
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical space guided discovery of antimicrobial bridged bicyclic peptides against Pseudomonas aeruginosa and its biofilms†
Ivan
Di Bonaventura
a,
Xian
Jin
a,
Ricardo
Visini
a,
Daniel
Probst
a,
Sacha
Javor
a,
Bee-Ha
Gan
a,
Gaëlle
Michaud
a,
Antonino
Natalello
 b,
Silvia Maria
Doglia
b,
Thilo
Köhler
c,
Christian
van Delden
c,
Achim
Stocker
a,
Tamis
Darbre
a and
Jean-Louis
Reymond
*a
b,
Silvia Maria
Doglia
b,
Thilo
Köhler
c,
Christian
van Delden
c,
Achim
Stocker
a,
Tamis
Darbre
a and
Jean-Louis
Reymond
*a
aDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: jean-louis.reymond@dcb.unibe.ch
bDepartment of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126 Milan, Italy
cDepartment of Microbiology and Molecular Medicine, University of Geneva, and Service of Infectious Diseases, University Hospital of Geneva, Geneva, Switzerland
First published on 13th July 2017
Abstract
Herein we report the discovery of antimicrobial bridged bicyclic peptides (AMBPs) active against Pseudomonas aeruginosa, a highly problematic Gram negative bacterium in the hospital environment. Two of these AMBPs show strong biofilm inhibition and dispersal activity and enhance the activity of polymyxin, currently a last resort antibiotic against which resistance is emerging. To discover our AMBPs we used the concept of chemical space, which is well known in the area of small molecule drug discovery, to define a small number of test compounds for synthesis and experimental evaluation. Our chemical space was calculated using 2DP, a new topological shape and pharmacophore fingerprint for peptides. This method provides a general strategy to search for bioactive peptides with unusual topologies and expand the structural diversity of peptide-based drugs.
Introduction
The emergence of multi-antibiotic resistance in pathogenic bacteria represents one of the major public health threats today, calling for new approaches to develop antibiotics, in particular against Gram-negative bacteria such as Pseudomonas aeruginosa and Acinetobacter baumannii.1–4 One promising approach consists in searching for new antimicrobial peptides (AMPs), a versatile class of antibiotics produced by animals, plants and microorganisms. AMPs act by various mechanisms such as perturbing the cytoplasmic bacterial membrane, interacting with intracellular targets and modulating the host immune system.5–12Besides linear peptides, a variety of cyclic, polycyclic and lasso topologies also occur in AMPs.13–26 The importance of such unusual topologies is highlighted by polymyxin (Pmx), a lipidated branched cyclic decapeptide currently used as a last resort antibiotic against Gram negative multidrug resistant (MDR) pathogens, but against which resistance is appearing.27 Following on reports of antimicrobial activities in polycationic dendrimers including peptides,28–30 we recently screened combinatorial libraries of peptide dendrimers prepared by solid-phase peptide synthesis (SPPS)31–33 using an antimicrobial activity assay originally developed to search for new analogs of cyclic AMPs,34 and discovered antimicrobial peptide dendrimers (AMPDs) interacting with the bacterial membrane,35,36 which we later optimized by sequence design to AMPDs (e.g.G3KL, Fig. 1) with potent activities against various strains of P. aeruginosa and A. baumannii.37,38 Inspired by the remarkable serum stability of our peptide dendrimers, we later expanded our exploration of multi-branched synthetic peptides by SPPS to bridged bicyclic peptides (e.g.BCP27c, Fig. 1).39–41 These bicyclic peptides showed comparable serum stabilities to our peptide dendrimers but better defined structures and favourable properties as drug scaffolds.42 Herein we report the identification of antimicrobial bridged bicyclic peptides (AMBPs) active against Gram negative P. aeruginosa and its biofilms as the first example of applying our bicyclic peptides to a specific biological activity.
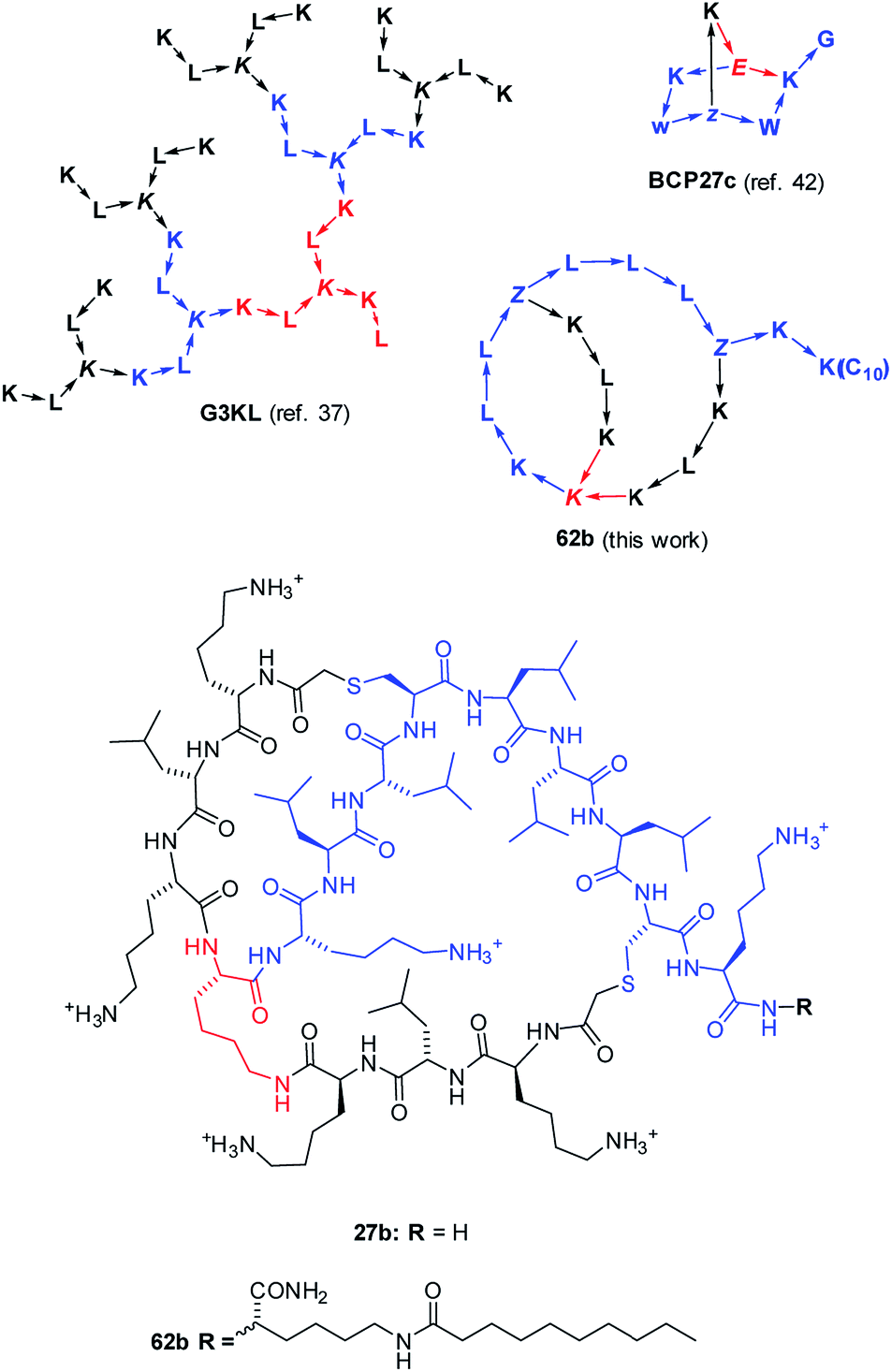 | ||
| Fig. 1 Structures of AMPD G3KL (ref. 37), bicyclic peptide BCP27c (calmodulin binder, ref. 42), and AMBPs 27b and 62b identified in this study. The peptides are shown with amino acids in one letter code with branching residues in italics and peptide bonds as arrows in C → N direction. Z = branching γ-homoglutamic acid (formed by ClAc ligation of cysteine). | ||
Because the synthesis of bridged bicyclic peptides requires purification of an intermediate prior to cyclization, a combinatorial chemistry and high-throughput screening approach was not readily applicable here as a tool for discovery.43–45 As an alternative compatible with the synthesis of only a few well-characterized compounds we used the idea of chemical space as a guide to select test compounds. A chemical space is a multi-dimensional virtual space in which dimensions correspond to different numerical descriptors of molecular structure, which together form a multi-dimensional feature vector, or fingerprint. Chemical space is a well-known concept in the area of small molecule drug discovery, where it is used to select small yet diverse subsets from large compound collections for experimental assays, as well as to optimize initial hits by searching for nearest neighbours.46–50 However it has not been used previously to design peptides.
As detailed below, using a chemical space encoding molecular shapes and pharmacophores to describe bicyclic peptides enabled us to logically explore their diversity using only a small number of test compounds, and later to optimize initial hits by focusing on nearest neighbours. This chemical space guided search led to the discovery of AMBP 27b, which we further optimized to its lipidated analog 62b acting on P. aeruginosa including several MDR clinical isolates (Fig. 1). We furthermore identified the related AMBPs 29b and 36b showing a potent anti-biofilm activity comparable to the recently reported D-enantiomeric peptide DJK5 (Scheme 1, Table 1).51 Our approach significantly extends previously reported computer-aided design methods for peptides, which are all based on extrapolating from the sequences of known linear AMPs to design new linear sequences,52–54 and might be generally useful to explore unusual peptide topologies and expand the structural diversity of peptide-based drugs.
