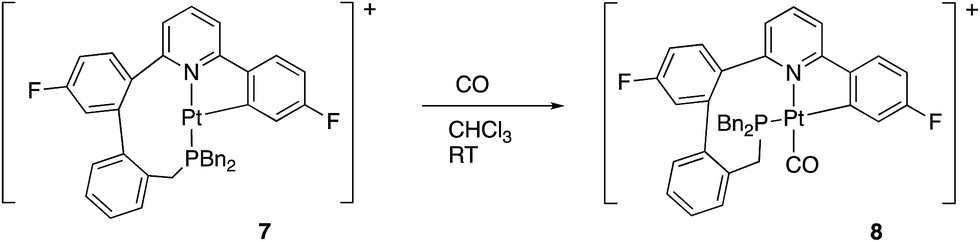 | ||
| Scheme 11 | ||
The coordination of CO to 7 to give 8 was immediately obvious from a colour change and from the IR spectrum, where a strong band at 2097 cm−1 indicates a coordinated carbonyl. A cis arrangement of the P and N donors was indicated from the solution NMR data, with a resolvable 5J of 6 Hz between the P and F nuclei. Such couplings are only visible when these two groups are trans to each other across the metal centre: for comparison, in all the other complexes described in this paper, the P and the fluorinated ring are cis to each other and no coupling is resolvable. Presumably this cis arrangement of the P and N donors is more strained than the trans arrangement seen in 5, otherwise it too would have this arrangement. The preferred geometry in 8 can be understood, though, on the basis of keeping two C donor ligands from coordinating trans to each other, a phenomenon sometimes referred to as “transphobia”.29 It is interesting to note that we have therefore observed three coordination modes for the monocyclometallated coupled ligand: a fac coordination in 6, and two mer coordination modes, one with the P and N cis, 8, but more commonly with the P and N trans, 5 and 7. In each of these modes there appears to be some difficulty in accommodating the nine-membered ring, and this ring ultimately proves to be the site where further reaction takes place, Scheme 13 below.
