
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of template-stabilized active and earth-abundant oxygen evolution catalysts in acid†
Michael
Huynh
,
Tuncay
Ozel
,
Chong
Liu
 ,
Eric C.
Lau
and
Daniel G.
Nocera
*
,
Eric C.
Lau
and
Daniel G.
Nocera
*
Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, USA. E-mail: dnocera@fas.harvard.edu
First published on 5th May 2017
Abstract
Oxygen evolution reaction (OER) catalysts that are earth-abundant and are active and stable in acid are unknown. Active catalysts derived from Co and Ni oxides dissolve at low pH, whereas acid stable systems such as Mn oxides (MnOx) display poor OER activity. We now demonstrate a rational approach for the design of earth-abundant catalysts that are stable and active in acid by treating activity and stability as decoupled elements of mixed metal oxides. Manganese serves as a stabilizing structural element for catalytically active Co centers in CoMnOx films. In acidic solutions (pH 2.5), CoMnOx exhibits the OER activity of electrodeposited Co oxide (CoOx) with a Tafel slope of 70–80 mV per decade while also retaining the long-term acid stability of MnOx films for OER at 0.1 mA cm−2. Driving OER at greater current densities in this system is not viable because at high anodic potentials, Mn oxides convert to and dissolve as permanganate. However, by exploiting the decoupled design of the catalyst, the stabilizing structural element may be optimized independently of the Co active sites. By screening potential–pH diagrams, we replaced Mn with Pb to prepare CoFePbOx films that maintained the high OER activity of CoOx at pH 2.5 while exhibiting long-term acid stability at higher current densities (at 1 mA cm−2 for over 50 h at pH 2.0). Under these acidic conditions, CoFePbOx exhibits OER activity that approaches noble metal oxides, thus establishing the viability of decoupling functionality in mixed metal catalysts for designing active, acid-stable, and earth-abundant OER catalysts.
Introduction
Adoption of solar energy is a desired low-risk climate pathway to meet growing global energy demand.1–6 Solar energy is the only renewable source scalable to the projected tens of terawatts of global energy consumption while remaining sustainable and cost-effective.1,2,5,7 Widespread solar implementation, however, requires inexpensive and high-density energy storage to manage the diurnal nature of the solar energy supply as well as to provide off-grid energy utilization.5,6 For large scale storage, batteries are ultimately limited by their energy density, thus providing an impetus for the solar-driven production of chemical fuels; central to this process is the splitting of water to oxygen and hydrogen.6,8–10 Of these two reactions, the oxygen evolution reaction (OER) is the more kinetically demanding half-reaction: the transfer of four protons is coupled to the loss of four electrons from two water molecules to produce a single oxygen molecule.11,12 We and others have sought to create catalysts that lower these kinetic barriers such that energy can be stored efficiently through water splitting. Inexpensive and active catalysts based on cobalt,13–16 nickel,17–20 and other earth-abundant metals21,22 for OER in neutral and alkaline pH have been developed, but these catalysts corrode in acidic solutions. Recent attempts to realize acid-stable systems from non-critical metals (such as Mn) suffer from low OER activity.23–28 Consequently, there are no acidic OER catalysts that are highly active, stable, and earth-abundant with only two out of the three criteria satisfied for known catalysts operating in acid.29–31The acidic pH regime is important for OER because of its applications in electrolysers and photoelectrochemical (PEC) devices as well as in fundamental mechanistic studies and catalyst design.30–32 Electrolysers based on acidic proton exchange membranes (PEM) are compact (sharing similar designs as PEM fuel cells), operate at high current and power densities, and achieve low gas crossover. However, their sulfonated Nafion membranes create an acidic local environment of pH ∼0–3 in water which necessitates corrosion-resistant components such as noble metal catalysts comprising Ir and Ru oxides for OER.33,34 Similarly, certain PEC photoelectrodes employ photovoltaic materials (such as silicon) that are more stable in acidic pH.35–38 Moreover, studying OER in acidic solutions contributes to a mechanistic understanding of how protonation influences OER kinetics when the solution is a poor proton acceptor thus opening avenues for designing and assessing, for example, catalysts with internal proton relays. Finally, the knowledge gained from optimizing catalysts for acid stability may be applied to other reactions at low pH including hydrohalic acid splitting,39–42 oxygen reduction (the opposite of OER where good proton donors facilitate the reaction),43 and formic/phosphoric acid oxidation (for fuel cell applications).44
We now demonstrate a pathway for the design of active, stable, and earth-abundant acidic OER catalysts comprising oxides of mixed metals. The experimental design is based on electrodeposited manganese oxide (MnOx), which is stable in acid but poorly catalyzes oxygen evolution.23 Increased activity for MnOx catalysts may be achieved through potential cycling deposition (alternating between anodic and cathodic potentials).25 To advance beyond activated MnOx, we have turned to a design principle to incorporate different co-interacting metals to fulfill specific roles to achieve desired functionality. To this end, OER activity and stability have been decoupled such that each property could be furnished by separate metals and optimized independently. Mixed metal oxide films have been constructed with Co as the catalytic metal and Mn as the structural metal (denoted CoMnOx). These films exhibit similar catalytic OER activity as electrodeposited cobalt oxide (CoOx) with Tafel slopes of ∼60 mV per decade in neutral pH and of ∼83 mV per decade in acidic conditions. Whereas CoOx fully dissolves within 3 h when operated at 0.1 mA cm−2 for OER at pH 2.5, we show that CoMnOx remains intact and stable for over 12 h of continuous operation. For the latter, stability at high anodic potential is limited by transformation to permanganate (MnO4−).45 Furthermore, by exploiting the ability to optimize the structural metal independently, Pb oxide was utilized to furnish high anodic stability at low pH. Mixed metal oxides were electrodeposited with Co as the catalytic component and Pb (with Fe dopant) as the structural element. These films exhibit similar Tafel behaviour as CoOx in neutral and acidic pH, yet they do not dissolve when operated at current densities of 1 mA cm−2 in acid continuously for over 12 h at pH 2.5 and over 50 h at pH 2.0. Under these conditions, the performance of these films begins to approach noble metal oxides, operating at ∼220 mV higher overpotential than Ir oxide. These results demonstrate that catalysts may be designed by mapping their target properties onto individual components of mixed metal films and show that an approach of using a stable metal oxide as a scaffold for active OER metals provides a promising path for the development of active, stable, and earth-abundant OER catalysts.