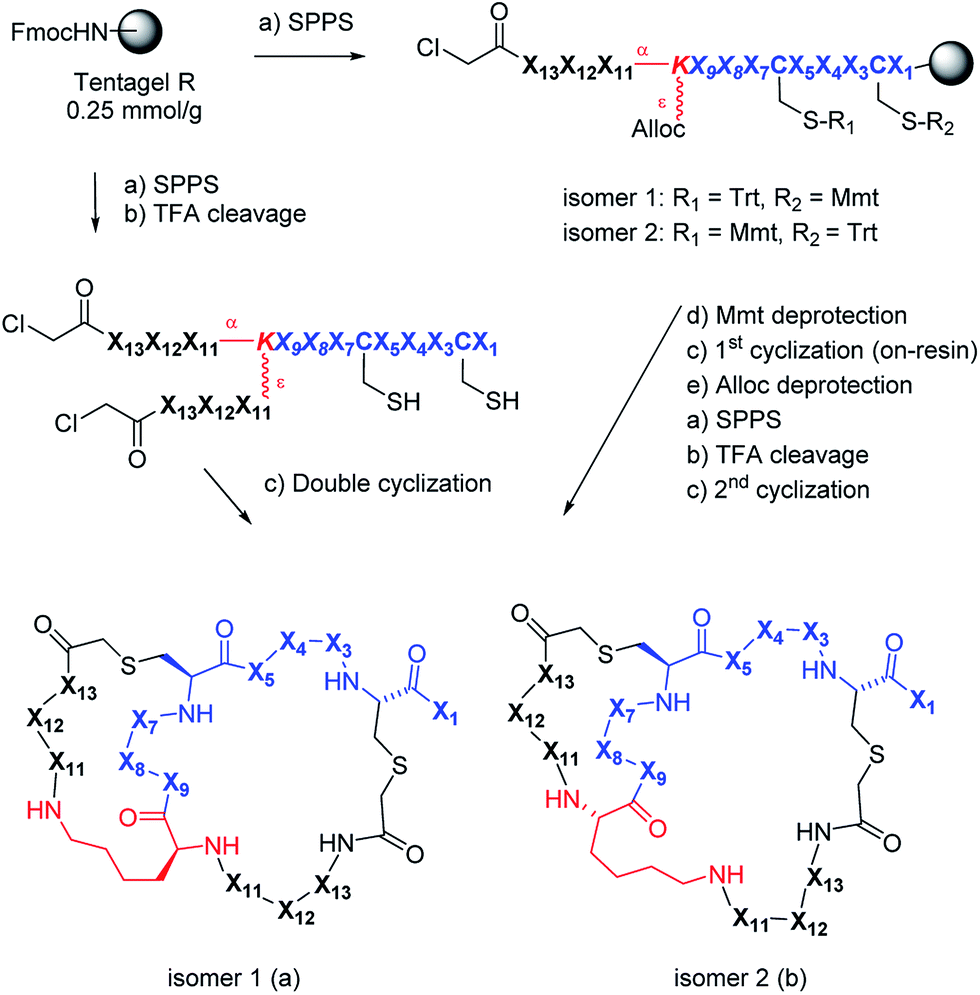 | ||
Scheme 1 Synthesis and structure of AMBPs containing a double thioether bridge. X1–X13 are variable L-amino acids or a deletion. Conditions: (a) (i) piperidine/DMF 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, (ii) FmocAAOH, oxyma/DIC, DMF or (ClCH2CO)2O, CH2Cl2; (b) TFA/TIS/DODT/H2O 94:2.5:2.5:1, v/v/v/v; (c) H2O/MeCN (50:50, v/v), KI (1 eq.), DIEA (5 eq.); (d) DCM/TES/TFA (96:3:1, v/v/v); (e) Pd(PPh3)4 (0.25 eq.), DCM, phenylsilane (25 eq.). :4, (ii) FmocAAOH, oxyma/DIC, DMF or (ClCH2CO)2O, CH2Cl2; (b) TFA/TIS/DODT/H2O 94:2.5:2.5:1, v/v/v/v; (c) H2O/MeCN (50:50, v/v), KI (1 eq.), DIEA (5 eq.); (d) DCM/TES/TFA (96:3:1, v/v/v); (e) Pd(PPh3)4 (0.25 eq.), DCM, phenylsilane (25 eq.). | ||
| No. | Sequencea | H | +c | MS calc./obs. [M] | MIC BR 151d | MIC PAO1d | MBICe | Dispersalf | MHCg |
|---|---|---|---|---|---|---|---|---|---|
| a Sequences use standard one-letter codes for amino acids, K = branching lysine, the peptide extended on the side chain is in parentheses, Z = γ-thia-homoglutamic acid (formed by ClAc ligation of cysteine), 1 and 2 indicate cyclization points using the SMILES formalism, B = diaminobutyric acid. All C-termini are carboxamides. b H = number of hydrophobic residues. c + = number of positive charges. d Minimal inhibitory concentration in μg mL−1 in Müller–Hinton (MH)-broth. e Minimal biofilm inhibitory conc. in μg mL−1. f Dispersal of preformed biofilm at 32 μg mL−1. MIC, MBIC and dispersal determinations were performed in independent triplicates with at least two experiments giving the same value. g Minimal hemolytic concentration in μg mL−1 on human red blood cells (hRBC). MHC determinations were performed in duplicate and averaged. h In this case [M + H]+ is observed together with TFA adducts, see ESI. i Measured with 8 μg mL−1 of 29b or 36b in the presence of 1.5 μg mL−1 of Pmx, FIC (fractional inhibitory concentration):6336b + Pmx = 0.155 (MBIC), 0.34 (dispersal), 29b + Pmx = 0.218 (MBIC), 0.34 (dispersal). Empty entries were not determined. | |||||||||
| 1a | 2 K(1)KZ1KLZ2L | 2 | 2 | 914.19/914.19 | >256 | >256 | |||
| 1b | 1 K(2)KZ1KLZ2L | 2 | 2 | 914.19/914.19 | 128 | >256 | |||
| 11a | L1LK(L2L)KLKZ2KKLZ1K | 6 | 5 | 1750.11/1750.11 | 64 | >256 | |||
| 11b | L1LK(L2L)KLKZ1KKLZ2K | 6 | 5 | 1750.11/1750.11 | 16 | >256 | |||
| 13a | K2LLK(K1LL)KLZ1KKLZ2 | 6 | 5 | 1750.11/1750.11 | 16 | >256 | |||
| 13b | K1LLK(K2LL)KLZ1KKLZ2 | 6 | 5 | 1750.11/1750.11 | 16 | >256 | |||
| 15 | L12KK(L12K)KLKZ21LLKZ12L | 6 | 5 | 1750.11/1750.11 | 16 | >256 | |||
| 19 | K12LKK(K12LK)LLLZ21KLKZ12L | 7 | 6 | 1991.29/1991.29 | 8 | >256 | |||
| 20 | 12LLKK(L12LK)LLLZ21KLKZ12L | 9 | 4 | 1961.27/1961.27 | 4 | 128 | 32 | 60% | |
| 24 | 12LLKK(L12LK)KLKZ21LLKZ12L | 8 | 5 | 1976.28/1976.28 | 4 | 128 | 16 | 50% | |
| 26a | 1KLLK(K2LL)KLLZ2KLKZ1K | 7 | 6 | 1991.29/1991.30 | 32 | >256 | 125 | ||
| 26b | 1KLLK(K2LL)KLLZ1KLKZ2K | 7 | 6 | 1991.29/1991.30 | 1 | 128 | 32 | 36% | 250 |
| 27a | 2KLKK(K1LK)KLLZ1LLLZ2K | 7 | 6 | 1991.29/1991.30 | 32 | 256 | 1000 | ||
| 27b | 1KLKK(K2LK)KLLZ1LLLZ2K | 7 | 6 | 1991.29/1991.30 | 1 | 32 | 32 | 12% | 2000 |
| 29a | 2KLLK(K1LL)KLLZ1KKKZ2L | 7 | 6 | 1991.29/1991.29 | 16 | >256 | 32 | 0% | 2000 |
| 29b | 1KLLK(K2LL)KLLZ1KKKZ2L | 7 | 6 | 1991.29/1991.29 | 2 | 256 | 8 | 100% | 2000 |
| 36a | 2KLKK(K1LK)KLLZ1LLKZ2K | 6 | 7 | 2006.30/2006.30 | 32 | >256 | >32 | 0% | 2000 |
| 36b | 1KLKK(K2LK)KLLZ1LLKZ2K | 6 | 7 | 2006.30/2006.30 | 16 | 64 | 8 | 100% | 2000 |
| 37a | 2KLKK(K1LK)KLLZ1KLKZ2L | 6 | 7 | 2006.30/2006.30 | 32 | >256 | 16 | 30% | |
| 37b | 1KLKK(K2LK)KLLZ1KLKZ2L | 6 | 7 | 2006.30/2006.30 | 32 | 256 | 16 | 100% | |
| 39a | 1LLKK(L2LK)KKLZ2LLLZ1K | 8 | 5 | 1976.28/1976.28 | 16 | 128 | |||
| 39b | 2LLKK(L1LK)KKLZ2LLLZ1K | 8 | 5 | 1976.28/1976.28 | 4 | 64 | >32 | ||
| 56a | 1KKKK(K2KK)KLLZ2LLLZ1K | 5 | 8 | 2021.31/2021.31 | 4 | 128 | >32 | 0% | |
| 56b | 2KKKK(K1KK)KLLZ2LLLZ1K | 5 | 8 | 2021.31/2022.31 | 2 | 32 | 32 | 75% | |
| 58a | 2KFKK(K1FK)KFFZ1FFFZ2K | 7 | 6 | 2229.18/2230.18 | 2 | >256 | 32 | 80% | |
| 58b | 1KFKK(K2FK)KFFZ1FFFZ2K | 7 | 6 | 2229.18/2229.18 | 2 | >256 | 32 | 0% | |
| 59 | 12KWKK(K12WK)KWWZ21WWWZ12K | 7 | 6 | 2502.25/2502.25 | 1 | 64 | 32 | 0% | |
| 60a | 2BLBK(B1LB)BLLZ1LLLZ2B | 7 | 6 | 1823.10/1824.10 | 4 | 64 | >32 | 0% | |
| 60b | 1BLBK(B2LB)BLLZ1LLLZ2B | 7 | 6 | 1823.10/1823.10 | 4 | 64 | >32 | 0% | |
| 61 | 12RLRK(R21LR)RLLZ21LLLZ12R | 7 | 6 | 2160.62/2160.52h | 1 | 64 | >32 | 0% | |
| 62a | 2KLKK(K1LK)KLLZ1LLLZ2KK(C10) | 8 | 6 | 2273.52/2273.52 | 4 | 32 | 128 | 15% | 125 |
| 62b | 1KLKK(K2LK)KLLZ1LLLZ2KK(C10) | 8 | 6 | 2273.52/2273.52 | 1 | 16–8 | 128 | 0% | 125 |
| Pmx | Polymyxin B | 4 | 5 | 1301.56/1301.56 | 4 | 2 | 16 | 100% | 2000 |
| +1 μg mL−129b | 1.5 | 100%i | |||||||
| +0.5 μg mL−136b | 1.5 | 100%i | |||||||
| DJK5 | Vqwrairvrvir | 7 | 4 | 1549.47/1549.47 | 16 | 4 | 100% | 2000 | |
Results and discussion
Fingerprint design and validation
We focused on the membrane disrupting properties of AMPs, which mostly depend on the number and relative position of cationic and hydrophobic residues in a peptide sequence. We envisioned that this information might be encoded as a topological atom pair fingerprint, a well-known method to describe shapes and pharmacophores in small molecules.55–59Our starting point was our recently reported 3DP fingerprint encoding the 3D-shape and pharmacophores of proteins from their experimental 3D-structure, which we used to create a web-based interactive map of the Protein Data Bank (PDB).60 The 3DP fingerprint comprises four atom categories: all atoms, cationic, anionic, and hydrophobic atoms. Distances between atoms are measured from the 3D-coordinates of the atoms in the experimental X-ray structures. To compute the fingerprint, each atom pair is converted to a Gaussian centered around the atom-pair distance. The sum of all Gaussians is then divided by HAC1.5 (HAC = heavy atom count, all non-hydrogen atoms) to reduce the dependency to molecule size, and sampled at 34 distances spanning the range 1.45–400 Å exponentially.
To obtain the corresponding atom-pair fingerprint 2DP based on topological distances, we used the formula for 3DP, but assigned all atoms to the α-carbon atom of their parent residue and counted distances between pairs of α-carbon atoms in bonds along the shortest path as determined from the 2D-structure of the peptides. The 2DP fingerprint comprised 136 values and defined a 136-dimensional chemical space, here called 2DP-space (Fig. 2a). Since 2DP used topological distances the fingerprint and its associated chemical space were well suited to explore peptides whose 3D-structure is yet unknown or poorly defined due to conformational flexibility.