Reaction with hydrogen gas is also rapid, with only one new species seen by NMR. The new species is clearly a hydride, and not a dihydrogen complex: in the 1H NMR spectrum there is a new resonance, relative integral one, at −24.34 ppm with 1JH–Pt = 1230 Hz. Further analysis of the NMR spectra shows the previously cyclometallated phenyl ring to be free (no coupling between 195Pt and 19F, two resonances ratio 2:2 in the 1H NMR spectrum), and we can formulate this compound as 9, Scheme 12. The new hydride species exchanged H for D on treatment with D2O, and then, more slowly, the two hydrogens in the fluorinated ring, ortho to the bond to the coordinated pyridine. Whilst the first process can simply be seen as reversible protonation reaction, the second process implies a reversible cyclometallation process.
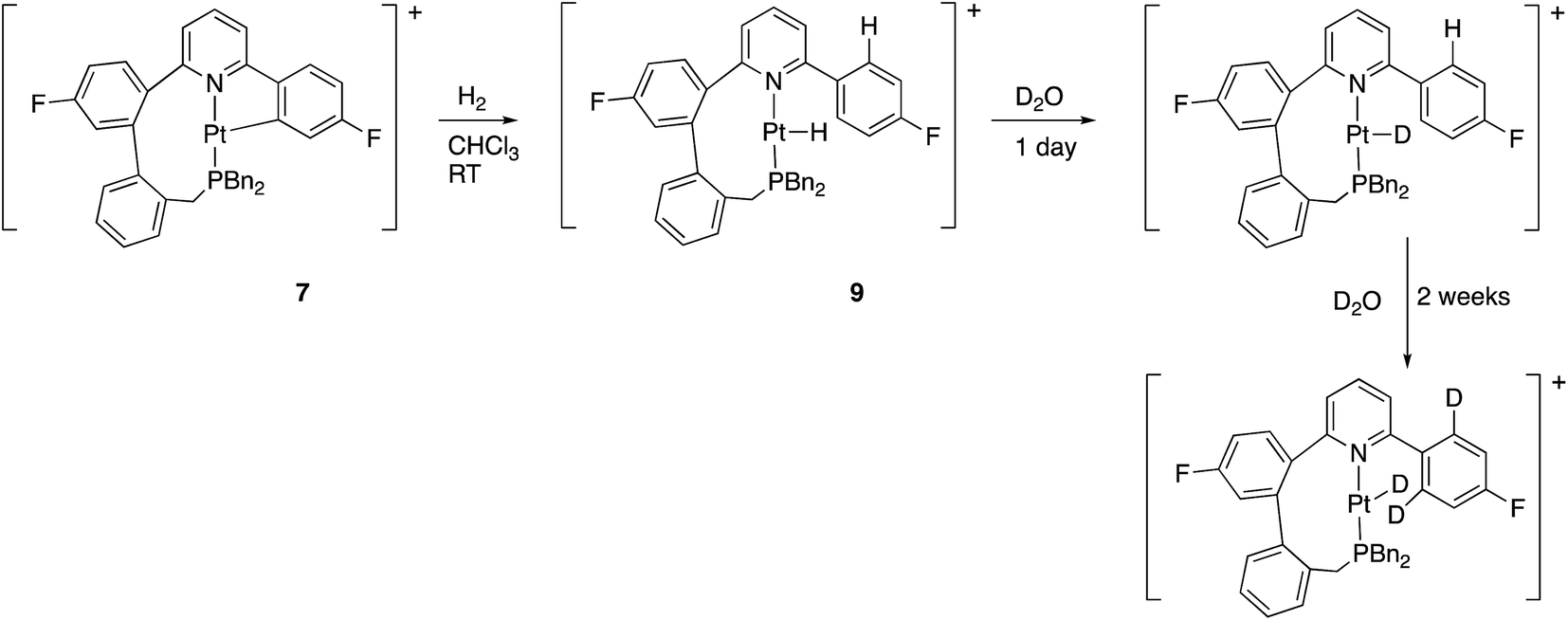 | ||
| Scheme 12 | ||
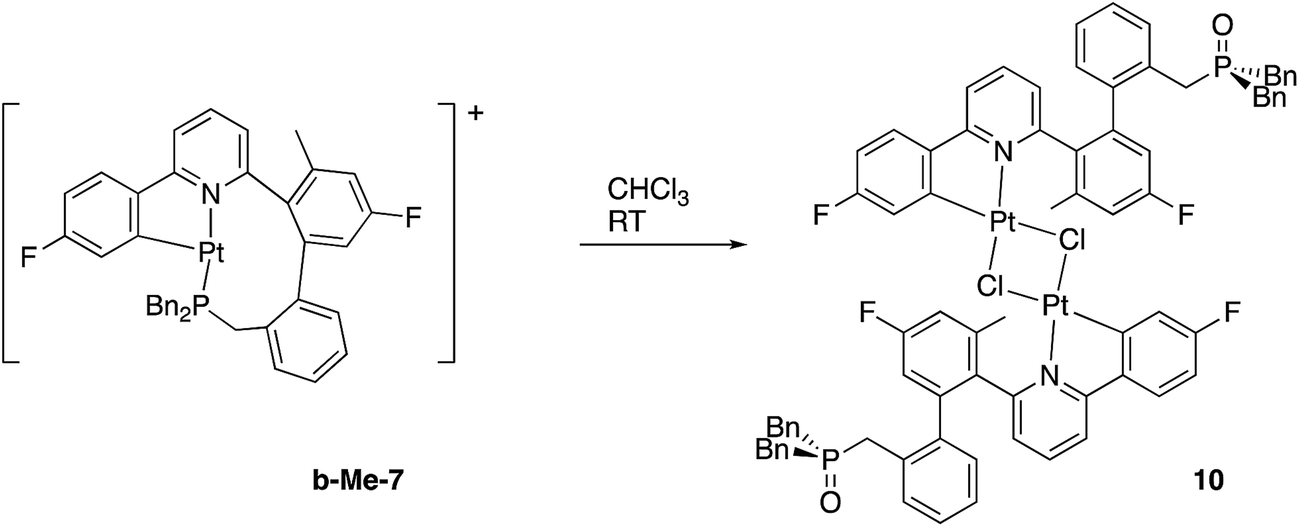
Though solutions of the all the coordinatively saturated complexes appear to survive for several weeks or more at room temperature and in air, the unsaturated Me-7 does change over that sort of timescale, in chloroform solution and in air. The reaction is not completely clean, with multiple peaks visible in the solution NMR spectra, but we were able to isolate crystals from