Results
Electrochemical deposition and Tafel analysis
The OER activity of the unary films was evaluated by measuring the steady-state current density as a function of applied potential in a Tafel analysis. Tafel plots for each film were collected in phosphate-buffered (Pi) solutions at neutral (pH 7.0, Fig. 1a) and acidic (pH 2.5, Fig. 1b) conditions. The slope of these plots served as the primary descriptor for comparing activity because the slope reflects the intrinsic kinetics of the catalyst's active site and is invariant to the amount of deposited catalyst.50 At pH 7.0, the Tafel slopes are: 60 mV per decade for CoOx, 90 mV per decade for NiOx, 45 mV per decade for FeOx, 125 mV per decade for MnOx, 130 mV per decade for PbOx, and 41 mV per decade for IrOx. Tafel slopes at pH 2.5 are: 82 mV per decade for CoOx, 51 mV per decade for FeOx, 650 mV per decade for MnOx, 121 mV per decade for PbOx (increasing to “infinite” slope at higher current densities), and 32 mV per decade for IrOx. A reliable Tafel plot could not be constructed for NiOx in acid since the film dissolved rapidly in solution. These slopes are consistent with our previous work on Co, Ni, and Mn oxides as well as literature on Fe, Pb, and Ir oxides.14,18,23,47,51–53
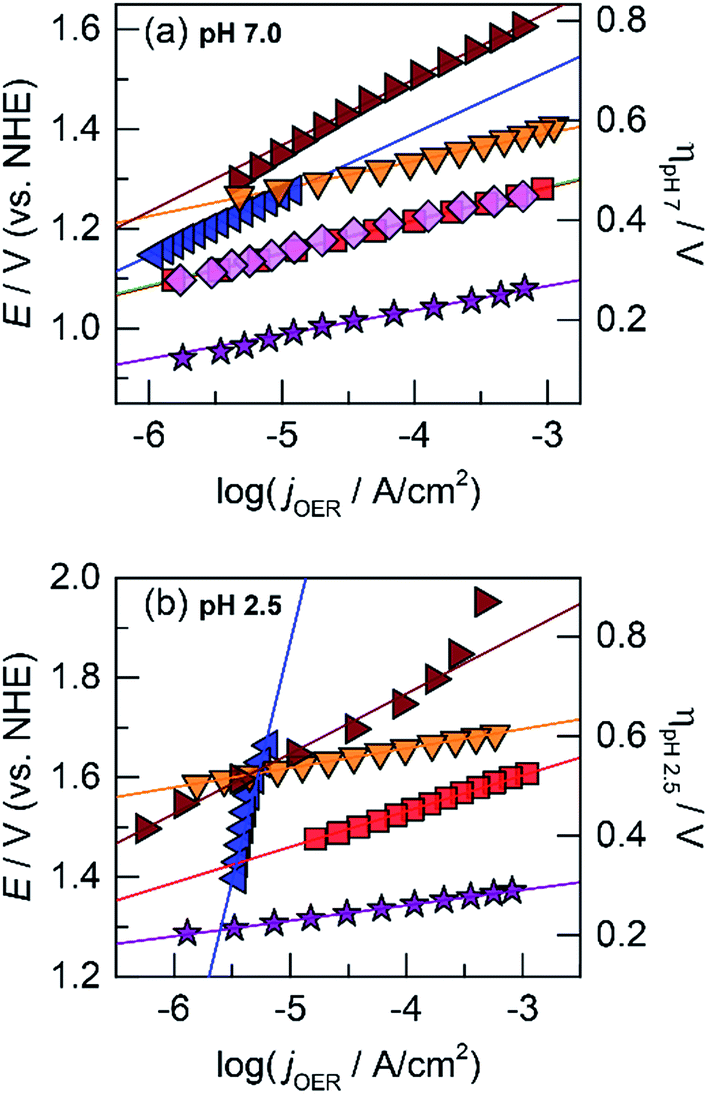 | ||
Fig. 1 Tafel plots of oxygen evolution for unary metal oxides in 0.10 M Pi and 1.0 M KNO3 at (a) pH 7.0 and (b) pH 2.5 of CoOx (red  , 60 and 82 mV per decade Tafel slope at pH 7.0 and 2.5, respectively), NiOx (light magenta , 60 and 82 mV per decade Tafel slope at pH 7.0 and 2.5, respectively), NiOx (light magenta  , 90 mV per decade at pH 7.0), FeOx (orange , 90 mV per decade at pH 7.0), FeOx (orange  , 45 and 51 mV per decade at pH 7.0 and 2.5, respectively), MnOx (blue , 45 and 51 mV per decade at pH 7.0 and 2.5, respectively), MnOx (blue  , 125 and 650 mV per decade at pH 7.0 and 2.5), PbOx (brown , 125 and 650 mV per decade at pH 7.0 and 2.5), PbOx (brown  , 130 and 121 mV per decade at pH 7.0 and 2.5), and IrOx (purple , 130 and 121 mV per decade at pH 7.0 and 2.5), and IrOx (purple  , 41 and 32 mV per decade at pH 7.0 and 2.5). , 41 and 32 mV per decade at pH 7.0 and 2.5). | ||
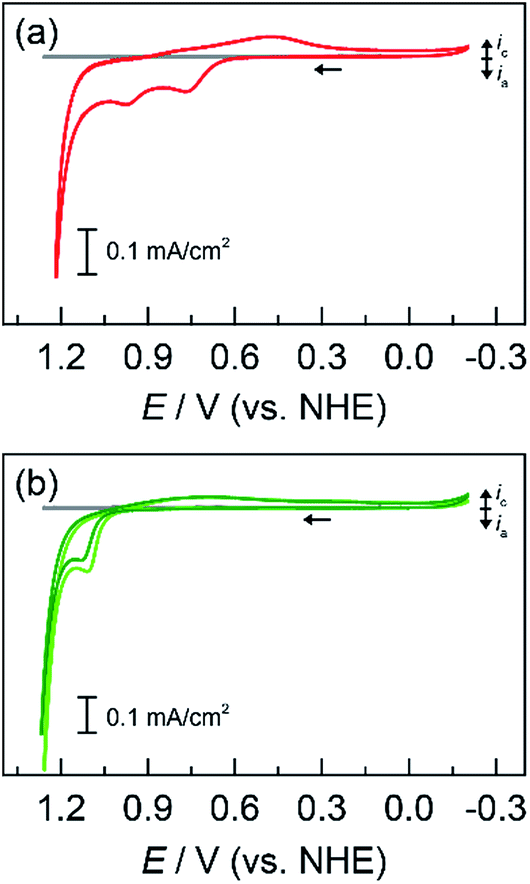 | ||
Fig. 2 Cyclic voltammograms (CVs) of a 1 cm2 FTO electrode at 50 mV s−1 in 50 mM MePi buffer at pH 8.0 with 0.25 mM of each metal: (a) Co2+ and Mn2+ (red  ); and (b) Co2+ and Pb2+ (light green ); and (b) Co2+ and Pb2+ (light green  ) with addition of Fe2+ (dark green ) with addition of Fe2+ (dark green  ). Background CV of metal-free MePi buffer (grey ). Background CV of metal-free MePi buffer (grey  ) included for comparison. ) included for comparison. | ||
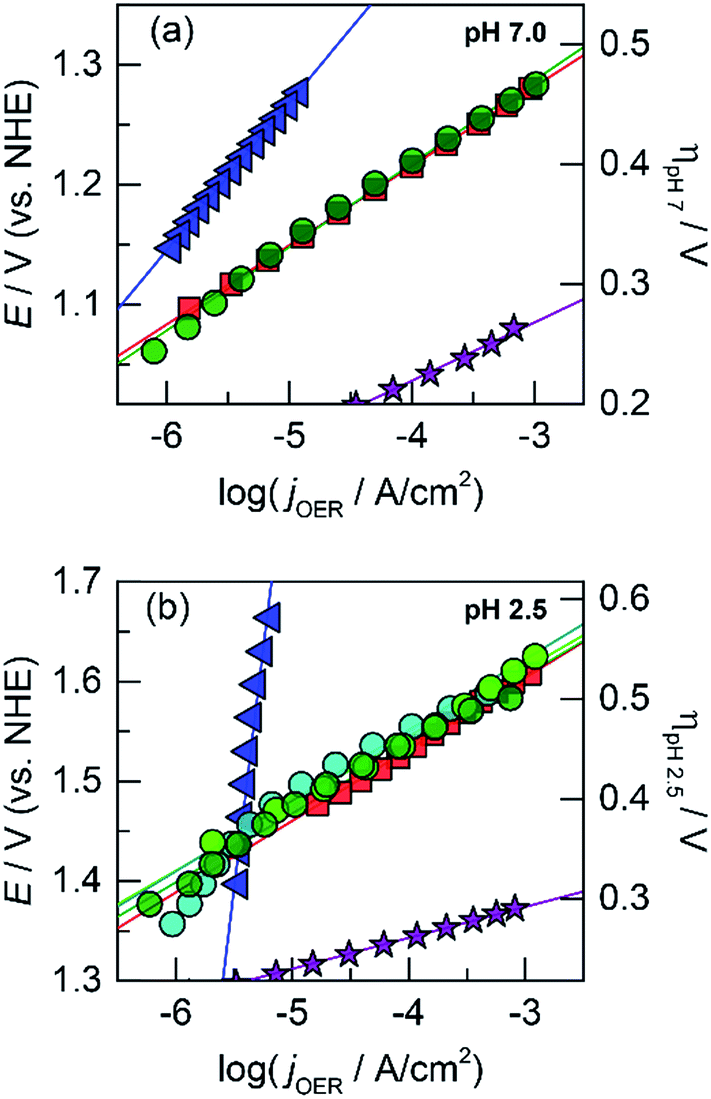 | ||
Fig. 3 Tafel plots of oxygen evolution for CoMnOx in 0.10 M Pi and 1.0 M KNO3 at (a) pH 7.0 and (b) pH 2.5 of: CoMnOx deposited at 0.90 (dark green  , 65 and 81 mV per decade at pH 7.0 and 2.5), 0.65 (light green , 65 and 81 mV per decade at pH 7.0 and 2.5), 0.65 (light green  , 85 mV per decade at pH 2.5), and 1.15 V (cyan , 85 mV per decade at pH 2.5), and 1.15 V (cyan  , 83 mV per decade at pH 2.5). Unary metal oxides provided for comparison with slopes defined in Fig. 1, CoOx (red , 83 mV per decade at pH 2.5). Unary metal oxides provided for comparison with slopes defined in Fig. 1, CoOx (red  ), MnOx (blue ), MnOx (blue  ), and IrOx (purple ), and IrOx (purple  ). ). | ||
Lead was substituted for Mn into mixed metal oxides as the structural metal. CVs were collected on solutions with equal concentrations of Co2+ with Pb2+ (Fig. 2b). For Co2+/Pb2+ solutions, a CoOx deposition pre-feature is observed at 1.15 V; PbOx deposition has a broader peak that underlies the pre-feature (>1.1 V according to the CV of PbOx in Fig. S1e†). Thus, a fixed constant potential of 1.15 V was employed to electrodeposit brown films of CoPbOx. Because the deposition of CoOx and PbOx occurs at similar potentials, it was not possible to control the ratio of mixed metal incorporation in the films by varying the deposition potential. Accordingly, the initial concentrations of the metal ions were adjusted in order to vary metal concentrations within the films. Tafel plots of CoPbOx for OER demonstrate kinetics that are similar to that of the unary CoOx film. In neutral and acidic pH (Fig. 4a and b), the Tafel slope of CoPbOx is ∼72 mV per decade, which is similar to that of CoOx and suggests that the mechanism of the active site is the same under a wide range of pH. These results are also consistent with Tafel analysis from CoMnOx films and demonstrate that incorporation of structural metals such as Mn or Pb with catalytic metals does not change the OER kinetics of active sites.
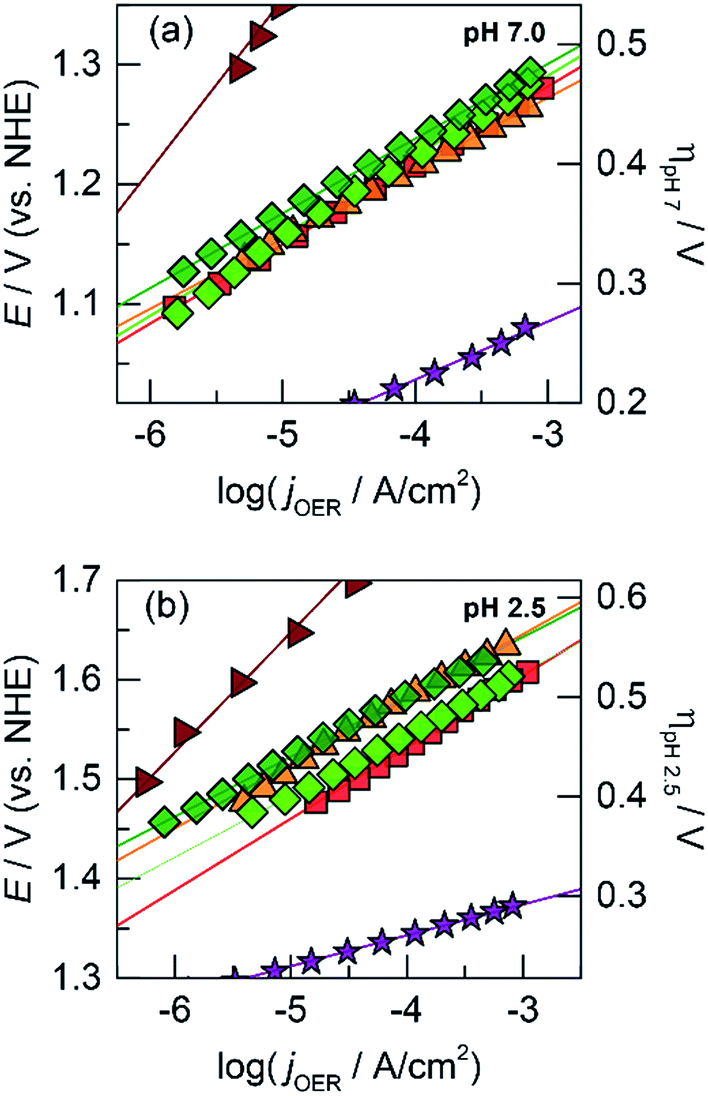 | ||
Fig. 4 Tafel plots of oxygen evolution in 0.10 M Pi and 1.0 M KNO3 at (a) pH 7.0 and (b) pH 2.5 for CoPbOx (light green  , ∼72 mV per decade at both pH 7.0 and 2.5) and CoFePbOx (dark green , ∼72 mV per decade at both pH 7.0 and 2.5) and CoFePbOx (dark green  , ∼70 mV per decade at both pH 7.0 and 2.5). CoFeOx (orange , ∼70 mV per decade at both pH 7.0 and 2.5). CoFeOx (orange  , ∼70 mV per decade at both pH 7.0 and 2.5) and unary metal oxides are provided for comparison with slopes defined in Fig. 1, CoOx (red , ∼70 mV per decade at both pH 7.0 and 2.5) and unary metal oxides are provided for comparison with slopes defined in Fig. 1, CoOx (red  ), PbOx (brown ), PbOx (brown  ), and IrOx (purple ), and IrOx (purple  ). ). | ||
Because addition of a structural metal does not interfere with catalytic activity, Pb ions were introduced during the deposition of CoFeOx films to assess if the low Tafel slopes of CoFeOx in base (∼30 mV per decade)54 could translate to neutral and acidic conditions when stabilized by Pb. Using similar deposition solution conditions, CVs were recorded on a solution containing equal concentrations of Co2+, Fe2+, and Pb2+ (Fig. 2b). The lack of a distinct Fe feature in the CVs is consistent with those obtained for Fe2+-only solutions (Fig. S1c†), which do not display deposition features due to poor kinetics for anodic FeOx formation (in the absence of higher temperatures). Although Fe deposition is not observable in the CV, a small percentage of Fe is trapped and incorporated into the mixed films during growth as supported by elemental analysis (vide infra) and studies on similar NiFeOx systems.20,55 Tafel plots of the Fe-incorporated mixed films (CoFePbOx films, Fig. 4a and b) at neutral and acidic pH exhibit similar slopes of ∼70 mV per decade as obtained for Fe-free films. The highly facile kinetics of CoFeOx in the alkaline regime with 30 mV per decade slope does not appear to persist in neutral and acidic solutions.