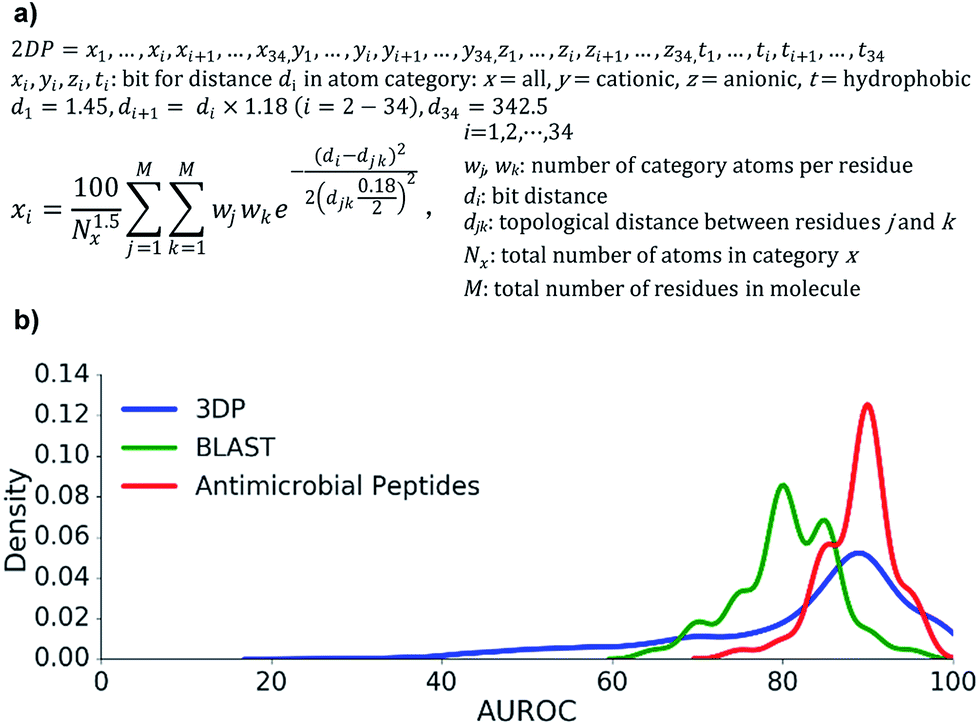 | ||
| Fig. 2 Design and benchmarking study of the 2DP fingerprint. (a) Equation for the calculation of 2DP entries for each atom category. (b) Performance of 2DP nearest neighbor searches for recovering various peptide types. Frequency histogram of AUROC values for recovering up to 100 3DP-nearest neighbours of each of the 2073 peptides up to 50 residues in the Protein Data Bank from all other entries within ±10% size of each query (blue curve), recovering 309 AMP sequences from 50 AMP sequences among 8815 decoys sequences in the UniProt databank (all limited to 50 residues, red curve), and recovering the closest BLAST analogs (with bit score > 19, 110-285 BLAST analogs) of 50 different 13-mer peptides from 8000 randomly scrambled peptide sequences of the query peptide (green curve). | ||
We performed three benchmarking studies to test the performance 2DP for virtual screening. Specifically, we tested if nearest neighbour searches in 2DP-space, using the city-block distance as similarity measure,61 could recover close analogs of linear peptides.
First, we extracted 2073 peptides up to 50 residues from the PDB, computed their 3DP from their X-ray structures and their 2DP from their sequences, respectively, and tested the recovery of 3DP-nearest neighbours of each peptide among a subset of peptides of similar size using 2DP similarity. The performance of each of the 2073 recoveries was quantified by the area under the receiver operating characteristic curve (AUROC). An AUROC > 80% indicative of good recovery characteristics78 was observed with over 70% of the peptides (Fig. 2b, blue line). This showed that 2DP partially perceived the 3D-shape and pharmacophore of peptides despite of only reading information from their 2D-structure.
Second, 2DP similarity searches were also performed to recover AMPs from decoys among linear peptides up to 50 residues in the UniProt database.79 The recovery characteristic were mostly excellent (AUROC > 80%, Fig. 2b red line), suggesting that 2DP was well suited to search for AMPs. Finally, 2DP was tested for recovering BLAST sequence analogs from scrambled sequences of short 13-mer peptides. Here also 2DP performed acceptably well (AUROC > 70%, Fig. 2b green line), showing that our fingerprint partly encoded peptide sequences even without direct encoding of amino acid residues.
Taken together, these retrospective analyses showed that 2DP-space offered a meaningful representation of linear peptides in terms of molecular shape, antimicrobial activities and sequences. In the absence of validation datasets for topologically diverse peptides, the above data with linear peptides combined with the similarity of 2DP with atom-pair fingerprints for encoding the shape and pharmacophores of small molecules, which are topologically diverse, convinced us to apply 2DP to explore bridged bicyclic peptides.
Synthesis design and 2DP-space analysis
We used 2DP-space to analyze a family of bridged bicyclic peptides resulting from a double chloroacetyl thioether (ClAc) ligation62 of a first generation peptide dendrimer containing two cysteine residues in its stem. The two possible isomers formed by cyclization were accessible by SPPS either as mixture, or as single isomers using an orthogonal protecting group strategy (Scheme 1).Using six possible residues typical for membrane active AMPs (lysine, arginine, diaminobutyric acid, leucine, phenylalanine, tryptophan) or a deletion at the 10 variable positions defined 243235706 possible bicyclic peptides. Focusing on lysine and leucine reduced the enumeration to only 20250 peptides (subset 1), 6230 of which had a ratio Lys/Leu ≈ 1 favorable for antimicrobial activity (subset 2).37 Subset 2 was sampled by centroid clustering to select 19 pairs of bicyclic peptide isomers between 8 and 16 residues in size for synthesis (subset 3). The three subsets spanned 2DP-diversity as broadly as the entire library as measured by a histogram of pairwise 2DP-distances (Fig. 3a). Nevertheless, visualization of the different subsets in the principal component analysis volume (PC1, PC2, PC3) color-coded by charge, molecular size and hydrophobic atoms showed that the two subsets with Lys/Leu ≈ 1 covered a somewhat more restricted region of 2DP-space compared with the complete library (Fig. 3b–e).
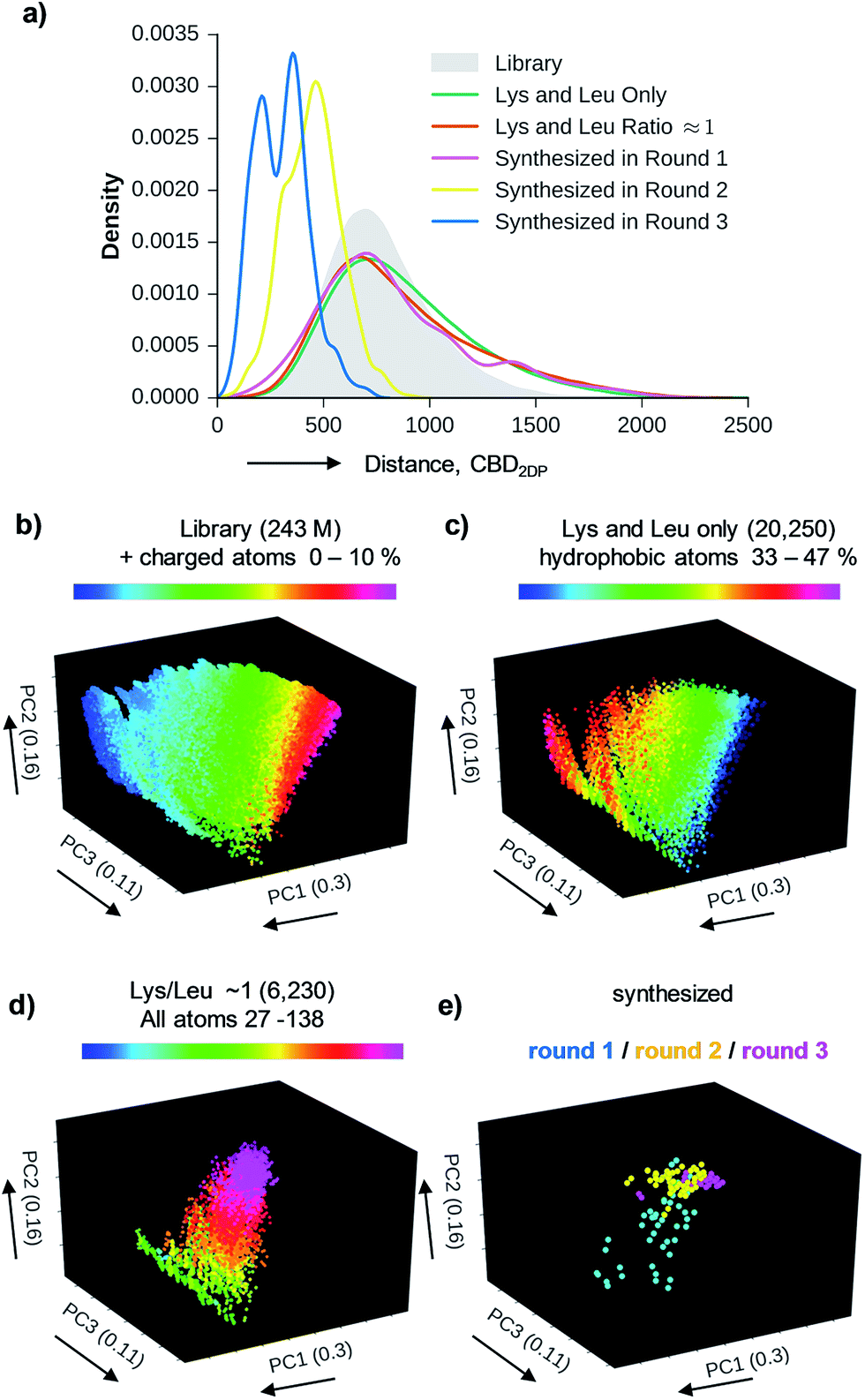 | ||
| Fig. 3 Chemical space analysis of bridged bicyclic peptides. (a) Distribution of pairwise 2DP-distances between compounds in the entire library and its three subsets. Distances are calculated as city-block distance in the 136-D 2DP space. (b)–(e) Color-coded images of the various sets of bicyclic peptides in the (PC1, PC2, PC3)-space obtained by PCA of 2DP-space. Variance covered: PC1 (30%), PC2 (16%), PC3 (11%). | ||
Discovery of AMBPs and biofilm inhibitors
The synthesis of the bicyclic peptides in subset 3 was performed by SPPS, purification of the chloroacetylated peptide dendrimer intermediate, double cyclization in solution and finally purification of the bicyclic peptide by preparative HPLC (Scheme 1, left). The syntheses yielded 28 bicyclic peptides as pure isomers separated by preparative HPLC, and 5 bicyclic peptides as inseparable mixtures of two isomers (1–19, Table S1,† active AMPDs shown in Table 1).While this initial series was inactive against P. aeruginosa (PAO1), we found several active bicyclic peptides against Bacillus subtilis (BR 151), a Gram-positive bacterium often sensitive to membrane disrupting compounds. Although an antimicrobial effect was already observable in 1b, the smallest bicyclic peptide in the series, activity generally increased with size and was strongly sequence and partly isomer dependent, with 19 showing the best activity in the series.