one reaction mixture, Fig. 8. The X-ray structure determined from the crystals revealed a molecule that no longer has the phosphine coordinated (it has become oxidised), but with the original cyclometallated ring still intact, the addition of chloride and a dimeric structure completes the square planar coordination at platinum, Scheme 13. Such a material is completely consistent with the dominant peaks in the NMR spectra and can be thought of as being favoured by the release of strain from breaking the nine-membered ring. The X-ray structure of 10 shows the central core of the molecule to have bond lengths and angles very similar to those found in similar analogues.9a,9c,16b,30
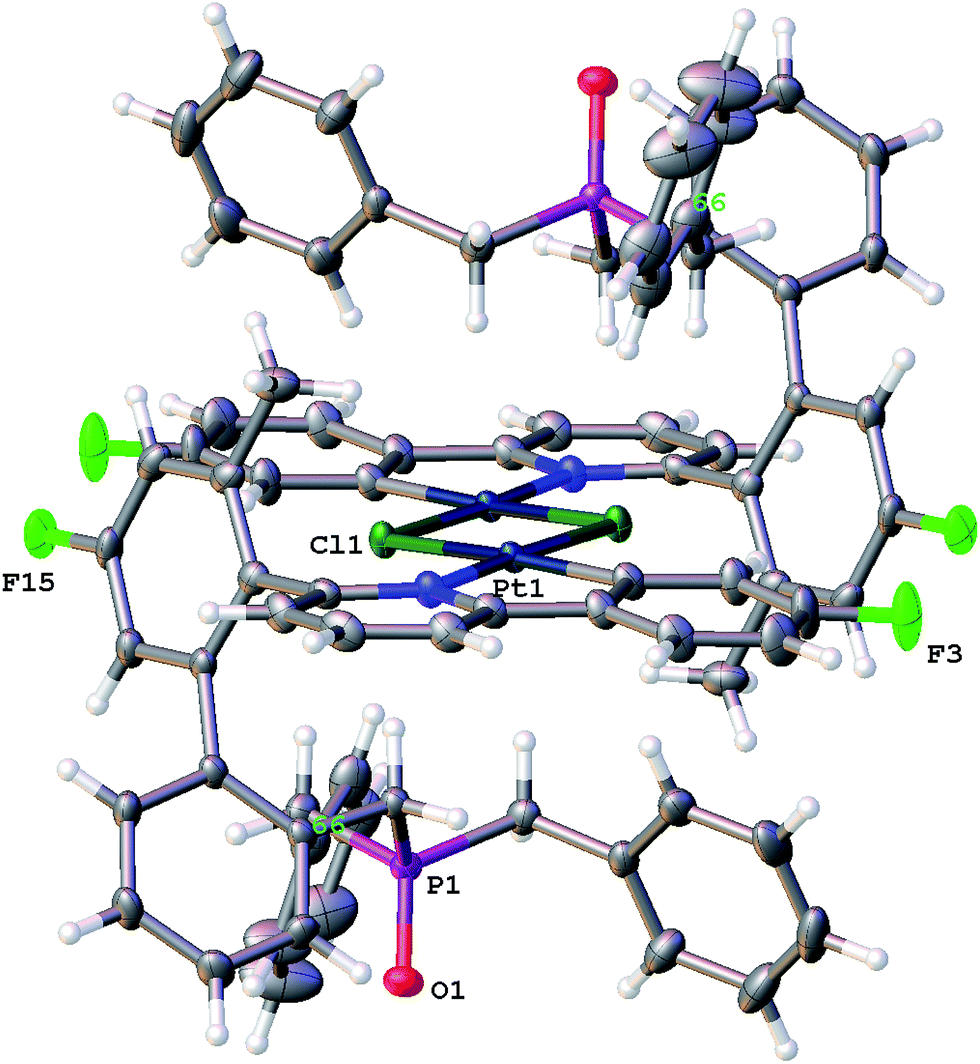 | ||
| Fig. 8 The solid-state structure of 10, thermal ellipsoids drawn at 50% probability level. Selected bond lengths (Å) and angles (°): C1–Pt1 1.968(4); C1–C2 1.406(6); C1–C6 1.401(5); O1–P1 1.490(3); Pt1–Cl11 2.4607(9); Pt1–Cl1 2.2982(9); Pt1–N7 2.052(3); C1–Pt1–Cl1 93.50(12); C1–Pt1–Cl11 171.00(13); C1–Pt1–N7 81.59(15); Cl1–Pt1–Cl11 79.15(3); N7–Pt1–Cl11 105.73(9); N7–Pt1–Cl1 175.09(9). | ||
 | ||
| Scheme 13 | ||
Though we did not isolate and purify a similar material from the non-methylated 7, it too degrades to something with similar NMR spectra and we can expect the majority product to be the non-methylated analogue of 10.