Acid stability and faradaic efficiency
The acidic stability of unary and mixed metal films was evaluated by using long-term chronopotentiometry. Solution conditions were the same as that employed for Tafel analysis (i.e., phosphate buffer with nitrate supporting electrolyte at pH 2.5). Electrodeposited films were immersed in stirred solutions and initially held at a constant current density of 0.1 mA cm−2 where after a brief capacitance period, only OER is sustained (as confirmed by direct O2 measurement, vide infra). OER on blank FTO occurs at a steady potential of 2.3 V (shown for ∼12 h in Fig. 5a). Whereas the substrate oxide is highly acid stable, it is an exceptionally poor OER catalyst. NiOx films dissolve quickly at pH 2.5. The applied potential required to maintain 0.1 mA cm−2 rises immediately and undergoes inflection at 40 min to correspond to the potential of that for blank FTO, indicating complete film dissolution. CoOx films have slightly longer stability than NiOx and catalyze OER at ∼1.6 V for almost 3 h before potential inflection, a sign of full dissolution. We note that dissolved Co2+ ions can still promote OER56 and thus the potential plateaus about 300 mV below that of blank FTO. Moving to the left on the periodic table, we have shown that MnOx can perform OER in acid with limited degradation by trading off activity for stability.23 Indeed whereas MnOx requires a high anodic potential of 2.0 V to maintain 0.1 mA cm−2, the current remains stable for over 12 h of testing. As a control, IrOx is included to represent a catalyst with both high activity and stability. OER from IrOx occurs at 1.4 V sustaining 0.1 mA cm−2 over the 12 h testing duration.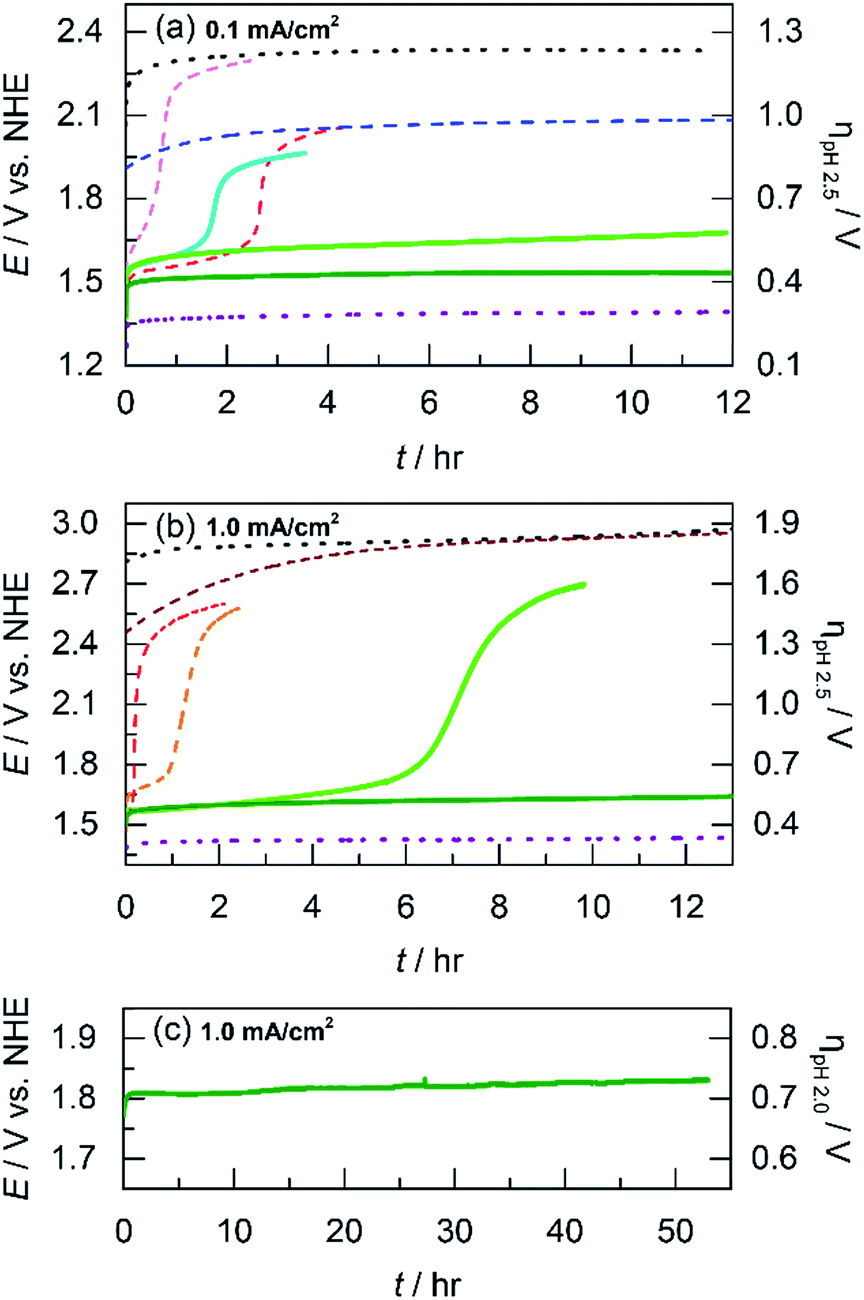 | ||
Fig. 5 Electrochemical stability for acidic OER measured by sustained chronoamperometry at: (a) 0.1 mA cm−2 in pH 2.5 Pi for CoMnOx deposited at 0.65 (light green  ), 0.90 (dark green ), 0.90 (dark green  ), and 1.15 V (cyan ), and 1.15 V (cyan  ) along with CoOx (red ) along with CoOx (red  ), NiOx (light magenta ), NiOx (light magenta  ), MnOx (blue ), MnOx (blue  ), IrOx (purple ), IrOx (purple  ), and FTO (grey ), and FTO (grey  ) for comparison; (b) 1.0 mA cm−2 in pH 2.5 Pi for CoPbOx (light green ) for comparison; (b) 1.0 mA cm−2 in pH 2.5 Pi for CoPbOx (light green  ) and CoFePbOx (dark green ) and CoFePbOx (dark green  ) with CoFeOx (orange ) with CoFeOx (orange  ), CoOx (red ), CoOx (red  ), and PbOx (brown ), and PbOx (brown  ) for comparison; and (c) 1.0 mA cm−2 in pH 2.0 sulfate for CoFePbOx (dark green ) for comparison; and (c) 1.0 mA cm−2 in pH 2.0 sulfate for CoFePbOx (dark green  ). The inflection of potential in the plots indicates film dissolution. ). The inflection of potential in the plots indicates film dissolution. | ||
To investigate if a structural metal could stabilize unary metal oxides against dissolution, Co (the catalytic element) was co-deposited with Mn (the structural component) at three different potentials (0.65, 0.90, and 1.15 V) to create CoMnOx films with different Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn ratios. Stability tests were repeated for these films under the same conditions as for unary catalysts (Fig. 5b). For OER at 0.1 mA cm−2, CoMnOx electrodeposited at 0.90 V exhibited a low OER potential of 1.5 V (similar to that of CoOx before dissolution) while remaining intact over 12 h of operation. Similarly, for CoMnOx deposited at 0.65 V, the films remained stable but exhibited a slight increase of 100 mV in its potential to sustain OER. In contrast, films deposited at 1.15 V were not stable and dissolved in ∼2 h, similar to the CoOx-only film. Overall, mixed metal films containing Co as the catalytic and Mn as the structural element can improve acid stability, extending it to over 12 h, while retaining the OER activity of CoOx.
:Mn ratios. Stability tests were repeated for these films under the same conditions as for unary catalysts (Fig. 5b). For OER at 0.1 mA cm−2, CoMnOx electrodeposited at 0.90 V exhibited a low OER potential of 1.5 V (similar to that of CoOx before dissolution) while remaining intact over 12 h of operation. Similarly, for CoMnOx deposited at 0.65 V, the films remained stable but exhibited a slight increase of 100 mV in its potential to sustain OER. In contrast, films deposited at 1.15 V were not stable and dissolved in ∼2 h, similar to the CoOx-only film. Overall, mixed metal films containing Co as the catalytic and Mn as the structural element can improve acid stability, extending it to over 12 h, while retaining the OER activity of CoOx.
While CoMnOx is stable for acidic OER at 0.1 mA cm−2, higher current densities are desired for practical applications and so the unary and mixed metal films were re-evaluated at 1 mA cm−2 under the same solution conditions (Fig. S2†). As a baseline, blank FTO is stable but requires a high potential of ∼2.9 V for OER. CoMnOx films (deposited at 0.90 V) initially evolve oxygen at ∼1.7 V but fully dissolve after 30 min. After dissolution, OER is catalyzed by free Co2+ ions at ∼2.5 V. For comparison, CoOx exhibits similar behavior but fully degrades slightly faster at 20 min. MnOx reaches dissolution in 10 min, and the solution color turns slightly pink, which is consistent with the oxidation of the film to permanganate (MnO4−) as predicted by the Pourbaix diagram of manganese.45 The MnO4− ions appear to catalyze OER at ∼2.4 V. In contrast, IrOx is both stable and highly active, performing OER without degradation at 1.43 V sustaining 1 mA cm−2 over 12 h.
The dissolution of Mn at high anodic potentials (caused by operating at higher current densities) prompted its replacement by another structural metal that is not prone to molecular decomposition. Lead was selected as an alternative for manganese after evaluating the potential–pH properties of many corrosion-resistant elements (search process presented in the Discussion). As a control, acidic stability tests at 1 mA cm−2 were performed on PbOx, which demonstrated long-term acidic stability over 12 h at 2.9 V (Fig. 5b). Because Pb was a promising structural metal, mixed metal films incorporating Co and Pb were prepared and evaluated under similar conditions at higher current densities. Indeed, the presence of Pb in CoPbOx films extended the operation of “CoOx” from 20 min to 7 h at ∼1.65 V before eventually dissolving (Fig. 5b). Pb thus slowed the rate of corrosion in CoPbOx films significantly.
Incorporation of lead was also investigated in cobalt oxide films doped with Fe. As a control, CoFeOx degrades quickly during acidic OER at 1 mA cm−2 as indicated by the rising potential around 1.7 V that inflects to indicate dissolution after 1 h (Fig. 5b). The unary FeOx is also unstable at higher current densities, degrading after 2 h (Fig. S2†). However, the slight addition of Fe to CoPbOx produced CoFePbOx films that could sustain OER at ∼1.65 V for over 12 h. Moreover, CoFePbOx operated at 1 mA cm−2 in sulfate buffer at pH 2.0 (Fig. 5c) remained stable at ∼1.8 V for over 50 h of continuous electrolysis, demonstrating that these films are both active and exhibit superior stability under laboratory test conditions. Overall, Pb appears to fulfill its role as a structural element and confers acidic corrosion resistance when incorporated in mixed metal oxide films.
To confirm that acid-stable mixed metal films are performing OER, the evolved oxygen was directly measured by gas chromatography in a gas-tight electrochemical cell and compared to the charge passed to the electrode during electrolysis. Employing the same solution conditions as that used for stability tests, CoMnOx was operated at 0.1 mA cm−2 and CoFePbOx at 1.0 mA cm−2, and the O2 concentration was measured every 20 min (Fig. S3†). The faradaic efficiency was calculated by dividing the moles of detected oxygen by the moles of transferred electrons. The average efficiency for CoMnOx is 91% and that for CoFePbOx is 97%. The slight decrease in efficiency of CoMnOx may result from the lower current densities of operation that correspond to a lower O2 signal-to-noise ratio and consequently decreased accuracy. Overall, both systems demonstrate faradaic efficiencies near unity for long-term OER in acid.