Assuming that AMBP 19 marked a bioactive region in 2DP-space we selected 100 of its nearest neighbours in 2DP space from the 20250 bicyclic peptides in subset 1 (Fig. 3). We then clustered these nearest neighbours in five groups and cherry-picked 3 to 4 sequences from each group for synthesis and experimental evaluation. These analogs had the same size as 19 but different sequences and ratios of leucine to lysine (20–38, Table S2,† the most active AMPDs are shown in Table 1). Synthesis and testing revealed additional hits against B. subtilis as well as 26b, 27b and 36b as the first AMBPs with significant activity against P. aeruginosa. Strikingly activity was systematically occurring in isomer b.
A third round of virtual screening, synthesis and testing of 2DP nearest neighbors of 27b (28 pure isomers and 5 isomeric mixtures, 39–57, Table S3,† active AMBPs shown in Table 1) did not yield improved activities, suggesting that 27b marked a local activity optimum. We therefore turned to designing specific analogs for further activity improvements. Analogs of 27b featuring exhaustive Leu → Phe (58b), Leu → Trp (59), Lys → Dab (60b) or Lys → Arg (61) replacements, which in principle probed compounds belonging to the larger virtual library of 243 M structures (Fig. 3a and b), were less active. On the other hand adding a side-chain decanoylated lysine at the C-terminus as a hydrophobic extension similar to that found in polymyxin gave AMBP 62b with increased activity against P. aeruginosa PAO1 (Table 1). AMBP 62b also showed moderate activity against several multidrug resistant P. aeruginosa clinical isolates, and Acinetobacter baumannii (Table S4†).
In view of the importance of biofilms in bacterial infections we tested all active AMBPs against P. aeruginosa biofilms using a protocol used earlier in our laboratory to investigate glycopeptide dendrimer biofilm inhibitors.64–66 These assays identified 29b and 36b as two particularly active AMBPs for biofilm inhibition and dispersal. Their activity was comparable to that of the recently reported linear all-D peptide biofilm inhibitor DJK5.51 As for DJK5, 29b and 36b were also less active against planctonic P. aeruginosa than against its biofilms. Remarkably 29b and 36b showed a synergistic effect with polymyxin on biofilm inhibition and dispersal (Table 1 and Fig. S4–S18†).
Extended activity profiling and membrane disruptive activity
The identified AMBPs were only moderately hemolytic (Table 1), and were generally resistant to serum degradation, presumably due to their branched cyclic structure limiting access of serum proteases to peptide bonds (Fig. 4a). Interestingly the more active isomers b were generally more resistant to serum degradation than isomers a. AMBP 27b efficiently released fluorescein from large unilamellar vesicles (LUVs) consisting of anionic phosphatidyl glycerol (PG), which mimic the bacterial membrane. The effect was stronger than with its less active isomer 27a, a trend also observed with the pair 26b/26a and 62b/62a (Fig. 4b and c and S19–S21†). Membrane perturbations were also visible in transmission electron microscopy (TEM) images of bacterial cells exposed to 27b or 62b showing the formation of multiple membrane protrusions similar to those observed with polymyxin (Fig. 4d, S22 and S23†).67 By contrast biofilm inhibitors 29b and 36b only weakly disrupted PG LUVs, suggesting that these might act by a different mechanism.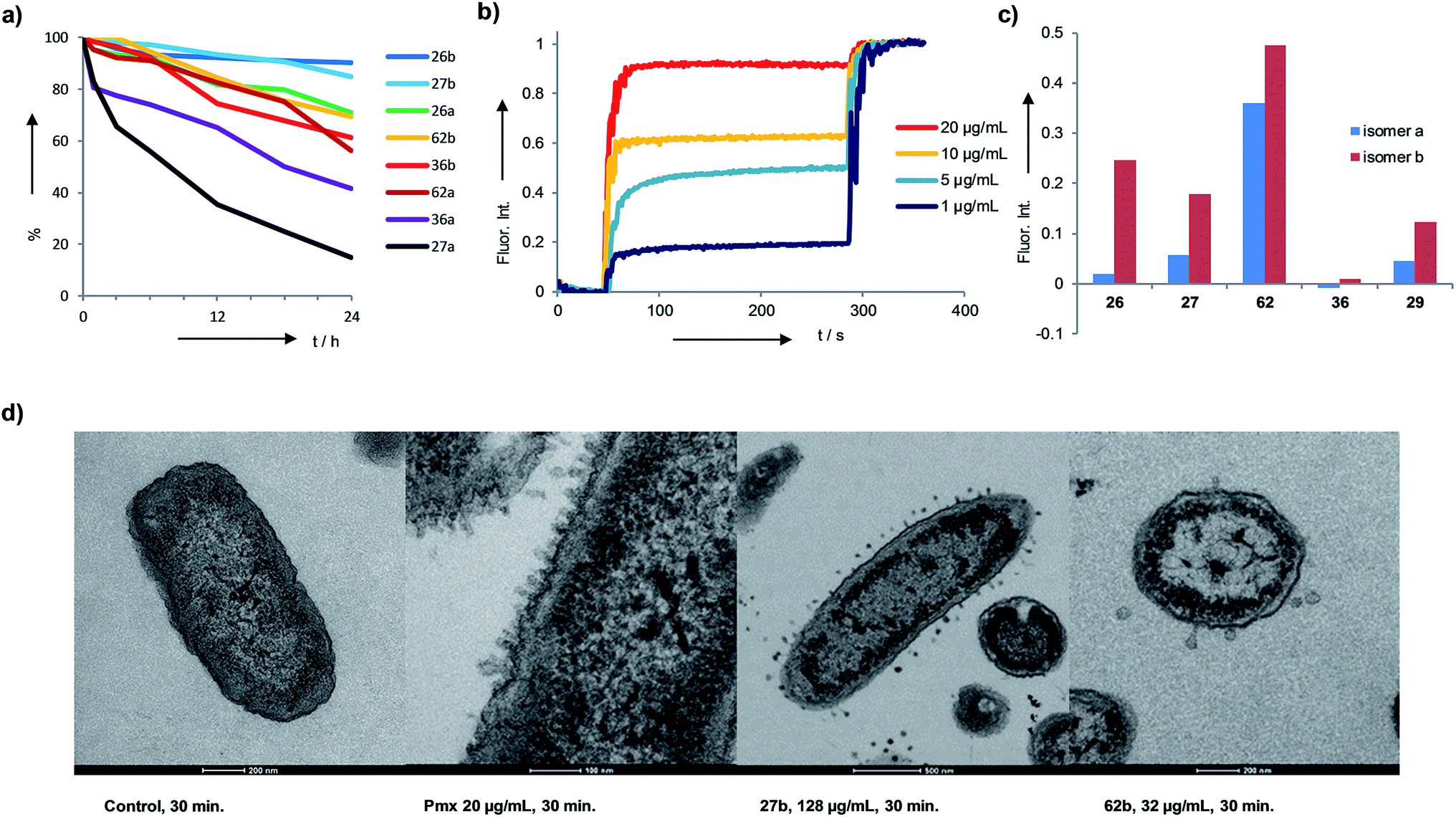 | ||
| Fig. 4 Serum stability and membrane disruptive properties of AMBPs. (a) Stability assay in human serum. AMBPs were incubated at 200 μM at 37 °C. The decrease of the peptide peak was followed by LC/MS. (b) Fluorescein leakage assay from PG vesicles with 62b. (c) Fluorescence intensity upon treatment of fluorescein loaded PG vesicles with 5 μg mL−1, t = 150 s, for the indicated AMBPs. (d) TEM images of P. aeruginosa cells treated with AMPs. | ||
Structural studies
We performed an isomer selective synthesis of 1a, 1b, 27a, and 27b using differentially protected cysteine building blocks and an ε-alloc protected lysine as branching point allowing sequential growth and cyclization of each branch independently (Scheme 1, right, see also Scheme S1† for details). These selective syntheses showed that for isomer b – in both cases the more active isomer eluting second during HPLC purification – the large dodecapeptide macrocycle runs through the ε-amino group of the branching lysine and is therefore longer than in isomer a (Scheme 1).We then turned to crystallography to gain additional structural insights into our AMBPs. While direct crystallization attempts with several AMBPs failed, we obtained the X-ray crystal structures of the smallest AMBPs 1a and 1b through their fucosylated derivatives of 63a and 63b in complex with P. aeruginosa lectin LecB. In 63a/b the sequence of 1a/b has been extended by an additional lysine residue at the C-terminus, whose side-chain is acylated with α-L-fucosylacetic acid. This fucoside appendage is used to anchor the peptide to the lectin, which crystallizes readily, a principle generally useful to access structural information on molecules otherwise difficult to crystallize.66,68,69 Here the crystal structures showed well defined peptide macrocycles for both AMBPs, which were immobilized in the crystal lattice not only via their anchoring fucosyl group, but also through several hydrogen bonds between the peptide backbone and the lectin (Table S5,†Fig. 5a and b). On the other hand, the amino acid side chains were mostly not visible indicating conformational flexibility. For both isomers the dihedral angles of the peptide backbone resided in allowed regions of the Ramachandran plot suggesting minimal strain (Fig. S24†), although a cis-peptide bond was present in the case of 63a. These structures also confirmed the isomer assignment derived from the selective synthesis. An additional X-ray structure was obtained for a LecB complex with 64a, an analog of 63a with a sequence designed to facilitate crystallization, and for which a well-resolved electron density was obtained including all residue side-chains, providing additional support for the structural assignment (PDB code 5NGQ, Table S5 and Fig. S25†).