Conclusions
The oxidation reactions of a doubly cylcometallated platinum(II) complex of diphenylpyridine with iodobenzenedichloride leads, as a first step, to the formation of a highly electrophilic metal centre. Given the presence of an adjacent aryl ring, this is followed by intramolecular electrophilic attack on that ring to give a triply cyclometallated species. In contrast, had the ligand had an alkyl group, a transcyclometallation13 reaction, as we saw with tributyl14a or tripropyl14b phosphine would have occurred. The cyclometallation itself generates a highly acidic arenium ion as an intermediate; this intermediate is sufficiently reactive to protonate unreacted starting platinum(II) material, faster than it can be oxidised, though this reaction can be supressed by the addition of an alternative substrate for protonation (in fact, simply adding water is sufficient).Once formed, the triply cyclometallated complex, with three five-membered metallacycles, can be induced to react, with two of those rings coupling to give a strained nine-membered ring. When the nine-membered ring spans trans across a square planar platinum with two further ligands, it appears to be conformationally rigid and not prone to any reversible cleavage. However, when it is part of a coordinatively unsaturated square plane, not only is the ring flipping from one side of the metal centre to the other, but the newly formed C–C bond is reversibly breaking to reform the two five-membered rings before reforming the nine-membered ring. A kinetic analysis from the 1H NMR data showed the reaction to be first order, with a rate constant of 0.0145 h−1 at 298 K. We can rationalise the reversible nature of the reaction in terms of the ring strain of the nine-membered ring reducing the favourability of the coupling to such an extent that it does not present an insurmountable barrier to its reverse.
A cis configuration of the nine-membered ring was seen with the octahedral coordination at the metal that results from oxidation of the square planar Pt(II). However all of the complexes with the nine-membered ring suffer from steric strain to some degree, though further reaction of the three coordinate complexes (7) with CO or hydrogen left the ring intact. Oxidative degradation did result in decomplexation of the phosphine donor, accompanied by formation of a PO group.
Conflict of interest
The authors declare no competing financial interests.Acknowledgements
We thank EPSRC for a DTG award to PAS and support from Advantage West Midlands (AWM) (part funded by the European Regional Development Fund) for the purchase of a high resolution mass spectrometer and the XRD system that was used to solve the crystal structures.References
- (a) Y. Chauvin, Adv. Synth. Catal., 2007, 349, 27–33 CrossRef CAS; (b) R. H. Grubbs, Adv. Synth. Catal., 2007, 349, 34–40 CrossRef CAS; (c) R. R. Schrock, Adv. Synth. Catal., 2007, 349, 41–53 CrossRef CAS.
- (a) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed; (b) C. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed.
- (a) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed; (b) H. Salem, Y. Ben-David, L. J. W. Shimon and D. Milstein, Organometallics, 2006, 25, 2292–2300 CrossRef CAS; (c) M. Montag, I. Efremenko, Y. Diskin-Posner, Y. Ben-David, J. M. L. Martin and D. Milstein, Organometallics, 2012, 31, 505–512 CrossRef CAS; (d) B. L. Edelbach, R. J. Lachicotte and W. D. Jones, J. Am. Chem. Soc., 1998, 120, 2843–2853 CrossRef CAS; (e) W. D. Jones, Mechanistic Studies of Transition Metal-Mediated C–C Bond Activation, in C–C Bond Activation, ed. G. Dong, Springer-Verlag: Berlin, 2014, Vol. 346, pp. 1–31 Search PubMed; (f) A. Barretta, F. G. N. Cloke, A. Feigenbaum, M. L. H. Green, A. Gourdon and K. Prout, J. Chem. Soc., Chem. Commun., 1981, 156–158 RSC; (g) R. H. Crabtree, R. P. Dion, D. J. Gibboni, D. V. McGrath and E. M. Holt, J. Am. Chem. Soc., 1986, 108, 7222–7227 CrossRef CAS; (h) Z. Lu, C. H. Jun, S. R. Degala, M. P. Sigalas, O. Eisenstein and R. H. Crabtree, Organometallics, 1995, 14, 1168–1175 CrossRef CAS; (i) A. B. Chaplin, J. C. Green and A. S. Weller, J. Am. Chem. Soc., 2011, 133, 13162–13168 CrossRef CAS PubMed; (j) M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef CAS PubMed.