Physical characterization
For mixed films containing Pb, ICP-MS shows that CoPbOx comprises 18% Co and 82% Pb, while CoFePbOx contains 15% Co, 2% Fe, and 83% Pb (Table S1†). In these films, the structural element (Pb) is the dominant component. Nevertheless, co-depositions include appreciable amounts of catalytic sites and sufficient Fe infiltrates the films during growth, despite the difficulty of directly electrodepositing FeOx at those same potentials. Overall, elemental analysis shows that anodic co-deposition is a viable technique for incorporating both catalytic and structural elements in the resulting mixed metal films.
 | ||
| Fig. 6 FESEM images of CoMnOx electrodeposited at (a) 0.65, (b) 0.90, and (c) 1.15 V with (d) CoOx and (e) MnOx for comparison. All samples were prepared on FTO substrate, and scale bars are 200 nm. | ||
 | ||
| Fig. 7 FESEM images of (a) CoPbOx and (b) CoFePbOx with (c) PbOx and (d) FTO for comparison. All samples were electro-deposited on FTO, and scale bars are 100 nm. | ||
 | ||
| Fig. 8 EDS elemental maps recorded through SEM of (a) CoMnOx (deposited at 0.90 V) and (b) CoFePbOx. Individual elemental channels for Co (red), Mn (blue), Fe (orange), and Pb (green) were combined and overlaid on the respective SEM image. All samples were prepared on FTO substrate, and scale bars are 200 nm for CoMnOx and 100 nm for CoFePbOx. | ||
 | ||
| Fig. 9 High-resolution EDS elemental maps recorded through STEM of (a) CoMnOx (deposited at 0.90 V) and (b) CoFePbOx. Individual elemental channels for Co (red), Mn (blue), Fe (orange), and Pb (green) were combined and overlaid on the respective image. Scale bars are 15 nm for a resolution of 7.4 Å per px. | ||
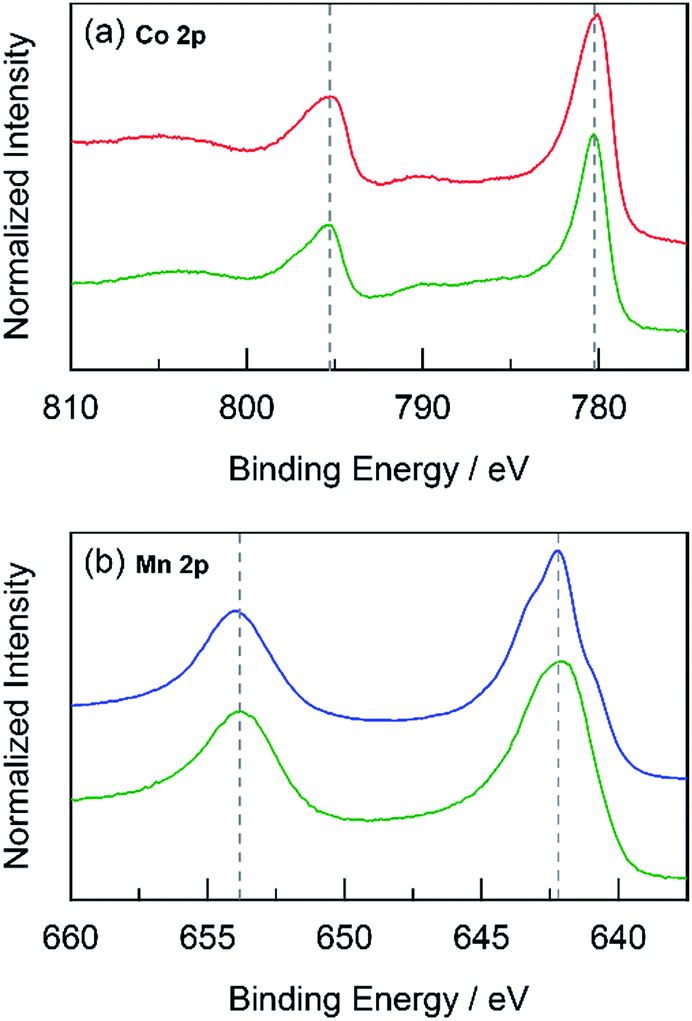 | ||
Fig. 10 High-resolution XPS spectra in the (a) Co 2p and (b) Mn 2p regions for: CoMnOx (deposited at 0.90 V, dark green  ) compared to CoOx (red ) compared to CoOx (red  ), and MnOx (blue ), and MnOx (blue  ). Grey dotted lines are presented as guides. ). Grey dotted lines are presented as guides. | ||
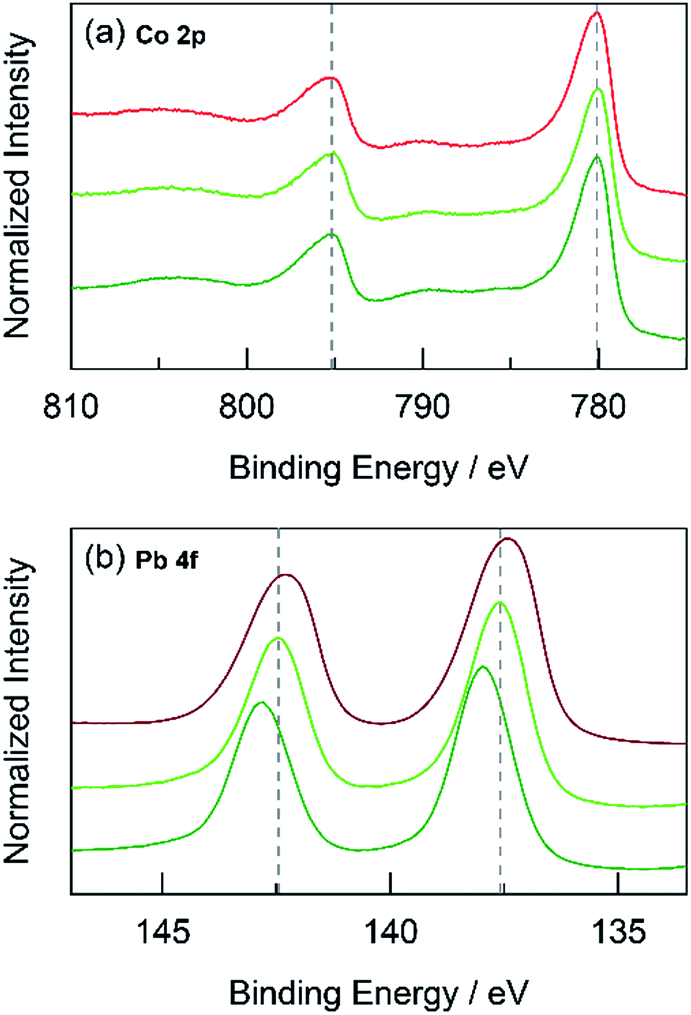 | ||
Fig. 11 High-resolution XPS spectra in the (a) Co 2p and (b) Pb 4f regions for: CoPbOx (light green  ) and CoFePbOx (dark green ) and CoFePbOx (dark green  ) compared to CoOx (red ) compared to CoOx (red  ), and PbOx (brown ), and PbOx (brown  ). Grey dotted lines are presented as guides. ). Grey dotted lines are presented as guides. | ||
Discussion
Heterogeneous OER catalysts based on metal oxides exhibit a propensity for corrosion in low pH solutions. Acids react with basic metal oxides to protonate the metal oxide framework, weakening metal–oxygen bonds and facilitating dissolution.60–62 For this reason, the only known viable acidic OER catalysts are based on platinum group metals, in particular, iridium and ruthenium.31,63,64 Consequently, performing OER in acid typically relies on using catalysts derived from Ir and Ru oxides with most studies focused on increasing the accessible active sites while decreasing the mass loading.65–69 Conversely, development of OER catalysts from earth-abundant metal oxides have targeted the alkaline regime, where stability is generally not a concern, with the use of multiple metals for the sole purpose of catalytic synergy (e.g. NiFe oxides) or for increasing the surface area of active catalyst material.70–74 OER in neutral solutions has been achieved through the development of self-healing unary catalysts,75,76 and in some cases, performance in acid may be achieved at low current density.23,25 Our interest to develop further design principles for active, stable, and earth-abundant acidic OER catalysts has led us to consider using mixed-metals as a framework for decoupling activity from stability by employing two different metals: one that acts as a catalytic element and the other that acts as a structural element. In assessing these mixed-metal catalysts, their performance is benchmarked against each other and to unary metal oxide films using techniques employed in our previous mechanistic studies14,19,25 as well as those outlined elsewhere.29,77–79 Benchmarking typically comprises measurements on elemental composition, surface area, faradaic efficiency, catalytic activity, and stability. Of these criteria, we exclude surface area because a rigorous measurement on porous thin films is unfeasible.80 Instead we rely on activity comparisons as defined by Tafel plots, where the slope is a measure of catalyst kinetics that is independent of catalyst loading or surface area.The iterative design path employed for this study is shown in Fig. 12. Metal oxidic films increase in stability from NiOx to CoOx and to MnOx, a trend which is consistent with an increase in metal–oxygen bond strength traversing from late to early first-row transition metals as embodied by the principles of the “oxo wall”.81 Indeed, the intrinsic strength of Mn–O bonds in MnOx results in resistance to corrosion of the oxide at low pH; this passive stability is augmented by the functional stability of the film derived from self-healing (the catalyst can re-form at the anodic potentials of OER23,24). Nonetheless, the greater stability of MnOx occurs at the expense of lower activity owing to the formation of MnO4− at the higher anodic potentials that accompany higher activity. The activity of conventionally electrodeposited MnOx films may be improved82–84 by driving a phase change from a birnessite-like (δ-MnO2) to hausmannite-like (α-Mn3O4) oxide that culminates in metastable turbostratic-disordered birnessite.25 This phase (denoted activated MnOx) is significantly more active for OER at pH 2.5 (than unactivated MnOx) with faster kinetics (i.e., a lower Tafel slope of ∼90 mV per decade and 100 times greater current density at η = 600 mV) while remaining stable for over 8 h of continuous operation at 0.1 mA cm−2. Spectroscopic and structural studies suggest that these phase changes trap catalytic MnIII sites within a robust MnIV oxidic matrix, and thus there are two types of Mn sites that allow both activity and stability to co-exist.25,85,86 However with structure and function embodied in the same metal, there is no clear path for independent optimization.