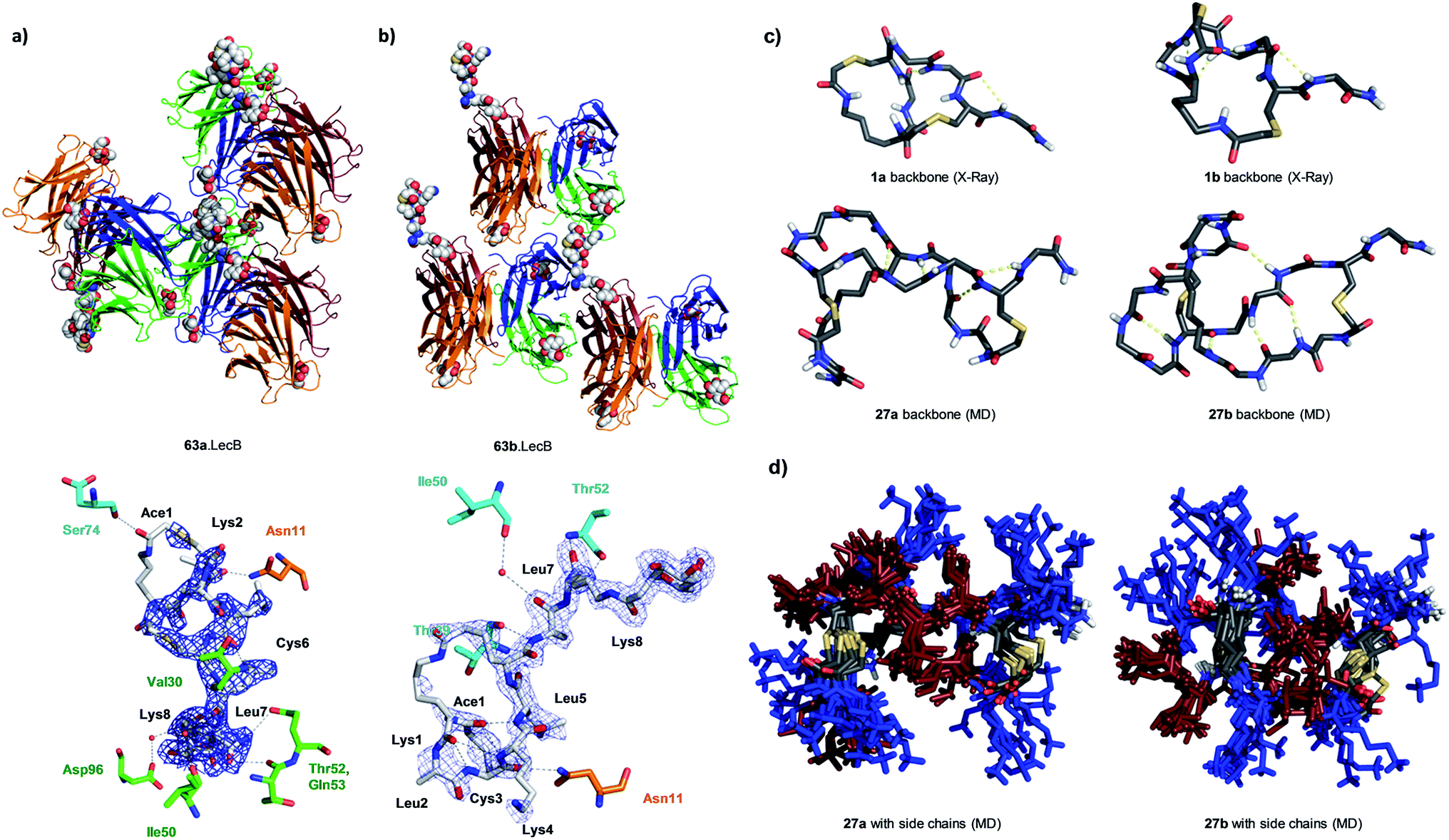 | ||
| Fig. 5 X-ray crystallography and molecular dynamics of AMBPs. (a) Overview (upper) and electron density with contacts (lower, distance cut-off 3.5 Å) of the X-ray structure the 63a·LecB complex (PDB code 518M). Each LecB subunit is shown in ribbon diagram with a different color and the visible ligands are shown in CPK models. Only one of the four fucose binding sites of the lectin tetramer is occupied by a fully resolved bicyclic peptide ligand, which is immobilized though contacts with other LecB units in the crystal lattice. In the lower image residues of the AMBP are written in black. LecB residues engaging in direct contact with the AMBP are drawn and labeled in the color of the corresponding subunit as shown in the upper figure. (b) Similar representation of 63b·LecB (PDB code 518X). (c) Structural model of the backbone for 1a/b from X-ray data of the fucosylated analogs 63a/b and for 27a/b from MD simulation. (d) 11 superimposed structures of the complete AMBP 27a/b from the last 100 ns of MD at 300 K after simulated annealing. Blue = Lys, brown = Leu. See also ESI Table S6, S7, Fig. S26 and S27.† | ||
LecB complexes with 65a or 65b, the fucosylated analogs of 27a and 27b, also gave well-diffracting crystals, however without observable electron densities for their ligand except for the anchoring fucosyl group, probably because these AMBP did not engage in immobilizing contacts with the lectin and retained an important conformational flexibility in the crystal lattice. We therefore performed molecular dynamics (MD) simulations in explicit water using the GROMACS molecular modeling package to gain a structural insight.70
A starting conformation was generated using the CORINA software.71 To access an unbiased set of stable conformations in solution, the model was first heated to 450 K to produce 11 snapshots of high energy structures that were progressively cooled to 300 K in a simulated annealing procedure. The low energy structures were then clustered and analyzed. These MD simulations indicated relatively stable and significantly different backbone geometries for each isomer, which resulted in significantly different overall structures despite of the flexibility of the lysine and leucine side chains (Fig. 5c and d).
In the structural model obtained by MD for 27a/b the hydrophobic leucine side chains and the positively charged lysine side chains formed distinct groups, partly as a result of the peptide sequence, which could be interpreted in terms of an amphiphilic structure that might explain membrane disruptive activity. However these models did not offer a convincing structural rationale for the stronger activity of b isomers compared to a isomers.
To gain additional insights we performed Fourier transform infrared (FTIR) spectroscopy in different media to probe whether the more active isomer 27b might undergo more pronounced conformational changes than 27a between water and the membrane environment since such changes are typically linked to antimicrobial and membrane disruptive activities of AMPs.5–12 FTIR spectra of 27a and 27b were recorded in aqueous PBS containing up to 90% trifluoroethanol (TFE) to mimic a transition from the aqueous environment to the membrane environment. Both isomers behaved similarly and showed a shift of the broad Amide I′ peak from 1638 cm−1 in aqueous PBS indicative of disordered hydrated α-helices to 1650 cm−1 in the presence of TFE characteristic for buried α-helices (Fig. 6). Similar FTIR data were observed with AMBPs 26a/b and biofilm inhibitor 29a/b (Fig. S28†).
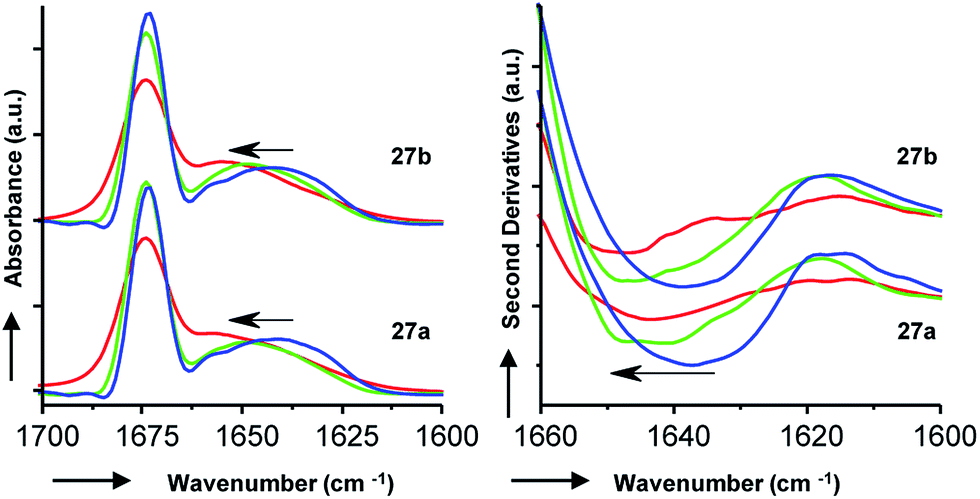 | ||
| Fig. 6 FTIR data with 27a/b, showing the Fourier self-deconvoluted spectra (left) and the second derivative (right). Bicyclic peptides were dissolved at a final concentration of 6 mg mL−1 in D2O based PBS (aq. 10 mM phosphate, 150 mM NaCl, pH 7.4) in the presence of 0% (blue lines), 50% (green lines), and 90% (red lines) of TFE at 25 °C. | ||
Since our AMBPs cannot fold into an α-helix due to their bicyclic topology we interpret the FTIR data in terms of a conformational rearrangement from a hydrated conformation in water to a more compact state in the membrane environment. However, the fact that both isomers undergo such conformational rearrangement to a similar extent implies that this effect cannot explain their different antimicrobial and vesicle leakage activities. We therefore propose that the stronger activity of b isomers versus a isomers results from differences in structure, as observed in the X-ray and MD studies above, rather than from differences in conformational flexibilities. Note that the chromatographic evidence that b isomers elute second and are therefore slightly more hydrophobic than a isomers suggests that they might engage in stronger interactions with the bacterial membrane.
Conclusion
In summary, the synthesis and evaluation of only a limited number of purified products guided by the concept of chemical space allowed us to discover 27b as the first AMBP active against P. aeruginosa. We optimized this AMBP to 62b, a lipidated analog with increased potency and activity against MDR clinical isolates. These AMBPs showed the hallmarks of a membrane disruptive activity. Screening of all active AMBPs for biofilm inhibition additionally led to the discovery of 29b and 36b as two potent P. aeruginosa biofilm inhibitors showing an interesting synergy with polymyxin. These discoveries suggest that bridged bicyclic peptides might represent a useful new class of antimicrobial peptides and biofilm inhibitors.To the best of our knowledge the chemical space guided approach demonstrated here with 2DP, a new shape and pharmacophore fingerprint inspired by computer-aided drug design methods for small molecules, is unprecedented for peptides. This method provides a general strategy to search for bioactive peptides with bicyclic or other unusual multi-branched topologies unsuitable for combinatorial synthesis and requiring careful evaluation of individually synthesized and purified compounds. Such exploratory approaches outside of standard topologies should help in expanding the structural diversity of peptide-based drugs. Although the sequences surveyed here only contain leucine and lysine, our 2DP fingerprint perceives detailed sequence elements (Fig. 2b, BLAST search) and should therefore be applicable to explore more diverse amino acid sequences and possibly other types of biological activities encountered with peptides.
Experimental
Cheminformatics
617853 bicyclic peptides, each of them giving two possible isomers. For each enumerated peptide sequence, the 2DP fingerprint was calculated for each isomer a and b separately.
235706 bicyclic peptides (including isomers) to reduce the dimensionality from the initial d = 136 to d = 3. The projection matrix yielded by the PCA of the exhaustively enumerated library was then also used to create the 3D plots of the subsets “Lys and Leu only”, “Lys/Leu” and “synthesized” (Fig. 2b–e).
Materials and reagents
All reagents were purchased from Fluorochem Ltd, Senn AG, Iris Biotech Gmbh, Sigma Aldrich, TCI (Tokyo Chemical Company). Chemicals were used as supplied and solvents were of analytical grade. Amino acids were used as the following derivatives: Fmoc-Cys(Trt)-OH, Fmoc-Cys(Mmt)-OH, Fmoc-Leu-OH, Fmoc-Lys(Boc)-OH, Fmoc-Lys(Fmoc)-OH, Fmoc-Lys(Alloc)-OH, Fmoc-Phe-OH, and Fmoc-Trp(Boc)-OH. Rink amide MBHA and Rink amide AM resins were purchased from Novabiochem, TentaGel Rink amide resin was purchased from Rapp Polymere. Cyclic peptide synthesis was performed manually and automatically by Biotage Initiator + Alstra and CEM Liberty Blue Automated Microwave Peptide Synthesizer.Analytics
Analytical RP-HPLC was performed with an Ultimate 3000 Rapid Separation LC-MS System (DAD-3000RS diode array detector) using an Acclaim RSLC 120 C18 column (2.2 μm, 120 Å, 3 × 50 mm, flow 1.2 mL min−1) from Dionex. Data recording and processing was done with Dionex Chromeleon Management System Version 6.80 (analytical RP-HPLC). All RP-HPLC were using HPLC-grade acetonitrile and Milli-Q deionized water. The elution solutions were: A MilliQ deionized water containing 0.05% TFA; D MilliQ deionized water/acetonitrile (10:90, v/v) containing 0.05% TFA. Preparative RP-HPLC was performed with a Waters automatic Prep LC Controller System containing the four following modules: Waters 2489 UV/Vis detector, Waters 2545 pump, Waters Fraction Collector III and Waters 2707 Autosampler. A Dr Maisch GmbH Reprospher column (C18-DE, 100 × 30 mm, particle size 5 μm, pore size 100 Å, flow rate 40 mL min−1) was used. Compounds were detected by UV absorption at 214 nm using a Waters 248 Tunable Absorbance Detector. Data recording and processing was performed with Waters ChromScope version 1.40 from Waters Corporation. All RP-HPLC were using HPLC-grade acetonitrile and Milli-Q deionized water. The elution solutions were: A MilliQ deionized water containing 0.1% TFA; D MilliQ deionized water/acetonitrile (10:90, v/v) containing 0.1% TFA. MS spectra, recorded on a Thermo Scientific LTQ OrbitrapXL, were provided by the MS analytical service of the Department of Chemistry and Biochemistry at the University of Bern (group PD Dr Stefan Schürch).