- (a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (b) A. S. K. Hashmi, L. Schwarz, J. H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285–2288 CrossRef CAS; (c) W. Shi, C. Liu and A. W. Lei, Chem. Soc. Rev., 2011, 40, 2761–2776 RSC; (d) P. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2769 CrossRef CAS PubMed; (e) S. Shekhar and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 13016–13027 CrossRef CAS PubMed; (f) K. I. Goldberg, J. Yan and E. M. Breitung, J. Am. Chem. Soc., 1995, 117, 6889–6896 CrossRef CAS; (g) J. Procelewska, A. Zahl, G. Liehr, R. van Eldik, N. A. Smythe, B. S. Williams and K. I. Goldberg, Inorg. Chem., 2005, 44, 7732–7742 CrossRef CAS PubMed; (h) R. Ghosh, X. W. Zhang, P. Achord, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2007, 129, 853–866 CrossRef CAS PubMed; (i) M. A. Bennett, S. K. Bhargava, D. C. R. Hockless, L. L. Welling and A. C. Willis, J. Am. Chem. Soc., 1996, 118, 10469–10478 CrossRef CAS; (j) E. G. Bowes, S. Pal and J. A. Love, J. Am. Chem. Soc., 2015, 137, 16004–16007 CrossRef CAS PubMed; (k) M. Crespo, C. M. Anderson, N. Kfoury, M. Font-Bardia and T. Calvet, Organometallics, 2012, 31, 4401–4404 CrossRef CAS; (l) C. M. Anderson, M. Crespo, N. Kfoury, M. A. Weinstein and J. M. Tanski, Organometallics, 2013, 32, 4199–4207 CrossRef CAS; (m) A. Ariafard, Z. Ejehi, H. Sadrara, T. Mehrabi, S. Etaati, A. Moradzadeh, M. Moshtaghi, H. Nosrati, N. J. Brookes and B. F. Yates, Organometallics, 2011, 30, 422–432 CrossRef CAS.
- (a) M. Albrecht, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 2001, 123, 7233–7246 CrossRef CAS PubMed; (b) C. Carmen, H. Atienza, C. Milsmann, S. P. Semproni, Z. R. Turner and P. J. Chirik, Inorg. Chem., 2013, 52, 5403–5417 CrossRef PubMed; (c) T. R. Dugan, E. Bill, K. C. MacLeod, G. J. Christian, R. E. Cowley, W. W. Brennessel, S. F. Ye, F. Neese and P. L. Holland, J. Am. Chem. Soc., 2012, 134, 20352–20364 CrossRef CAS PubMed; (d) J. J. Garcia, N. M. Brunkan and W. D. Jones, J. Am. Chem. Soc., 2002, 124, 9547–9555 CrossRef CAS PubMed; (e) M. J. Monreal and P. L. Diaconescu, J. Am. Chem. Soc., 2010, 132, 7676–7683 CrossRef CAS PubMed; (f) M. Montag, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2012, 134, 10325–10328 CrossRef CAS PubMed; (g) M. H. Shaw, N. G. McCreanor, W. G. Whittingham and J. F. Bower, J. Am. Chem. Soc., 2015, 137, 463–468 CrossRef CAS PubMed.
- (a) M. Mendez, M. P. Munoz, C. Nevado, D. J. Cardenas and A. M. Echavarren, J. Am. Chem. Soc., 2001, 123, 10511–10520 CrossRef CAS PubMed; (b) P. Y. Toullec, C. M. Chao, Q. Chen, S. Gladiali, J. P. Genet and V. Michelet, Adv. Synth. Catal., 2008, 350, 2401–2408 CrossRef CAS; (c) L. M. Zhang, J. W. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271–2296 CrossRef CAS.