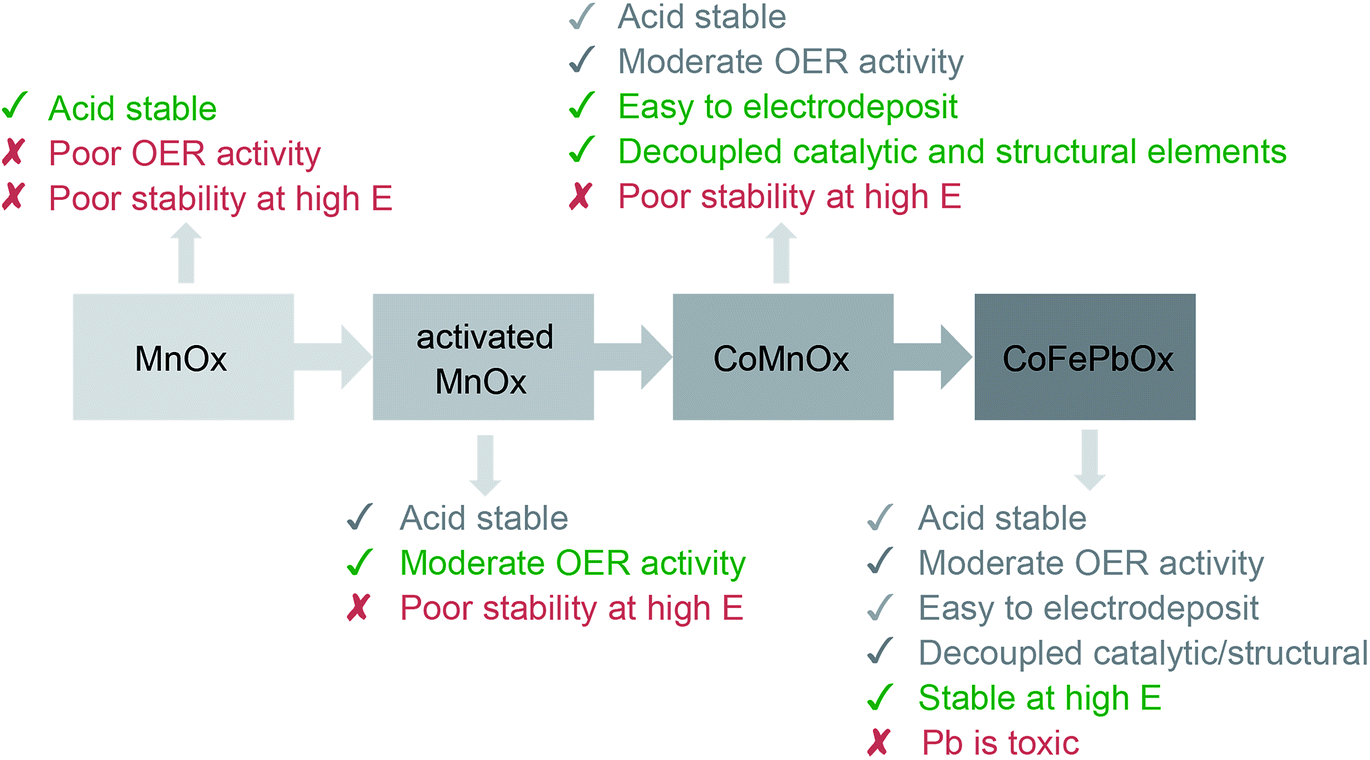 | ||
| Fig. 12 Progression of designing an active, stable, and earth-abundant acidic OER catalyst. The first generation system focused on demonstrating film stability at low pH with MnOx.23,24 The activity of MnOx was improved in the second generation by activating MnOx for OER.25 The catalyst was reformulated as a mixed metal oxide for the third generation, where functionality was separated into catalytic and structural elements comprising Co and Mn, respectively. Finally, the degradation of Mn oxides at high anodic potentials was solved by replacing it with a FePb oxide structural component to create the fourth-generation catalyst. | ||
CoMnOx
Accordingly, we turned to using manganese as the structural element and Co as the catalytic element as we have extensively characterized CoOx, which is among the most active materials for OER in neutral pH.87 An advantage of a mixed metal Mn and Co system is that the oxides can be easily electrodeposited at modest potentials in buffered solutions (MePi at pH 8) with known deposition kinetics.24,46 Furthermore, the ratio of Co to Mn in the films can be controlled by the deposition potential. In the CV of Co2+ and Mn2+ in MePi buffer at pH 8 (Fig. 2a), the deposition peak for MnOx occurs ∼250 mV to lower anodic potential (i.e., less positive) than that of CoOx deposition. This separation in potentials can be exploited to regulate the kinetics of the two reactions. When the electrode potential is near that of MnOx deposition, more Mn than Co will be incorporated into the catalyst. As the potential is raised nearer to that of CoOx deposition, more Co is assimilated into the final film (Table S1†). SEM and STEM elemental mapping, to resolution of ∼7.4 Å, indicates no segregated domains of CoOx and MnOx (Fig. 8a and 9a).Whereas CoMnOx has utility in battery and supercapacitor materials88,89 and has been studied for seawater90 and alkaline91 electrolysis, their application towards acidic OER has only recently begun to be explored through screening.78 For the studies performed herein, the performance of CoMnOx for oxygen evolution in neutral and acidic pH was evaluated using Tafel analysis (i.e., logj vs. E plots). In general, better catalysts are positioned lower on the Tafel plots (i.e., requiring less potential to achieve the same current density) and have lower slopes (i.e., produces more current density at the same potential). However, the position of the plot is dictated by the exchange current density (i.e., the y-intercept), which is dependent on the number of active sites in the catalyst films. Because there are no known methods to quantify active sites accurately for heterogeneous systems (with the exception of single crystal substrates),80 we prefer to rely on the Tafel slope, which reflects the kinetics or mechanism of the reaction and is governed by the type of active site rather than its abundance.50,92 Both CoMnOx and CoOx exhibit the same slope in neutral (∼65 mV per decade) and acidic pH (∼82 mV per decade) suggesting that it is the Co sites in the mixed films that are supporting OER catalysis. Furthermore, given that MnOx has worse OER kinetics at neutral (125 mV per decade) and acidic (∼650 mV per decade) solutions, it is likely that Mn sites in the mixed metal film do not contribute to OER. Overall, there appears to be no catalytic synergy between Co and Mn, as the addition of Mn does not appear to change the electrochemical properties of CoOx. The advantage of Mn emerges in its role as a structural metal. Stability tests for CoMnOx demonstrate continuous oxygen evolution (at 0.1 mA cm−2) in pH 2.5 for over 12 h without dissolution (Fig. 5a) and with an OER faradaic efficiency near unity (Fig. S3†). For these stability measurements, constant current density was maintained to ensure a steady rate of catalysis and provide an indicator for film degradation. For stable films, the OER potential remains steady over the course of the experiments. Conversely, as the film dissolves, the potential slowly rises until there is no solid catalyst remaining, whereupon a sharp inflection of potential is observed with the current density approaching that of a blank FTO. Whereas such an inflection is observed for CoOx and CoMnOx films with <50% Mn composition (which fully degrade within 3 h), MnOx and CoMnOx films with ≥50% Mn content show no such dissolution behavior as they remain intact over long-term acidic OER (Fig. 5a). Thus, the stability of CoMnOx appears to be derived from oxide films possessing a Mn:Co ratio that is equal or greater than unity. It is noteworthy that IrOx performs OER at 0.1 mA cm−2 in pH 2.5 at a potential that is only 140 mV lower than that of CoMnOx films (≥50% Mn).
CoPbOx and CoFePbOx
Although CoMnOx is active, stable, and composed of earth-abundant elements, Mn-based oxide films have the limitation in that they are oxidized to permanganate ions (MnO4−),45,93 which are soluble and lead to catalyst dissolution at high anodic potentials (see Fig. S8,† Pourbaix diagram). A workaround is to restrict the upper operating potential of these films by operating at low current densities and by increasing the amount of catalyst loading, which may be achieved with the use of porous electrodes.94–96 Nonetheless, it is also desirable to support higher operating current densities without resorting to physical surface area augmentation. To this end, we sought to replace Mn with another structural metal that is stable in acid at high anodic potentials. Experimentally45 and computationally97 derived Pourbaix diagrams of transition and post-transition metals guide in the selection of passivated oxide phases that persist at low pH and high potentials. The initial screen presented 16 candidates (Fig. 13) that were down-selected based on removing elements that are not earth-abundant (e.g., precious metals), experimentally known to be unstable in acid (e.g., Co), and difficult to electrodeposit (e.g., Nb, Ti, and Si). Of the four elements remaining (Bi, Pb, Sn, and W), Pb was the best candidate because it can be anodically electrodeposited easily and has a significant advantage in electrical conductivity owing to its oxygen defects.98,99 Acknowledging the toxicity of Pb, this concern is somewhat mitigated inasmuch as a small amount of the element is needed for the mixed metal catalyst; moreover for the purposes of this study, the focus was to assess the viability of an independent catalytic/structural mixed-metal approach. On its own, PbOx electrodeposits under similar conditions as CoOx but with faster deposition kinetics (as evidenced by the steepness of the Pb2+ oxidation wave in Fig. S1e†). PbOx exhibits ∼120–130 mV per decade Tafel slopes for OER in neutral and acidic pH.100 When compared to CoOx under the same conditions, PbOx is a relatively poor catalyst and also suffers from a sudden increase to “infinite” slope near 1 mA cm−2 for the acidic regime (Fig. 1b), suggesting that additional driving force has no effect on the rate of the reaction. Disregarding its inferior OER properties, PbOx is an excellent structural metal for long-term acidic OER stability at higher current densities (of 1 mA cm−2, Fig. 5b), with over 12 h of continuous operation without degradation. We note stability measurement conditions are comparable to those made on acidic OER catalysts near pH 0 at ∼3 mA cm−2 for at least 2 h. Here a slightly less acidic pH is offset by better buffering conditions (sulfate and phosphate at pH 2 and 2.5, respectively).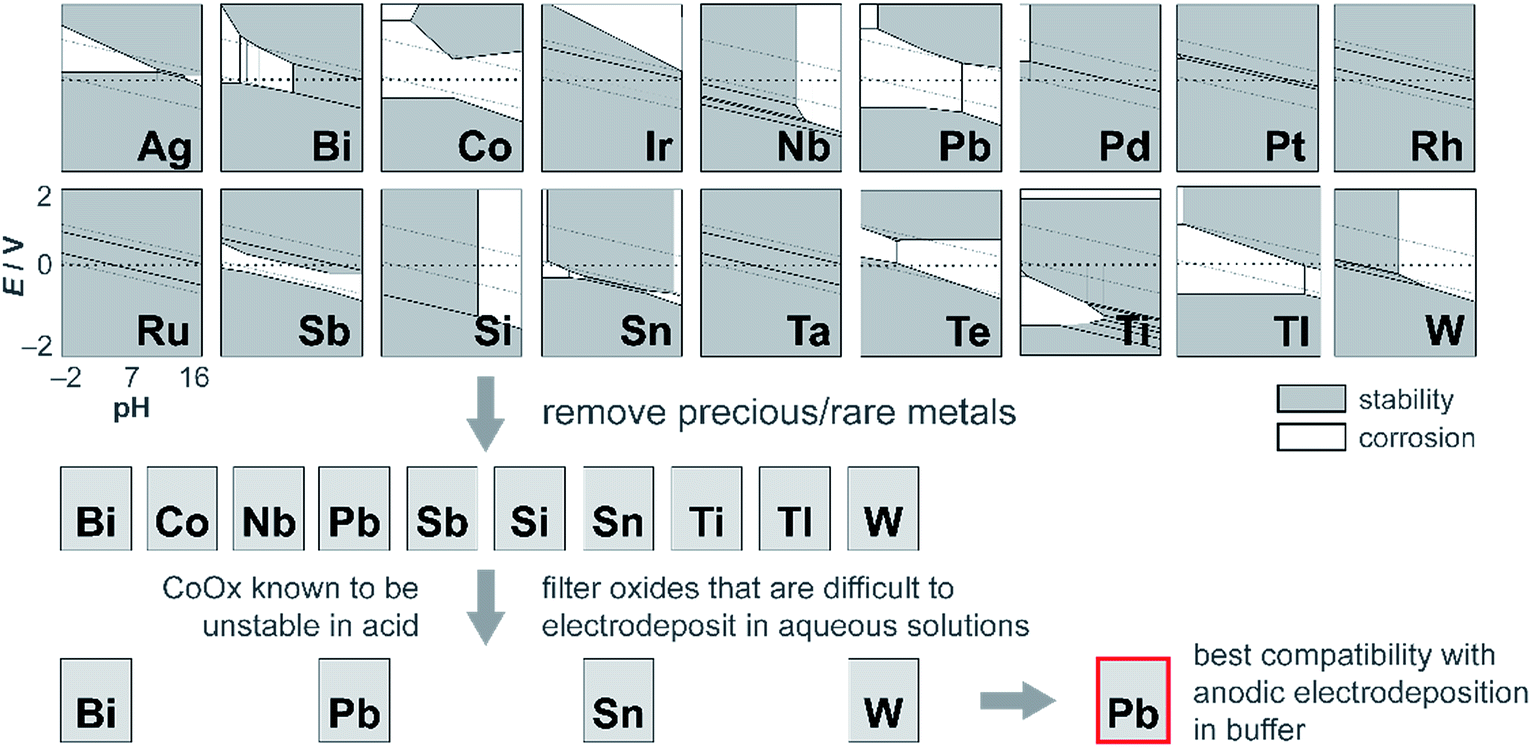 | ||
| Fig. 13 Process for independently optimizing the structural component of mixed metal films to discover a metal oxide that is both stable at high anodic potentials and at acidic pH. Pourbaix diagrams of metals (shown as simplified representations of stability and corrosion, generated from the Materials Project97 and experimental data45) were analysed for stability in the top left region of the plots (corresponding to high anodic potentials and low pH). The candidates were then filtered by removing precious and rare metals; then further refined by excluding oxides that were incompatible with anodic electrodeposition in buffer. In this manner, Pb was identified as a promising replacement for Mn for stabilizing OER catalysts in acid. | ||
Against this backdrop, Pb was employed as the structural element for Co as the catalytic component. Electrodeposition of CoPbOx was similar to that for CoMnOx with the precursor solution containing equal concentrations of Co2+ and Pb2+ in MePi buffer at pH 8. However, the potentials for CoOx and PbOx deposition are similar (∼1.15 V) unlike that for CoOx and MnOx deposition; thus, the composition of the mixed metal film could not be as easily controlled through varying the potential. Instead, the initial concentrations of Co2+ and Pb2+ can be relatively adjusted to change composition. For solutions containing equal concentrations of Co2+ and Pb2+, electrodeposited CoPbOx films contain 18% Co and 82% Pb (as measured by ICP-MS, Table S1†) with homogeneous distribution of the two metals throughout the film (as observed from EDS elemental maps from SEM, Fig. S4c†). The use of CoPbOx as a stable electrode has precedence in the electrosynthesis of oxidants, decomposition of organics, as well as electrowinning of metals.101–103 Although the application of CoPbOx for acidic oxygen evolution has largely been unexplored, its prior history in corrosive environments suggested that it was a promising target.