Solid-phase peptide synthesis
The peptides were synthesized as mixture of isomers a and b by SPPS, purified by preparative HPLC, and subsequently subjected to cyclization in solution. The final products were purified again, isomers a and b were separated during this second purification whenever possible.SPPS was performed either manually in polypropylene syringes (see below) or by automated SPPS using microwave synthesizers. The Fmoc-Rink amide resin (0.25 mmol g−1, 400 mg) was placed in a 10 mL polypropylene syringe. The resin was swollen in DCM for 60 min. After removal of DCM, the Fmoc protecting group was removed using a solution of piperidine in DMF (1:4, v/v). Stirring of the reaction mixture at any given step described below was performed by attaching the closed syringe to a rotating axis. The following conditions were used:
:4, v/v) for 20 min. After filtration the resin was washed with NMP (3 × 6 mL), MeOH (3 × 6 mL) and DCM (3 × 6 mL).
:20, v/v) were added to the resin. 5 eq. of DIEA were added and the reaction was stirred for 90 min. The resin was then washed with NMP (3 × 6 mL), MeOH (3 × 6 mL) and DCM (3 × 6 mL).
:20, v/v) were added to the resin. 6 eq. of DIEA were added and the reaction was stirred for 120 min. The resin was then washed with NMP (3 × 6 mL), MeOH (3 × 6 mL) and DCM (3 × 6 mL). The resulting coupling was checked by TNBS test.
:2.5:2.5:1, v/v/v/v) solution for 3 h. The peptide solutions were precipitated with 25 mL of TBME, centrifuged for 10 min at 3500 rpm, resuspended in TBME and centrifuged again; finally the precipitate was dried under high vacuum for 60 min. The crude was then dissolved in a water/acetonitrile mixture and purified by preparative RP-HPLC. The fractions containing the product (by LC-MS) were then lyophilized. Yields were calculated for the TFA salts of the products.
Cyclization
Bicyclic peptides were synthesized by submitting peptide dendrimers to high dilution conditions (1 mM) in a mixture of H2O/MeCN (50:50, v/v). The chloroacetylated peptide in H2O/MeCN was added dropwise to KI (1 eq.) and DIEA (10 eq.) in H2O/MeCN. The mixture was stirred for 60 min under argon atmosphere. The completion of the reaction was monitored by LC-MS after 60 min and lyophilized directly. The products, 2 isomers bicyclic peptides, were then subjected to RP-HPLC purification with 90 min gradient to achieve separation of the two isomers and all the fractions were analysed by LC-MS with a 7.5 min gradient and lyophilized twice. Yields were calculated for the TFA salts.
Isomer selective synthesis of bicyclic peptides
An orthogonal protecting groups strategy was used to synthesize selectively compounds 1a, 1b, 27a, 27b (Scheme S1†).The linear chloroacetylated peptide was first synthesized using the standard SPPS procedure described above, but using Fmoc-Lys(Alloc)-OH at the position of the branching lysine. The Mmt protecting group was then removed with 8 mL of DCM/TES/TFA (96:3:1, v/v/v) for 20 min. After filtration the resin was then washed with NMP (3 × 6 mL), MeOH (3 × 6 mL) and DCM (3 × 6 mL). The first thioether ligation was then performed on resin with a mixture of DMF/MeCN (80:20, v/v), KI (5 eq.), DIEA (5 eq.) overnight. After filtration, the resin was washed with NMP (3 × 6 mL), MeOH (3 × 6 mL) and DCM (3 × 6 mL).
The Alloc group of the branching lysine was then removed as follows: the polypropylene syringe was equipped with a septum and dried under vacuum for one hour. It was then swollen in dry DCM for 20 min under argon. After removal of the solvent, Pd(PPh3)4 (0.25 eq.) was dissolved in 8 mL of dry DCM and added to the resin under argon. Phenylsilane (25 eq.) was then added to the resin. The reaction was stirred under argon bubbling for 45 min (2×). The reagents were then removed by filtration and the resin washed with dry DCM (6 mL, 2 × 15 min), sodium diethylamino dithiocarboxylate (20 mM in DMF, 6 mL, 10 min) and finally with NMP (3 × 6 mL), MeOH (3 × 6 mL) and DCM (3 × 6 mL).
After Alloc deprotection the other branch of the peptide dendrimer was grown by SPPS, like mentioned above and last Fmoc deprotection and chloroacetylation were performed like usual. Cleavage, purification of monocyclic peptide and second cyclization were performed under standard conditions used previously.
Synthesis of bicyclic lipidated and fucosylated peptides
The bicyclic peptides 62a/b–65a/b were prepared as mixture of isomers a and b using the SPPS procedure described above starting with an additional Fmoc-Lys(Alloc)-OH building block as the first residue in the sequence.Before the last Fmoc deprotection the Alloc group was removed as described above, and the lysine side chain of the first residue was coupled with either decanoic acid or peracetylated α-L-fucosyl-acetic acid as follows: the acid (5 eq.), HATU (4 eq.) and DIPEA (10 eq.) were dissolved in 4.5 mL of NMP and 1.5 mL of DCM and added to the syringe. The mixture was stirred overnight. The solvent was filtrated and the washing step, NMP (2 × 6 mL), MeOH (2 × 6 mL) and DCM (2 × 6 mL), was performed.
The two N-terminal Fmoc groups were then removed and the two N-termini were chloroacetylated as described above. The dendrimer was then cleaved from the resin, purified, and cyclized in solution. For the fucose containing compounds the acetyl groups were removed prior to HPLC purification as follows: the cyclization solution was lyophilized, and the crude bicyclic glycopeptide was dissolved and stirred 24 h in a mixture of MeOH/H2O/NH3 (8:1:1). The MeOH was removed under vacuum and the solution was lyophilized. Finally, the bicyclic peptides were purified by preparative HPLC as above. Isomers a and b could be separated in all cases.
Antimicrobial activity by broth microdilution method
A colony of bacteria (Pseudomonas aeruginosa strain PA01 or Bacillus subtilis strain BS168) from glycerol stock was grown in LB medium overnight at 37 °C and 180 rpm shaking. The compounds were prepared as stock solutions of 8 mg mL−1 in sterilized MilliQ deionized water, added to the first well of 96-well sterile, polypropylene round bottom microtiter plates (TPP, untreated) and diluted serially by 1/2. The concentration of the bacteria was quantified by measuring absorbance at 600 nm and diluted to an OD600 of 0.022 in MH medium. The sample solutions (150 μL) were mixed with 4 μL diluted bacterial suspension with a final inoculation of about 5 × 105 CFU. For each test, two columns of the plate were kept for sterility control (MH medium only), growth control (MH medium with bacterial inoculum, no compound). The positive control, polymyxin B (starting with a concentration of 64 μg mL−1) in MH medium with bacterial inoculums, was introduced in the two first lines of the plate. The plates were incubated at 37 °C for ∼18 hours under static conditions. 15 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (1 mg mL−1 in sterilized MilliQ deionized water) were added to each well and the plates were incubated for 20–30 minutes at room temperature. The minimal inhibitory concentration (MIC) was defined as the lowest concentration of the dendrimer that inhibits the visible growth of the tested bacteria (yellow) with the unaided eye. For broth microdilution assay, polymyxin B was used as references. See also ESI Fig. S1–S3.†Pseudomonas aeruginosa biofilm inhibition and dispersal
96-well sterile, U-bottomed polystyrene microtiter plates (TPP Switzerland) were prepared by adding 200 μL of sterile deionized water to the peripheral wells to decrease evaporation from test wells. Aliquots of 180 μL of culture medium (0.25% (w/v) nutrient broth no. 2, Oxoid) containing desired concentration of the test compound were added to the internal wells. Compound containing solutions were sterile filtered (pore size 0.22 μm) prior to addition to the wells. Inoculum of Pseudomonas aeruginosa strain PAO1 was prepared from 5 mL overnight culture grown in LB broth overnight at 37 °C and 180 rpm shaking. Aliquots of 20 μL of overnight cultures, pre-washed in 0.25% (w/v) nutrient broth and normalized to an OD600 of 1, were inoculated into the test wells. Plates were incubated in a humid environment for 24–25 hours at 37 °C under static conditions. Wells were washed twice with 200 μL sterile deionized water before staining with 200 μL 0.25% (w/v) nutrient broth containing 0.5 mM WST-8 and 20 μM phenazine ethosulfate for 2.5–3 hours at 37 °C under static conditions. Afterwards, the well supernatants were transferred to a polystyrene flat bottomed 96-well plate (TPP Switzerland) and the absorbance was measured at 450 nm with a plate reader (SpectraMax250 from Molecular Devices).For biofilm dispersal, biofilm was formed as described above but in the absence of compound for 24 hours. Wells were washed twice with 200 μL sterile deionized water before adding 200 μL 0.25% (w/v) nutrient broth containing the desired concentration of compound. Compound containing solutions were sterile filtered (pore size 0.22 μm) prior to addition to the wells. After another 24 hours of incubation at 37 °C under static conditions, the well supernatants were removed and the wells were washed twice with 200 μL sterile deionized water. The biofilm was stained with 200 μL of 0.25% (w/v) nutrient broth containing 0.5 mM WST-8 and 20 μM phenazine ethosulfate for 2.5–3 hours at 37 °C under static conditions. The resulting absorbance was measured as in the biofilm inhibition experiment.