- (a) M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471–2526 CrossRef CAS PubMed; (b) L. Johansson, M. Tilset, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2000, 122, 10846–10855 CrossRef CAS; (c) J. S. Owen, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2006, 128, 2005–2016 CrossRef CAS PubMed; (d) H. A. Zhong, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2002, 124, 1378–1399 CrossRef CAS PubMed; (e) B. L. Madison, S. B. Thyme, S. Keene and B. S. Williams, J. Am. Chem. Soc., 2007, 129, 9538–9539 CrossRef CAS PubMed; (f) D. M. Crumpton-Bregel and K. I. Goldberg, J. Am. Chem. Soc., 2003, 125, 9442–9456 CrossRef CAS PubMed; (g) M. P. Jensen, D. D. Wick, S. Reinartz, P. S. White, J. L. Templeton and K. I. Goldberg, J. Am. Chem. Soc., 2003, 125, 8614–8624 CrossRef CAS PubMed; (h) B. S. Williams, A. W. Holland and K. I. Goldberg, J. Am. Chem. Soc., 1998, 121, 252–253 CrossRef; (i) G. S. Hill and R. J. Puddephatt, Organometallics, 1998, 17, 1478–1486 CrossRef CAS; (j) J. D. Scott and R. J. Puddephatt, Organometallics, 1983, 2, 1643–1648 CrossRef CAS; (k) F. Zhang, E. M. Prokopchuk, M. E. Broczkowski, M. C. Jennings and R. J. Puddephatt, Organometallics, 2006, 25, 1583–1591 CrossRef CAS.
- (a) R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560–564 CrossRef CAS PubMed; (b) A. Caballero and P. J. Perez, Chem. Soc. Rev., 2013, 42, 8809–8820 RSC.
- (a) S. H. Crosby, G. J. Clarkson, R. J. Deeth and J. P. Rourke, Organometallics, 2010, 29, 1966–1976 CrossRef CAS; (b) S. H. Crosby, G. J. Clarkson, R. J. Deeth and J. P. Rourke, Dalton Trans., 2011, 40, 1227–1229 RSC; (c) J. Mamtora, S. H. Crosby, C. P. Newman, G. J. Clarkson and J. P. Rourke, Organometallics, 2008, 27, 5559–5565 CrossRef CAS.
- (a) S. H. Crosby, H. R. Thomas, G. J. Clarkson and J. P. Rourke, Chem. Commun., 2012, 48, 5775–5777 RSC; (b) S. H. Crosby, G. J. Clarkson and J. P. Rourke, Organometallics, 2012, 31, 7256–7263 CrossRef CAS; (c) P. A. Shaw and J. P. Rourke, Dalton Trans., 2017, 46, 4768–4776 RSC; (d) P. A. Shaw, G. J. Clarkson and J. P. Rourke, Organometallics, 2016, 35, 3751–3762 CrossRef CAS.
- L. M. Rendina and R. J. Puddephatt, Chem. Rev., 1997, 97, 1735–1754 CrossRef CAS PubMed.
- (a) H. R. Thomas, R. J. Deeth, G. J. Clarkson and J. P. Rourke, Organometallics, 2011, 30, 5641–5648 CrossRef CAS; (b) S. H. Crosby, G. J. Clarkson and J. P. Rourke, Organometallics, 2011, 30, 3603–3609 CrossRef CAS.
- (a) A. D. Ryabov and A. K. Yatsimirsky, Inorg. Chem., 1984, 23, 789–790 CrossRef CAS; (b) A. D. Ryabov, Inorg. Chem., 1987, 26, 1252–1260 CrossRef CAS; (c) M. Albrecht, P. Dani, M. Lutz, A. L. Spek and G. v. Koten, J. Am. Chem. Soc., 2000, 122, 11822–11833 CrossRef CAS.
- (a) P. A. Shaw, J. M. Phillips, C. P. Newman, G. J. Clarkson and J. P. Rourke, Chem. Commun., 2015, 51, 8365–8368 RSC; (b) P. A. Shaw, J. M. Phillips, G. J. Clarkson and J. P. Rourke, Dalton Trans., 2016, 45, 11397–11406 RSC.
- (a) K. L. Hull, E. L. Lanni and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 14047–14049 CrossRef CAS PubMed; (b) S. R. Whitfield and M. S. Sanford, Organometallics, 2008, 27, 1683–1689 CrossRef CAS; (c) J. M. Racowski, N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18022–18025 CrossRef CAS PubMed.
- (a) G. W. V. Cave, N. W. Alcock and J. P. Rourke, Organometallics, 1999, 18, 1801–1803 CrossRef CAS; (b) G. W. V. Cave, F. P. Fanizzi, R. J. Deeth, W. Errington and J. P. Rourke, Organometallics, 2000, 19, 1355–1364 CrossRef CAS.
- P. S. Pregosin and R. W. Kunz, 31P and 13C NMR of Transition Metal Phosphine Complexes, Springer-Verlag: Berlin, 1979 Search PubMed.