Like CoMnOx, the OER performance of CoPbOx resembled that of CoOx with similar Tafel slope of ∼72 mV per decade in neutral and acidic pH (Fig. 4). While there is no catalytic synergism between Co and Pb, there is no discord either: the presence of Pb does not hinder the OER activity of Co sites. Interestingly, given that the mass loading of the films are comparable, the number of total Co active sites (i.e., 18% Co) in CoPbOx should be less than that of CoOx. However, Tafel plots of CoPbOx overlaid with those of CoOx, which indicates a similar level of activity. This effect could manifest from: first, the difference in morphology of the films where CoOx has a smooth surface that may limit the accessibility of active sites while CoPbOx is a rougher film comprised of small nanoparticles; and secondly, the high electrical conductivity of PbOx may enable a greater number of Co sites to be electrically addressed in CoPbOx with low internal resistance. Four-point probe electrical measurements of PbO2 exhibit metallic-like bulk resistivity from 10−3 to 10−5 ohm cm.104,105 Conversely, early transition metal oxides generally behave as insulators or semiconductors with high resistivity (in units of ohm cm): Co3O4 on the order of 104,106,107 MnO2 from 102 (electrolytic manganese dioxide, EMD) to 105 (birnessite),108,109 and Fe2O3 and FeOOH from 104 to 108.110,111 PbO2 is orders of magnitude more conductive than Co3O4 although its exact influence on CoOx when co-deposited as CoPbOx requires direct conductivity measurements by electrochemical impedance spectroscopy or via an interdigitated electrode array. Thus, Pb in the mixed metal film appears to act as a structural metal while Co plays the catalytic role.
The acidic OER stability of CoPbOx was evaluated at intermediate (1 mA cm−2) current densities (Fig. 5b) where the presence of Pb enhanced the corrosion resistance of the films. For OER at 1 mA cm−2, both CoMnOx and CoOx films fully dissolve within 30 min while CoPbOx resists degradation to 7 h. While the addition of Pb does not confer long-term stability against acid corrosion, it does slow the rate of degradation. Thus, the role of Pb as an anodically stable metal in acid was promising and encouraged us to test variants of CoPbOx.
One such variant involved the use of Co and Fe as the catalytic metal components with Pb remaining as the structural element. The motivation for CoFe stemmed from recent studies demonstrating a synergistic effect that increases the kinetics for OER when Fe is doped into Co or Ni oxides.51,54,55,70 We were curious if the low Tafel slope of 30 mV per decade in alkaline pH could translate to neutral and acidic solutions with the aid of Pb for stabilization at lower pH. As a control, Tafel plots of CoFeOx (ref. 54) were recorded at neutral and acidic pH; here, Co and Fe did not show any catalytic synergy, as the Tafel plots (with 65–70 mV per decade slope) overlaid with those of CoOx (Fig. 4). Stability tests of CoFeOx at higher current densities of 1 mA cm−2 show improvement in stability with film dissolution at ∼1.5 h instead of at ∼0.5 h for CoOx (Fig. 5b). This demonstrates that Fe has a minor structural role, which is consistent with a trend of trading off activity for stability when moving to earlier first-row transition metals. Next, Pb-stabilized CoFeOx was anodically electrodeposited in a solution containing equal concentrations of Co2+, Fe2+, and Pb2+. The resulting film exhibited a composition of 15% Co, 2% Fe, and 83% Pb (by ICP-MS, Table S1†) with homogeneous distribution of the three metals (as determined to ∼7.4 Å resolution by EDS elemental maps from SEM and STEM, Fig. 8 and 9). Tafel analysis of CoFePbOx in neutral and acidic solutions shows no difference from that of CoOx films (slopes of 65–70 mV per decade, Fig. 4). However, unlike CoOx and CoFeOx catalyst films, CoFePbOx exhibits long-term stability in acid, even at higher current densities. While CoPbOx, CoFeOx, and CoOx dissolve in pH 2.5 buffer when performing OER at of 1 mA cm−2, CoFePbOx can maintain a steady ∼1.65 V for over 12 h of operation (Fig. 5b) with a faradaic efficiency near unity (Fig. S3†). To further demonstrate the robustness of these films, CoFePbOx was continuously operated at 1 mA cm−2 for over 50 h in sulfate buffer at pH 2.0 (Fig. 5c) and at only ∼220 mV higher potential than IrOx operating in the same conditions (Fig. 5b); little degradation was observed at OER operating voltages of ∼1.8 V.
CoMnOx (at 0.1 mA cm−2) and CoFePbOx (at 1 mA cm−2) exhibit chemical stability when performing acidic OER near pH 2.5. There are two important considerations for their operation. First their stability depends on application of an anodic bias above ∼1.3 V during operation to maintain its oxidic state. As known for CoOx and as apparent in the Pourbaix diagrams of Co and Pb (Fig. S8†), the Co3O4 and PbO2 states are only maintained at high anodic potentials (above the equilibrium potential for OER) under acidic pH. When voltage is switched off, there is a slow decay of the film back to its original Co2+ and Pb2+ ions in solution. Second, whereas these films are chemically stable, they not indefinitely stable because they lack the ability to self-heal.75 Over long period of operation, mechanical stress on the film from the evolution of O2 gas slowly removes catalyst material from the electrode. Because CoFePbOx (and also CoMnOx) cannot be electrodeposited in acidic conditions (their electrosynthesis occurs at near-neutral pH), mechanical losses cannot be repaired during operation. Discovering an OER catalyst that can both electrodeposit and operate efficiently under acidic pH will furnish self-healing and allow mechanical challenges to be overcome, as is the case for native CoPi and NiBi catalysts.
Nature of stability
The foregoing results demonstrate that structural metals stabilize catalytic sites in mixed metal oxide films. Guidelines emerge for achieving stability while maintaining OER activity. First, mixed metal films should be composed of at least 50% structural metal to maintain an appreciable stability. For example, CoMnOx films prepared with ≥50% Mn do not dissolve in pH 2.5 solution (for OER at 0.1 mA cm−2) while the film with only 17% Mn degraded quickly, similar to a CoOx-only film (Fig. 5a). Given the homogeneous distribution of metals in these catalysts, the requirement that at least half of the film is composed of structural metal suggests that stabilization of active sites requires sufficient scaffolding to exist around those sites. Second, Tafel analyses of mixed metal films show no improvement in OER kinetics when compared to the unary catalytic metal oxide films. This is consistent with XPS spectra indicating negligible differences in the electronic features of Co when comparing CoMnOx and CoFePbOx to CoOx (Fig. 10a and 11a), suggesting that the Co catalytic sites are unchanged from the unary system and obey the same catalytic mechanism (i.e., same Tafel slope) in all three systems. As a corollary, the structural metals do not significantly participate in catalysis, as both MnOx and PbOx have poor OER kinetics and do not affect the Tafel slope of Co catalytic sites in the mixed metal films. However, the structural metal does appear to engage in internal transport of electrons (and possibly protons) inasmuch as even when the number of catalytic sites diminishes in mixed films, its absolute activity (i.e., position of the Tafel plot) remains similar, suggesting that the scaffolding provided by the structural element permeates throughout the film and provides an electronic pathway for addressing active sites.These results are consistent with the contention that the structural metal provides an oxide that embeds the catalytic element and prevents it from dissolution. After OER, the metal–oxygen framework is disrupted with the elimination of O2, and the formal oxidation state of the catalytic metal is reduced, resulting in a weakening of the metal–oxygen framework owing to population of metal–oxygen antibonding orbitals and an attendant decrease in the ligand field strength afforded by moving from oxo to hydroxyl/water.60–62,112,113 The presence of structural metals helps fortify the oxygen framework about the catalytic active site thus minimizing its degradation and leading to enhanced stability of the mixed-metal oxide. This fortification of the oxygen framework stems from the lack of catalysis at structural sites and thus preservation of the structural metal at a high formal charge, as indicated by XPS spectra on CoMnOx (Fig. 10) and CoPbOx/CoFePbOx (Fig. 11). For the former, XPS spectra indicates a slight broadening of the Mn 2p peaks to incorporate features of higher binding energies relative to that of MnOx (Fig. 10b). Given that as-deposited MnOx is birnessite-like (δ-MnO2), which has an average Mn oxidation state of +3.8, the shifting of Mn 2p peaks to higher binding energies suggests an increase in Mn oxidation state nearer to +4.0, which may reflect a pyrolusite-like phase (β-MnO2).25,82,83 Similarly, for CoPbOx and CoFePbOx, the shift of the Pb 4f peaks to higher binding energies when compared to PbOx implies the stabilization of the PbO2 phase.59 It is important to maintain this phase because the reduction of PbO2 to Pb2+ can occur readily at low pH (as governed by its Pourbaix diagram).45 Thus the increase in binding energy, especially from the doping of Fe, corresponds to a slightly higher average formal oxidation state of Pb, which provides additional anodic protection of PbOx against corrosion. We believe that while catalytic sites undergo redox state changes during OER that can momentarily weaken the metal–oxo bond, the oxidation state and bonding environment of the structural metal remains consistent and provides the electron and proton conductivity to facilitate re-oxidization of catalytic species before dissolution.