Hemolysis assay (MHC)
The approval for using the blood for hemolysis assay was given by the Blutspendedienst SRK Bern AG (Switzerland). For this experiment approval from an independent Ethics Committee was not required. To determine the minimal hemolytic concentration (MHC) stock solutions of 8 mg mL−1 of the peptide in H2O were prepared and 50 μL were diluted serially by 1/2 in 50 μL PBS (pH 7.4) in 96-well plate (Costar or Nunc, polystyrene, untreated). Human red blood cells (hRBC) were obtained by centrifugation of 1.5 mL of whole blood, from the blood bank of Bern, at 3000 rpm for 15 minutes at 40 °C. Plasma was discarded and the pellet was re-suspended in a 15 mL falcon tube up to 5 mL of PBS. The washing was repeated three times and the remaining pellet was re-suspended in 10 mL of PBS at a final hRBC concentration of 5%. The hRBC suspension (50 μL) was added to each well and the plate was incubated at room temperature for 4 hours. Minimal hemolytic concentration (MHC) end points were determined by visual determination of the wells after the incubation period. Controls on each plate included a blank medium control (50 μL PBS + 50 μL of hRBC suspension) and a hemolytic activity control (mQ-deionized water 50 μL + 50 μL hRBC suspension).Serum stability assay
Human serum was diluted in DMEM (1:4, v/v). Selected peptide was diluted in Tris buffer to a concentration of 400 μM. Aliquots of peptide solution (50 μL) were added to aliquots of serum (50 μL) in sterile Eppendorf tubes, to reach a peptide concentration of 200 μM during the assay. Samples were incubated at 37 °C under gentle stirring (350 rpm). Different samples (triplicates) were quenched at different time points (0/1/3/6/24 h) by precipitating serum proteins through the addition of (0.1 M) ZnSO4·7H2O/ACN (1:1) (0.1 M, 100 μL) and cooling down in ice bath. Protein precipitates were pelleted under centrifugation and the supernatants were sampled and evaporated to dryness in a centrifugal evaporator. Samples were re-suspended in a H2O/ACN (4:1, v/v) mixture and centrifuged again to remove residual protein precipitate. Supernatants were then sampled and analyzed by LC-MS. Experiment controls included a precipitation control for each peptide, to test their resistance to the protein precipitation conditions, and serum blanks, to check reproducibility over different serum batches. Two peaks originating from DMEM, surviving the incubation in serum and the protein precipitation conditions were used as internal standard.
Membrane interaction experiment
TEM transmission electron microscopy
Exponential phase of Pseudomonas aeruginosa PAO1 were washed with PBS and treated with 4× MIC of the corresponding compound in M63 minimal medium. Each time, 1 mL of the bacteria were centrifuged after 15, 30 and 60 min at 12000 rpm for 3 min and fixed overnight with 2.5% glutaraldehyde in 0.15 M HEPES with an osmolarity of 670 mOsm and adjusted to a pH of 7.35. The next day, PAO1 were washed with 0.15 M HEPES three times for 5 min, postfixed with 1% OsO4 in 0.1 M Na-cacodylate-buffer at 4 °C for 1 h. Thereafter, bacteria cells were washed in 0.1 M Na-cacodylate-buffer three times for 5 min and dehydrated in 70, 80, and 96% ethanol for 15 min each at room temperature. Subsequently, they were immersed in 100% ethanol three times for 10 min, in acetone two times for 10 min, and finally in acetone–Epon (1:1) overnight at room temperature. The next day, bacteria cells were embedded in Epon and hardened at 60 °C for 5 days. Sections were produced with an ultramicrotome UC6, first semithin sections (1 μm) for light microscopy that were stained with a solution of 0.5% toluidine blue O and then ultrathin sections (70–80 nm) for electron microscopy. The sections, mounted on single slot copper grids, were stained with uranyl acetate and lead citrate with an ultrastainer. Sections were then examined with a Tecnai Spirit transmission electron microscope equipped with two digital cameras.
X-ray crystallography
LecB lectin was expressed, purified and dialyzed as previously described.72 Co-crystallization of 63a/b with LecB lectin was carried out by the sitting drop method. The lyophilized protein was dissolved in water (5 mg mL−1) in the presence of salts (6 mM CaCl2 and MgCl2). The compounds were added to the protein at a 20:1 molar excess, taking into account that the biological unit of LecB is a homo-tetramer. Crystals were obtained within five days after mixing 1.5 μL of LecB ligand-complex with 1.5 μL of reservoir solution at 18 °C. Primary crystallization conditions were found in Crystal screens I/II (Hampton Research, Laguna Niguel, CA, USA). Crystals of highest diffraction were obtained from condition 0.2 M ammonium acetate, 0.1 M sodium citrate tribasic dehydrate pH 5.6 and 30% w/v polyethylene glycol 4000, Crystal Screen I 9 in the case of 63b. In the case of 63a the best condition was 0.2 M sodium acetate trihydrate, 0.1 M Tris hydrochloride pH 8.5 and 30% w/v polyethylene glycol 4000, which is Crystal Screen I 22. The structures were solved using the XDS,73 CCP4,74 the phenix75 program suite and the Coot76 graphical program. Pictures were done using the PyMol software.77
The crystallization of 64a succeeded with the soaking method. The apo crystals were grown in the same way as described above for the co-crystallization. Then different amounts of ligand were added. The best result was observed in the Crystal Screen I condition 47 which contain 0.1 M sodium acetate trihydrate pH 4.6 and 2.0 M ammonium sulfate. The ligand was added in a volume of 2 μL which results in a final concentration factor of 12.3 for the ligand compared with the protein concentration. All crystallization attempts with 64b and 65a/b either gave no crystals or well-resolved crystals in which only the fucosylated linker gave observable electron density.
Molecular dynamics
Molecular dynamics (MD) simulations were performed for bicyclic peptide isomers 27a and 27b using GROMACS software version 2016.1 and the Gomos53a6 force field.70 The starting topology was built by adding the non-natural bonds to the corresponding linear peptide and the initial coordinates were generated using CORINA.71 A dodecahedral box was created around the protein 1.0 nm from the edge of the protein and filled with extended simple point charge water molecules. Sodium and chloride ions were added to produce an electroneutral solution at a final concentration of 0.15 M NaCl.The energy was minimized using a steepest gradient method to remove any close contacts before the system was subjected to a two-phase position-restrained MD equilibration procedure. The system was first allowed to evolve for 1.0 ns in a canonical NVT (N is the number of particles, V the system volume, and T the temperature) ensemble at 300 K before pressure coupling was switched on and the system was equilibrated for an additional 50 ns in the NPT (P is the system pressure) ensemble at 1.0 bar.
This pre-equilibrated system was used as the starting point for the simulated annealing molecular dynamics (SA-MD) run. The system was heated to 450 K and high energy conformers were sampled at 100, 150, 200, 250 and 300 ns. Each of the conformers was the cooled from 450 K to 300 K over 50 ns and allowed to evolve for an additional 200 ns. All bond lengths were constrained to their equilibrium values by using the LINCS algorithm. The neighbor list for the calculation of nonbonded interactions was updated every five time steps with a cutoff of 1.0 nm with a step size of 2 fs. A twin range cutoff of 1.0 nm was used for both Coulomb and Lennard-Jones interactions. The system was split into two groups, “Protein” and “Non-Protein”, which were coupled separately to a temperature bath using the V-rescale algorithm with a time constant of 0.1 ps while the pressure coupling was conducted using an isotropic Parrinello–Rahman barostat with a time constant of 2.0 ps.
The last 100 ns (150 → 250 ns) of each SA-MD run were used in the subsequent analysis. To obtain a representative conformer for each SA-MD run, the last 100 ns (10001 frames) of each run were clustered using an RMSD cut-off to 0.12 nm. The large number of clusters combined with the very large percentage of structures in the top cluster is an indication of the stability of the one main conformer in each case (Table S6†). The PyMol Molecular Graphics System, version 1.8 (Schrödinger, LLC), was used to create structural models.
FTIR spectroscopy
Lyophilized bridged bicyclic peptides were dissolved at a final concentration of 6 mg mL−1 in D2O based PBS (aq. 10 mM phosphate, 150 mM NaCl, pH 7.4) in the presence of 0%, 50%, and 90% of TFE. An aliquot of 15 μL of the peptide solution was placed in a home-made transmission cell with two BaF2 windows and a Teflon spacer of 100 μm. FTIR spectra were acquired in transmission mode by the Varian 610-IR infrared microscope coupled to the Varian 670-IR FTIR spectrometer and equipped with a mercury cadmium telluride nitrogen-cooled detector. The variable microscope aperture was adjusted to 200 μm diameter. Measurements were performed under the following conditions: 2 cm−1 of spectral resolution, 25 kHz of scan speed, triangular apodization, and 2000 scan coadditions. The peptide FTIR spectra were obtained after subtraction of the solvent spectra, collected under identical conditions, and were normalized at the Amide I′ band area.The Fourier-self-deconvolution (FSD) of the Amide I′ band was performed by the Resolutions-Pro software (Varian Australia Pty Ltd.) by using an enhancement factor K = 2.4 and half-bandwidth HW = 13 cm−1. Second derivative spectra were calculated following the Savitsky–Golay method, after a binomial 11 points smoothing of the spectra by the GRAMS-32 software (Galactic Industries, Salem, NH).
Acknowledgements
This work was supported financially by the Swiss National Science Foundation. We thank Maurane Robadey for technical help in the synthesis of fucosylated bicyclic peptides, and the staff at the Swiss Light Source, Beamline X06DA (PXIII), Villigen, Switzerland, for support during data collection. We thank ChemAxon Pvt. Ltd. for providing a free academic license for their products.Notes and references
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- P. D. Cotter, R. P. Ross and C. Hill, Nat. Rev. Microbiol., 2013, 11, 95–105 CrossRef CAS PubMed.
- Y. Y. Liu, Y. Wang, T. R. Walsh, L. X. Yi, R. Zhang, J. Spencer, Y. Doi, G. Tian, B. Dong, X. Huang, L. F. Yu, D. Gu, H. Ren, X. Chen, L. Lv, D. He, H. Zhou, Z. Liang, J. H. Liu and J. Shen, Lancet Infect. Dis., 2016, 16, 161–168 CrossRef CAS PubMed.
- P. Nordmann and L. Poirel, Clin. Microbiol. Infect., 2016, 22, 398–400 CrossRef CAS PubMed.
- P. M. Hwang and H. J. Vogel, Biochem. Cell Biol., 1998, 76, 235–246 CrossRef CAS PubMed.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- H. Jenssen, P. Hamill and R. E. Hancock, Clin. Microbiol. Rev., 2006, 19, 491–511 CrossRef CAS PubMed.
- L. T. Nguyen, E. F. Haney and H. J. Vogel, Trends Biotechnol., 2011, 29, 464–472 CrossRef CAS PubMed.
- K. M. G. O'Connell, J. T. Hodgkinson, H. F. Sore, M. Welch, G. P. C. Salmond and D. R. Spring, Angew. Chem., Int. Ed. Engl., 2013, 52, 10706–10733 CrossRef PubMed.
- S. C. Mansour, O. M. Pena and R. E. Hancock, Trends Immunol., 2014, 35, 443–450 CrossRef CAS PubMed.
- J. Lakshmaiah Narayana and J. Y. Chen, Peptides, 2015, 72, 88–94 CrossRef CAS PubMed.
- L. Angelique, W. J. Frederik, J. Garmi and P. L. Hester du, Molecules, 2015, 20, 15392–15433 CrossRef CAS PubMed.
- L. H. Kondejewski, M. Jelokhani-Niaraki, S. W. Farmer, B. Lix, C. M. Kay, B. D. Sykes, R. E. Hancock and R. S. Hodges, J. Biol. Chem., 1999, 274, 13181–13192 CrossRef CAS PubMed.
- M. Wu and R. E. Hancock, J. Biol. Chem., 1999, 274, 29–35 CrossRef CAS PubMed.