- (a) P. S. Pregosin, Coord. Chem. Rev., 1982, 44, 247–291 CrossRef CAS; (b) E. Gabano, E. Marengo, M. Bobba, E. Robotti, C. Cassino, M. Botta and D. Osella, Coord. Chem. Rev., 2006, 250, 2158–2174 CrossRef CAS.
- S. Fornarini and M. E. Crestoni, Acc. Chem. Res., 1998, 31, 827–834 CrossRef CAS.
- (a) A. S. K. Hashmi, J. W. Bats, J. H. Choi and L. Schwarz, Tetrahedron Lett., 1998, 39, 7491–7494 CrossRef CAS; (b) G. W. Kabalka and R. M. Pagni, Tetrahedron, 1997, 53, 7999–8065 CrossRef CAS.
- (a) S. B. Zhao, G. Wu and S. Wang, Organometallics, 2008, 27, 1030–1033 CrossRef CAS; (b) K. A. Grice, M. L. Scheuermann and K. I. Goldberg, Top. Organomet. Chem., 2011, 35, 1–28 CrossRef CAS; (c) A. Vigalok, Acc. Chem. Res., 2015, 48, 238–247 CrossRef CAS PubMed.
- (a) K. Tatsumi, R. Hoffman, A. Yamamoto and J. K. Stille, Bull. Chem. Soc. Jpn., 1981, 54, 1857 CrossRef CAS; (b) T. A. Albright, J. K. Burdett and M. H. Whangbo, Orbital Interactions in Chemistry, Wiley and Sons Inc, 1985 Search PubMed.
- (a) S. Li, S.-F. Zhu, J.-H. Xie, S. Song, C.-M. Zhang and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 1172–1179 CrossRef CAS PubMed; (b) S.-F. Zhu, Y.-B. Yu, S. Li, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2012, 51, 8872–8875 CrossRef CAS PubMed; (c) C. Guo, D.-W. Sun, S. Yang, S.-J. Mao, X.-H. Xu, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2015, 137, 90–93 CrossRef CAS PubMed; (d) Y.-B. Yu, L. Cheng, Y.-P. Li, Y. Fu, S.-F. Zhu and Q.-L. Zhou, Chem. Commun., 2016, 52, 4812–4815 RSC; (e) S.-F. Zhu, J.-B. Xie, Y.-Z. Zhang, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2006, 128, 12886–12891 CrossRef CAS PubMed.
- C.-H. Chen and W.-Y. Yeh, Dalton Trans., 2013, 42, 2488–2494 RSC.
- (a) R. Corbo, D. C. Georgiou, D. J. D. Wilson and J. L. Dutton, Inorg. Chem., 2014, 53, 1690–1698 CrossRef CAS PubMed; (b) N. Wang, T. M. McCormick, S.-B. Ko and S. Wang, Eur. J. Inorg. Chem., 2012, 2012, 4463–4469 CrossRef CAS.
- (a) R. Tan, P. Jia, Y. Rao, W. Jia, A. Hadzovic, Q. Yu, X. Li and D. Song, Organometallics, 2008, 27, 6614–6622 CrossRef CAS; (b) R. Tan and D. Song, Inorg. Chem., 2011, 50, 10614–10622 CrossRef CAS PubMed.
- O. Saavedra-Díaz, R. Cerón-Camacho, S. Hernández, A. D. Ryabov and R. Le Lagadec, Eur. J. Inorg. Chem., 2008, 2008, 4866–4869 CrossRef.
- S. R. Whitfield and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 15142–15143 CrossRef CAS PubMed.
- (a) J. Vicente, A. Arcas, D. Bautista and P. G. Jones, Organometallics, 1997, 16, 2127–2138 CrossRef CAS; (b) J. Vicente, J. A. Abad, A. D. Frankland and M. C. R. de Arellano, Chem.–Eur. J., 1999, 5, 3066–3075 CrossRef CAS.
- C. P. Newman, K. Casey-Green, G. J. Clarkson, G. W. V. Cave, W. Errington and J. P. Rourke, Dalton Trans., 2007, 3170–3182 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details of synthetic procedures, variable temperature NMR spectra for 7 and a comparison of those spectra with those of 5 are available. The data from which the analysis of the equilibrium between a-Me7 and b-Me7 was made is included. Full details and discussions of the X-ray structures are available. CCDC 1540378–1540384. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01361b |
| This journal is © The Royal Society of Chemistry 2017 |