Conclusion
The current status of acidic OER catalyst properties can be summarized by picking any two properties from highly active, stable, and comprised of earth-abundant materials. Commercial catalysts use Ir or Ru oxides, which are expensive and not scalable, while the best earth-abundant catalysts (such as Co and Ni oxides) dissolve in acid. We demonstrate here that catalysts may be iteratively designed to fulfill the three properties for acidic OER. Unary metal oxides trade activity for acid stability. This trade-off may be circumvented by decoupling catalytic and structural sites as separate elements in mixed metal oxide films. Combining Co as the catalytic metal with Mn as the structural element engenders fast OER kinetics for CoMnOx in acid (∼82 mV per decade Tafel slope in pH 2.5) with sustained activity of 0.1 mA cm−2 for over 12 h; this behavior contrasts CoOx, which dissolves in minutes under the same conditions. To operate at higher current densities, Pb serves as the structural element in CoPbOx and CoFePbOx films, which maintain the OER activity of Co while displaying resistance to corrosion in pH 2.5 solution. Whereas CoOx dissolves within 30 min, CoPbOx is stable for 7 h and CoFePbOx does not degrade after 12 h. Furthermore, CoFePbOx can sustain OER current densities of 1 mA cm−2 for 50 h at pH 2.0. This result is noteworthy in the context that the activity of CoFePbOx comprising earth-abundant elements occurs at ∼220 mV higher overpotential than that of IrOx operating at 1 mA cm−2 in pH 2.5 solution. In this regard, the principle of using different metals to fulfil independent functional components within extended lattices may be useful for designing catalysts that need to fulfil several different criteria simultaneously.Experimental
General electrochemical details
Electrochemical experiments were conducted on a CH Instruments 760D potentiostat. A three-electrode configuration (working, reference, and auxiliary) in glass H-cells was employed where a porous glass frit separated the working and auxiliary compartments. Glassware was pre-cleaned by soaking in aqua regia followed by type I reagent water (EMD Millipore, 18 MΩ cm resistivity). The working electrode was fluorine-doped, tin oxide-coated glass (FTO) with 7 Ω sq−1. surface resistivity (TEC-7, precut 1 cm × 2.5 cm slides from Hartford Glass). Prior to use, FTO slides were cleaned by sonication in acetone and then rinsing with type I water; a 1 cm2 geometric electrode area was created by masking the FTO with Scotch tape, which was removed immediately after film electrodeposition. A Ag/AgCl reference electrode (BASi, filled with saturated KCl) was positioned close to the FTO in the working compartment, and a Pt mesh (99.9%, Alfa Aesar) electrode in the auxiliary side of the H-cell was used to complete the circuit. All experiments were conducted at ambient temperature (∼23 °C). Uncompensated resistance was first measured on a blank FTO electrode in the same cell setup and then corrected through automatic positive feedback during cyclic voltammograms or by subtracting the ohmic potential drop from applied potentials in Tafel and stability measurements after data collection. Potentials were converted to the NHE scale by the following relation: ENHE = EAg/AgCl + 0.197 V, and overpotentials (η) for the oxygen evolution reaction from water were calculated by: η = EOER − (1.23 V − 0.059 V × pH). Positive potentials are oxidizing while negative potentials are reducing.Electrodeposition of catalyst films
Catalysts were prepared by anodic electrodeposition. Generally, a constant anodic potential is applied to 1 cm2 FTO for a fixed amount of time in a 50 mM methylphosphonate (MePi) solution buffered at pH 8.0 containing a total of 0.5 mM metal ion(s). MePi was prepared from methylphosphonic acid (98%, Alfa Aesar) that was purified by recrystallizing twice from boiling acetonitrile (HPLC grade, Aldrich). Although MePi is a preferred buffer since it stabilizes metal ions in solution (yet does not bind too strongly to precipitate the complex) and buffers well in slightly alkaline pH (a regime that accommodates the anodic electrodeposition of a wide range of candidate metal ions), alternative buffers such as acetate at neutral pH also suffice. pH of the buffer solutions was adjusted with KOH (<0.001% Ni, Fe, and other heavy metals; from EMD Millipore). After deposition, films were briefly immersed in type I water to remove any lingering trace metal ions and subsequent electrochemical characterization was performed immediately to prevent films from drying.Electrodeposition conditions for unary metal oxide films employed previous literature procedures in the presence of 50 mM MePi at pH 8.0: MnOx from 0.5 mM Mn2+ (MnCl2·4H2O, 99.995% trace metal basis, Strem) at 0.54 V;24 cobalt oxide (CoOx) from 0.5 mM Co2+ (using Co(NO3)2·6H2O, 99.999% trace metal basis, Strem) at 1.05 V;46 nickel oxide (NiOx) from 0.5 mM Ni2+ (using Ni(NO3)2·6H2O, 99.9988% trace metal basis, Strem) at 1.25 V;18 and lead oxide (PbOx) from 0.5 mM Pb2+ (using Pb(NO3)2, 99.999% trace metal basis, Strem) at 1.35 V.47 Iron oxide (FeOx) was electrodeposited at 1.20 V in a solution of 0.5 mM Fe2+ (using (NH4)2Fe(SO4)2·6H2O, 99.997% trace metal basis, Aldrich) with 1.0 M KNO3 (99.0–100.5%, Macron) heated to 75 °C.48 For comparison, iridium oxide (IrOx) was prepared following literature procedures at 0.85 V in 2 mM Ir3+ (from K3IrCl6, Aldrich) with 15 mM oxalate (from oxalic acid, 98.9–101.0%, Aldrich) in 100 mM carbonate buffer (from K2CO3, >99.0%, Aldrich) at pH 10.5.49 Cobalt-iron oxide (CoFeOx) films were cathodically electrodeposited from ca. 1 mM Co2+ and Fe2+ at −0.3 V, similar to literature procedures.54 Mixed metal oxide films were also anodically co-deposited in MePi buffer at pH 8.0: CoMnOx at 0.65, 0.90, and 1.15 V in ca. 0.25 mM of Co2+ and Mn2+; CoPbOx at 1.15 V in ca. 0.25 mM Co2+ and Pb2+; and CoFePbOx at 1.15 V in ca. 0.125 mM Co2+ and Pb2+ with 0.25 mM Fe2+. All deposition protocols aimed to achieve roughly similar mass loading of films.
Tafel data collection
The oxygen evolution activity of catalyst films was evaluated by measuring the steady-state current density (j) as a function of applied potential (E) in buffered solutions of 100 mM phosphate (Pi, prepared from phosphoric acid, 99.99% trace metal basis, Aldrich) and 1.0 M KNO3 at neutral (pH 7.0) and acidic (pH 2.5) conditions. Steady-state conditions were achieved by holding films at each discrete potential for at least 30 s to allow the current to converge, and measurements were initiated at the highest potential (the first point in the series) for at least 100 s to further minimize any pseudocapacitance. Solutions were stirred at ∼600 rpm with a Teflon stir bar (sufficient to remove mass transport limitations) and applied potentials were post-corrected for uncompensated resistance by subtracting iR (measured on a blank FTO in same solution conditions) from each point where typical values of uncompensated resistance are ∼17 Ω at pH 7 and 2.5. Further precautions were exercised by targeting Tafel data collection at current densities between 1 μA cm−2 and 1 mA cm−2 where these lower currents minimize the impact of any ohmic drop. The current–potential data were plotted as logj vs. E (or η, the overpotential) to construct Tafel plots, where the position and slope (within 5 mV per decade) of independently prepared films under the same conditions were reproducible.
Acid stability during oxygen evolution
Catalyst degradation during oxygen evolution in acidic pH was assessed by long-term chronoamperometry. Similar conditions were employed as for Tafel measurements but with higher concentration of buffer to decrease local pH gradients in the H-cell during prolonged electrolysis: 0.5 M Pi at pH 2.5 and stirred at ∼600 rpm. Chronoamperometry on catalyst films was performed at 0.1 mA cm−2 and 1.0 mA cm−2, and the potential was recorded over time until films were completely dissolved or until 12 h had passed. Dissolution was indicated visually (i.e., the FTO becomes transparent) and also by the inflection in potential that sharply increases to resemble the catalytic properties of blank FTO or catalysis by metal ions in solution. As a demonstration, CoFePbOx was also evaluated over longer time and at slightly lower pH in 0.5 M sulfate (from sulfuric acid, 99.99% trace metal basis, Aldrich) at pH 2.0 for over 50 h of continuous operation. Independently prepared samples following the same protocol were reproducible within 20 min of stability time.Faradaic efficiency of oxygen evolution
The faradaic efficiency of oxygen evolution on CoMnOx and CoFePbOx was determined in 0.5 M Pi at pH 2.5 using a gas chromatograph. The films were mounted in a custom-built two-compartment electrochemical cell where a cation-exchange membrane (Nafion 117, Sigma Aldrich) was used to separate the two chambers. A Ag/AgCl-based leak-free reference electrode (LF-1, Warner Instruments) was used as the reference electrode and a Pt wire was the counter electrode. A Viton O-ring was applied to define the area of working electrode and OER was sustained at constant current densities of 0.1 and 1 mA cm−2 for CoMnOx and CoFePbOx, respectively. While stirring, a constant flow of Ar gas (20 sccm) was bubbled through the chamber of working/reference electrodes. The gas outlet was connected to a gas chromatograph equipped with a thermal conductivity detector (multiple gas analyzer #3, SRI Instruments). The amount of O2 in the out-fluxing Ar gas was quantified, based on the calibration with known O2 concentrations. Initial control experiments were performed to ensure that the O2 in the air has no contribution to the measured O2 signals. The detected O2 concentrations were compared to the theoretical yield of O2 (calculated by dividing the charge passed by 4F) to obtain the faradaic efficiency.Inductively coupled plasma mass spectrometry (ICP-MS)
Trace elemental analysis on catalyst films was performed with a quadrupole ICP-MS (Thermo Electron, X-Series ICP-MS with CCT). Because these films are resistant to passive dissolution in acid, they were digested by voltage cycling (between 1.3 and −0.4 V) in 12 mL of 2% v/v nitric acid (TraceSELECT, Fluka). Co, Mn, Pb, and Fe calibration standards were prepared from corresponding ICP standard solutions (TraceCERT, Fluka), which enabled the construction of a calibration curve to convert the detected counts for 59Co, 55Mn, 208Pb, and 57Fe to concentrations (in ppm).Electron microscopy
The compositional morphology of catalyst films was observed by field emission scanning electron microscopy (SEM, Zeiss Supra 55VP) operated at a beam voltage of 15.0 kV, working distance of ∼8.5 mm, a 30 μm aperture, and an InLens detector. Elemental quantification was determined at a beam voltage of 14 kV with an energy dispersive X-ray spectrometer (EDS from EDAX Inc.) using EDAX ZAF correction factors. Homogeneity on the 20 nm per px scale of mixed metal films were evaluated by EDS elemental maps using characteristic X-rays at the K-edge for Co, Mn, and Fe as well as the M-edge for Pb.CoMnOx and CoFePbOx samples were subjected to higher resolution examination of crystallinity and homogeneity by using spherical aberration corrected high angle annular dark field scanning transmission electron microscopes (JEOL ARM200F HAADF STEM and Hitachi HD-2300 STEM). Samples were directly electrodeposited on 400-mesh pure carbon film supported on Au TEM grids (Electron Microscopy Sciences) by partially immersing the grid in the deposition solution as the working electrode. Samples were then dipped in type I water to remove any residual electrolyte and dried at room temperature. Images were taken at a beam accelerating voltage of 200 kV in the scanning transmission electron mode. Brightness and contrast of the images were processed using Gatan DigitalMicrograph software. Homogeneity on the 7.4 Å per px scale of mixed metal films were evaluated by EDS elemental maps using signals corresponding to K line energy values for cobalt, manganese, and iron; and L line energy values for lead.
X-ray photoelectron spectroscopy (XPS)
Comparison of oxidation state and chemical environment between unary and mixed metal catalyst films employed XPS (using a Thermo Scientific K-alpha XPS system). Catalyst films were electrodeposited on FTO as described above for: CoFePbOx, CoPbOx, CoMnOx, CoOx, PbOx, and MnOx. All samples were illuminated using a monochromated Al Kα X-ray source (1486.6 eV energy and 0.85 eV line width)114 with a 400 μm spot size. Surface charging was compensated by a low-energy (0–14 eV) electron flood gun. The system was pre-calibrated with Au, Ag, and Cu standards built into the sample stage using an automated routine. High-resolution spectra for Co 2p, Mn 2p, Pb 4f, and Fe 2p were measured with a step size of 0.1 eV. All spectra were then calibrated to the C 1s peak at 284.8 eV.57Powder X-ray diffraction (XRD)
The crystallinity of CoMnOx and CoFePbOx samples were tested by XRD. To maximize the possibility of observing diffraction, powder samples of the catalysts were prepared by large-scale electrodeposition (using the same protocols described earlier) on multiple 20 × 8 cm2 FTO plates. The thin films were briefly immersed in type I water to remove any residual solution, dried, carefully stripped from the FTO, and ground into a fine powder. The powders were loaded in a small cavity in a Si zero-diffraction plate (MTI Corporation) and inserted into a Bruker D2 Phaser powder diffractometer equipped with a Cu Kα X-ray source (generated at 20 kV and 20 mA; passed through a 1 mm slit) and LynxEye detector. A knife-edge attachment was used to reduce scattered signal, and the stage was rotated by 3° min−1. Samples were examined in Bragg–Brentano mode from 2θ = 10 to 75° in 0.02° increments with 0.5 s per point scan rate.Acknowledgements
We thank Nancy Li and Zhongxing Chen for help with ICP-MS, Jules Gardener for assistance with STEM imaging, and Mircea Dincă for discussions. C. L. acknowledges Prof. X. Ling at Nanyang Technological University and financial support from Lee Kuan Yew Postdoctoral Fellowship. This material is based upon work supported under the Solar Photochemistry Program of the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences of the U. S. Department of Energy. SEM, TEM, and XPS were performed at Harvard University's Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Infrastructure Network (NNIN), which is supported by the National Science Foundation under ECS-0335765.References
- D. G. Nocera, Daedalus, 2006, 135, 112–115 CrossRef
.