- S. Fernandez-Lopez, H. S. Kim, E. C. Choi, M. Delgado, J. R. Granja, A. Khasanov, K. Kraehenbuehl, G. Long, D. A. Weinberger, K. M. Wilcoxen and M. R. Ghadiri, Nature, 2001, 412, 452–455 CrossRef CAS PubMed.
- A. Wessolowski, M. Bienert and M. Dathe, J. Pept. Res., 2004, 64, 159–169 CrossRef CAS PubMed.
- V. Dartois, J. Sanchez-Quesada, E. Cabezas, E. Chi, C. Dubbelde, C. Dunn, J. Granja, C. Gritzen, D. Weinberger, M. R. Ghadiri and T. R. Parr, Antimicrob. Agents Chemother., 2005, 49, 3302–3310 CrossRef CAS PubMed.
- D. I. Chan, E. J. Prenner and H. J. Vogel, Biochim. Biophys. Acta, 2006, 1758, 1184–1202 CrossRef CAS PubMed.
- M. Jelokhani-Niaraki, L. H. Kondejewski, L. C. Wheaton and R. S. Hodges, J. Med. Chem., 2009, 52, 2090–2097 CrossRef CAS PubMed.
- D. Oh, J. Sun, A. Nasrolahi Shirazi, K. L. LaPlante, D. C. Rowley and K. Parang, Mol. Pharm., 2014, 11, 3528–3536 CrossRef CAS PubMed.
- A. C. Conibear and D. J. Craik, Angew. Chem., Int. Ed. Engl., 2014, 53, 10612–10623 CrossRef CAS PubMed.
- S. Finger, A. Kerth, M. Dathe and A. Blume, Biochim. Biophys. Acta, 2015, 1848, 2998–3006 CrossRef CAS PubMed.
- B. Mojsoska and H. Jenssen, Pharmaceuticals, 2015, 8, 366–415 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef CAS PubMed.
- J. P. Tam, S. Wang, K. H. Wong and W. L. Tan, Pharmaceuticals, 2015, 8, 711–757 CrossRef CAS PubMed.
- M. Montalban-Lopez, A. J. van Heel and O. P. Kuipers, FEMS Microbiol. Rev., 2017, 41, 5–18 CrossRef PubMed.
- A. O. Olaitan, S. Morand and J. M. Rolain, Front. Microbiol., 2014, 5, 643 Search PubMed.
- J. P. Tam, Y. A. Lu and J. L. Yang, Eur. J. Biochem., 2002, 269, 923–932 CrossRef CAS PubMed.
- A. W. Young, Z. Liu, C. Zhou, F. Totsingan, N. Jiwrajka, Z. Shi and N. R. Kallenbach, MedChemComm, 2011, 2, 308–314 RSC.
- M. A. Mintzer, E. L. Dane, G. A. O'Toole and M. W. Grinstaff, Mol. Pharm., 2012, 9, 342–354 CrossRef CAS PubMed.
- A. Clouet, T. Darbre and J. L. Reymond, Angew. Chem., Int. Ed., 2004, 43, 4612–4615 CrossRef CAS PubMed.
- N. Maillard, A. Clouet, T. Darbre and J. L. Reymond, Nat. Protoc., 2009, 4, 132–142 CrossRef CAS PubMed.
- J.-L. Reymond and T. Darbre, Org. Biomol. Chem., 2012, 10, 1483–1492 CAS.
- V. S. Fluxa, N. Maillard, M. G. Page and J. L. Reymond, Chem. Commun., 2011, 47, 1434–1436 RSC.
- M. Stach, N. Maillard, R. U. Kadam, D. Kalbermatter, M. Meury, M. G. P. Page, D. Fotiadis, T. Darbre and J.-L. Reymond, MedChemComm, 2012, 3, 86–89 RSC.
- H. K. Ravi, M. Stach, T. A. Soares, T. Darbre, J. L. Reymond and M. Cascella, Chem. Commun., 2013, 49, 8821–8823 RSC.
- M. Stach, T. N. Siriwardena, T. Kohler, C. van Delden, T. Darbre and J. L. Reymond, Angew. Chem., Int. Ed., 2014, 53, 12827–12831 CrossRef CAS PubMed.
- J. Pires, T. N. Siriwardena, M. Stach, R. Tinguely, S. Kasraian, F. Luzzaro, S. L. Leib, T. Darbre, J. L. Reymond and A. Endimiani, Antimicrob. Agents Chemother., 2015, 59, 7915–7918 CrossRef CAS PubMed.
- M. Bartoloni, R. U. Kadam, J. Schwartz, J. Furrer, T. Darbre and J.-L. Reymond, Chem. Commun., 2011, 47, 12634–12636 RSC.
- J. L. Reymond and T. Darbre, Chimia, 2013, 67, 864–867 CrossRef CAS PubMed.
- M. Bartoloni, S. Waltersperger, M. Bumann, A. Stocker, T. Darbre and J. L. Reymond, ARKIVOC, 2014, III, 113–123 Search PubMed.
- M. Bartoloni, X. Jin, M. J. Marcaida, J. Banha, I. Dibonaventura, S. Bongoni, K. Bartho, O. Grabner, M. Sefkow, T. Darbre and J.-L. Reymond, Chem. Sci., 2015, 6, 5473–5490 RSC.
- A. Nefzi, J. M. Ostresh and R. A. Houghten, Chem. Rev., 1997, 97, 449–472 CrossRef CAS PubMed.
- R. Fleeman, T. M. LaVoi, R. G. Santos, A. Morales, A. Nefzi, G. S. Welmaker, J. L. Medina-Franco, M. A. Giulianotti, R. A. Houghten and L. N. Shaw, J. Med. Chem., 2015, 58, 3340–3355 CrossRef CAS PubMed.
- C. Heinis and G. Winter, Curr. Opin. Chem. Biol., 2015, 26, 89–98 CrossRef CAS PubMed.
- T. I. Oprea and J. Gottfries, J. Comb. Chem., 2001, 3, 157–166 CrossRef CAS PubMed.
- S. Renner, M. Popov, A. Schuffenhauer, H. J. Roth, W. Breitenstein, A. Marzinzik, I. Lewis, P. Krastel, F. Nigsch, J. Jenkins and E. Jacoby, Future Med. Chem., 2011, 3, 751–766 CrossRef CAS PubMed.
- T. Scior, A. Bender, G. Tresadern, J. L. Medina-Franco, K. Martinez-Mayorga, T. Langer, K. Cuanalo-Contreras and D. K. Agrafiotis, J. Chem. Inf. Model., 2012, 52, 867–881 CrossRef CAS PubMed.
- K. Heikamp and J. Bajorath, Chem. Biol. Drug Des., 2013, 81, 33–40 CAS.
- J. L. Reymond, Acc. Chem. Res., 2015, 48, 722–730 CrossRef CAS PubMed.
- C. de la Fuente-Nunez, F. Reffuveille, S. C. Mansour, S. L. Reckseidler-Zenteno, D. Hernandez, G. Brackman, T. Coenye and R. E. Hancock, Chem. Biol., 2015, 22, 196–205 CrossRef CAS PubMed.
- A. Cherkasov, K. Hilpert, H. Jenssen, C. D. Fjell, M. Waldbrook, S. C. Mullaly, R. Volkmer and R. E. Hancock, ACS Chem. Biol., 2009, 4, 65–74 CrossRef CAS PubMed.
- C. D. Fjell, J. A. Hiss, R. E. Hancock and G. Schneider, Nat. Rev. Drug Discovery, 2012, 11, 37–51 CAS.
- E. Y. Lee, B. M. Fulan, G. C. Wong and A. L. Ferguson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13588–13593 CrossRef CAS PubMed.
- R. E. Carhart, D. H. Smith and R. Venkataraghavan, J. Chem. Inf. Comput. Sci., 1985, 25, 64–73 CrossRef CAS.
- R. P. Sheridan, M. D. Miller, D. J. Underwood and S. K. Kearsley, J. Chem. Inf. Comput. Sci., 1996, 36, 128–136 CrossRef CAS.
- G. Schneider, W. Neidhart, T. Giller and G. Schmid, Angew. Chem., Int. Ed. Engl., 1999, 38, 2894–2896 CrossRef CAS PubMed.
- M. Awale and J. L. Reymond, J. Chem. Inf. Model., 2014, 54, 1892–1897 CrossRef CAS PubMed.
- M. Awale, X. Jin and J. L. Reymond, J. Cheminf., 2015, 7, 3 Search PubMed.
- X. Jin, M. Awale, M. Zasso, D. Kostro, L. Patiny and J. L. Reymond, BMC Bioinf., 2015, 16, 339 CrossRef PubMed.
- A. A. Khalifa, M. Haranczyk and J. Holliday, J. Chem. Inf. Model., 2009, 49, 1193–1201 CrossRef PubMed.
- N. A. Uhlich, T. Darbre and J. L. Reymond, Org. Biomol. Chem., 2011, 9, 7071–7084 CAS.
- J. Meletiadis, S. Pournaras, E. Roilides and T. J. Walsh, Antimicrob. Agents Chemother., 2010, 54, 602–609 CrossRef CAS PubMed.
- E. M. Johansson, S. A. Crusz, E. Kolomiets, L. Buts, R. U. Kadam, M. Cacciarini, K. M. Bartels, S. P. Diggle, M. Camara, P. Williams, R. Loris, C. Nativi, F. Rosenau, K. E. Jaeger, T. Darbre and J. L. Reymond, Chem. Biol., 2008, 15, 1249–1257 CrossRef CAS PubMed.
- J. L. Reymond, M. Bergmann and T. Darbre, Chem. Soc. Rev., 2013, 42, 4814–4822 RSC.
- G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty, A. Stocker, T. Darbre and J.-L. Reymond, Chem. Sci., 2016, 7, 166–182 RSC.
- M. Koike, K. Iida and T. Matsuo, J. Bacteriol., 1969, 97, 448–452 CAS.
- E. Mitchell, C. Houles, D. Sudakevitz, M. Wimmerova, C. Gautier, S. Perez, A. M. Wu, N. Gilboa-Garber and A. Imberty, Nat. Struct. Biol., 2002, 9, 918–921 CrossRef CAS PubMed.
- P. Roethlisberger, A. Istrate, M. J. Marcaida Lopez, R. Visini, A. Stocker, J. L. Reymond and C. J. Leumann, Chem. Commun., 2016, 52, 4749–4752 RSC.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- J. Sadowski and J. Gasteiger, Chem. Rev., 1993, 93, 2567–2581 CrossRef CAS.
- H. Funken, K. M. Bartels, S. Wilhelm, M. Brocker, M. Bott, M. Bains, R. E. Hancock, F. Rosenau and K. E. Jaeger, PLoS One, 2012, 7, e46857 CAS.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- Schrodinger The Pymol Molecular Graphics System, Version 1.8, LLC, 2015 Search PubMed.
- A. N. Jain and A. Nicholls, J. Comput.-Aided Mol. Des., 2008, 22, 133–139 CrossRef CAS PubMed.
- Uniprot: The Universal Protein Knowledgebase, Nucleic Acids Res., 2017, 45, D158–D169 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of all compounds, crystallography tables, and characterization of all compounds. See DOI: 10.1039/c7sc01314k |
| This journal is © The Royal Society of Chemistry 2017 |