- D. Abbott, Proc. IEEE, 2010, 98, 42–66 CrossRef CAS
- D. G. Nocera, Energy Environ. Sci., 2010, 3, 993–995 Search PubMed
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed
-
N. S. Lewis and D. G. Nocera, The Bridge, 2015, vol. 46, pp. 41–47 Search PubMed
- D. G. Nocera, Acc. Chem. Res., 2017, 50, 616–619 CrossRef CAS
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. I. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS PubMed
- C. Liu, B. Colón, M. Ziesack, P. A. Silver and D. G. Nocera, Science, 2016, 352, 1210–1213 CrossRef CAS PubMed
- F. Liu, J. J. Concepcion, J. W. Jurss, T. Cardolaccia, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2008, 47, 1727–1752 CrossRef CAS PubMed
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS PubMed
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS PubMed
- K. Klingan, F. Ringleb, I. Zaharieva, J. Heidkamp, P. Chernev, D. Gonzalez-Flores, M. Risch, A. Fischer and H. Dau, ChemSusChem, 2014, 7, 1301–1310 CrossRef CAS PubMed
- D. A. Corrigan, J. Electrochem. Soc., 1987, 134, 377–384 CrossRef CAS
- M. Dincă, Y. Surendranath and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10337–10341 CrossRef PubMed
- D. K. Bediako, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2013, 135, 3662–3674 CrossRef CAS PubMed
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed
- D. Kang, T. W. Kim, S. R. Kubota, A. C. Cardiel, H. G. Cha and K. S. Choi, Chem. Rev., 2015, 115, 12839–12887 CrossRef CAS PubMed
- I. Roger and M. D. Symes, J. Mater. Chem. A, 2016, 4, 6724–6741 CAS
- M. Huynh, D. K. Bediako and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 6002–6010 CrossRef CAS PubMed
- M. Huynh, D. K. Bediako, Y. Liu and D. G. Nocera, J. Phys. Chem. C, 2014, 118, 17142–17152 CAS
- M. Huynh, C. Shi, S. J. L. Billinge and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 14887–14904 CrossRef CAS PubMed
- L. G. Bloor, P. I. Molina, M. D. Symes and L. Cronin, J. Am. Chem. Soc., 2014, 136, 3304–3311 CrossRef CAS PubMed
- R. Frydendal, E. A. Paoli, I. Chorkendorff, J. Rossmeisl and I. E. L. Stephens, Adv. Energy Mater., 2015, 5, 1500991 CrossRef
- R. J. R. Jones, A. Shinde, D. Guevarra, C. Xiang, J. A. Haber, J. Jin and J. M. Gregoire, ACS Comb. Sci., 2015, 17, 71–75 CrossRef CAS PubMed
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed
- B. Seger, K. Vinodgopal and P. V. Kamat, Langmuir, 2007, 23, 5471–5476 CrossRef CAS PubMed
- N. Bunkin, P. Ignatiev, V. Kozlov, A. Shkirin, S. Zakharov and A. Zinchenko, Water, 2013, 4, 129–154 Search PubMed
- C. R. Cox, M. T. Winkler, J. J. H. Pijpers, T. Buonassisi and D. G. Nocera, Energy Environ. Sci., 2013, 6, 532–538 CAS
- R. Costi, E. R. Young, V. Bulović and D. G. Nocera, ACS Appl. Mater. Interfaces, 2013, 5, 2364–2367 CAS
- M. J. Kenney, M. Gong, Y. Li, J. Z. Wu, J. Feng, M. Lanza and H. Dai, Science, 2013, 342, 836–840 CrossRef CAS PubMed
- M. F. Lichterman, K. Sun, S. Hu, X. Zhou, M. T. McDowell, M. R. Shaner, M. H. Richter, E. J. Crumlin, A. I. Carim, F. H. Saadi, B. S. Brunschwig and N. S Lewis, Catal. Today, 2016, 262, 11–23 CrossRef CAS
- D. G. Nocera, Inorg. Chem., 2009, 48, 10001–10007 CrossRef CAS PubMed
- A. F. Heyduk and D. G. Nocera, Science, 2001, 293, 1639–1641 CrossRef CAS PubMed
- T. S. Teets and D. G. Nocera, Chem. Commun., 2011, 47, 9268–9274 RSC
- D. C. Powers, S. J. Hwang, S. Zheng and D. G. Nocera, Inorg. Chem., 2014, 53, 9122–9128 CrossRef CAS PubMed
- A. A. Gewirth and M. S. Thorum, Inorg. Chem., 2010, 49, 3557–3566 CrossRef CAS PubMed
-
R. O'Hayre, S. W. Cha, W. Colella and F. B. Prinz, Fuel Cell Fundamentals, Wiley, Hoboken, NJ, 2nd edn, 2009 Search PubMed
-
M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, National Association of Corrosion Engineers, New York, NY, 2nd edn, 1974 Search PubMed
- Y. Surendranath, D. A. Lutterman, Y. Liu and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6326–6336 CrossRef CAS PubMed
- X. Li, D. Pletcher and F. C. Walsh, Chem. Soc. Rev., 2011, 40, 3879–3894 RSC
- R. L. Spray and K.-S. Choi, Chem. Mater., 2009, 21, 3701–3709 CrossRef CAS
- M. A. Petit and V. Plichon, J. Electroanal. Chem., 1998, 444, 247–252 CrossRef CAS
-
E. Gileadi, Physical Electrochemistry: Fundamentals, Techniques and Applications, Wiley-VCH, Weinheim, 1st edn, 2011 Search PubMed
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed
- Y. Zhao, N. M. Vargas-Barbosa, E. A. Hernandez-Pagan and T. E. Mallouk, Small, 2011, 7, 2087–2093 CrossRef CAS PubMed
- D. Pavlov and T. Rogachev, Electrochim. Acta, 1978, 23, 1237–1242 CrossRef CAS
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CAS
- H. Sánchez and Y. Meas, React. Kinet. Catal. Lett., 1991, 45, 67–71 CrossRef
- T. L. Barr and S. Seal, J. Vac. Sci. Technol., A, 1995, 13, 1239–1246 CAS
- S. C. Petitto and M. A. Langell, J. Vac. Sci. Technol., A, 2004, 22, 1690–1696 CAS
- R. W. Hewitt and N. Winograd, Surf. Sci., 1978, 78, 1–14 CrossRef CAS
-
W. Stumm and G. Furrer, in Aquatic Surface Chemistry: Chemical Processes at the Particle-Water Interface, ed. W. Stumm, Wiley, New York, 1987, pp. 197–204 Search PubMed
- W. H. Casey, J. Colloid Interface Sci., 1991, 146, 586–589 CrossRef CAS
- W. Stumm, Colloids Surf., A, 1997, 120, 143–166 CrossRef CAS
-
A. C. C. Tseung, S. Yeh, X. Liu, G. H. Kelsall and P. Dykstra, Novel Acid-Resistant Oxygen Evolution Electrodes, EUR/European Commission; Commission of the European Communities, Luxembourg, 1991 Search PubMed
- S. Song, H. Zhang, X. Ma, Z. Shao, R. T. Baker and B. Yi, Int. J. Hydrogen Energy, 2008, 33, 4955–4961 CrossRef CAS
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS
- D. Seley, K. Ayers and B. A. Parkinson, ACS Comb. Sci., 2013, 15, 82–89 CrossRef CAS PubMed
- M. Datta, K. Kadakia, O. I. Velikokhatnyi, P. H. Jampani, S. J. Chung, J. A. Poston, A. Manivannan and P. N. Kumta, J. Mater. Chem. A, 2013, 1, 4026–4037 CAS
- K. B. Kokoh, E. Mayousse, T. W. Napporn, K. Servat, N. Guillet, E. Soyez, A. Grosjean, A. Rakotondrainibé and J. Paul-Joseph, Int. J. Hydrogen Energy, 2014, 39, 1924–1931 CrossRef CAS
- W. Hu, H. Zhong, W. Liang and S. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 12729–12736 CAS
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. v. Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486–1491 CrossRef CAS PubMed
- M. Woodhouse and B. A. Parkinson, Chem. Mater., 2008, 20, 2495–2502 CrossRef CAS
- J. B. Gerken, J. Y. C. Chen, R. C. Massé, A. B. Powell and S. S. Stahl, Angew. Chem., Int. Ed., 2012, 51, 6676–6680 CrossRef CAS PubMed
- K. Izumiya, E. Akiyama, H. Habazaki, A. Kawashima, K. Asami and K. Hashimoto, J. Appl. Electrochem., 1997, 27, 1362–1368 CrossRef CAS
- J. Rosen, G. S. Hutchings and F. Jiao, J. Am. Chem. Soc., 2013, 135, 4516–4521 CrossRef CAS PubMed
- D. K. Bediako, A. M. Ullman and D. G. Nocera, Top. Curr. Chem., 2015, 371, 173–214 CrossRef
- D. A. Lutterman, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2008, 131, 3838–3839 CrossRef PubMed
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed
- A. Shinde, R. J. R. Jones, D. Guevarra, S. Mitrovic, N. Becerra-Stasiewicz, J. A. Haber, J. Jin and J. M. Gregoire, Electrocatalysis, 2015, 6, 229–236 CrossRef CAS
- R. J. R. Jones, A. Shinde, D. Guevarra, C. Xiang, J. A. Haber, J. Jin and J. M. Gregoire, ACS Comb. Sci., 2015, 17, 71–75 CrossRef CAS PubMed
- S. Trasatti and O. A. Petrii, J. Electroanal. Chem., 1992, 327, 353–376 CrossRef CAS
-
J. R. Winkler and H. B. Gray, in Structure and Bonding, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer, Berlin, Heidelberg, 2012, vol. 142, p. 17 Search PubMed
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Energy Environ. Sci., 2012, 5, 7081–7089 CAS
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494–3501 CrossRef CAS PubMed
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Energy Environ. Sci., 2012, 5, 7081–7089 CAS
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494–3501 CrossRef CAS PubMed
- Y. Surendranath, M. Dincă and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 2615–2620 CrossRef CAS PubMed
- P.-Y. Chuang and C.-C. Hu, Mater. Chem. Phys., 2005, 92, 138–145 CrossRef CAS
- P. Lavela, J. L. Tirado and C. Vidal-Abarca, Electrochim. Acta, 2007, 52, 7986–7995 CrossRef CAS
- K. Izumiya, E. Akiyama, H. Habazaki, N. Kumagai, A. Kawashima and K. Hashimoto, Mater. Trans. JIM, 1997, 38, 899–905 CrossRef CAS
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed
-
Y. Surendranath and D. G. Nocera, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Inc., Hoboken, NJ, 2011, vol. 57, pp. 505–560 Search PubMed
-
K. Pisarczyk, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2000; pp. 1–78 Search PubMed
- M. R. Wixom, D. J. Tarnowski, J. M. Parker, J. Q. Lee, P.-L. Chen, I. Song and L. T. Thompson, MRS Proceedings, 1997, 496, 643–653 CrossRef
- S. Park, J.-W. Lee and B. N. Popov, Int. J. Hydrogen Energy, 2012, 37, 5850–5865 CrossRef CAS
- A. J. Forman, Z. Chen, P. Chakthranont and T. F. Jaramillo, Chem. Mater., 2014, 26, 958–964 CrossRef CAS
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef
- D. J. Payne, R. G. Egdell, W. Hao, J. S. Foord, A. Walsh and G. W. Watson, Chem. Phys. Lett., 2005, 411, 181–185 CrossRef CAS
- D. O. Scanlon, A. B. Kehoe, G. W. Watson, M. O. Jones, W. I. F. David, D. J. Payne, R. G. Egdell, P. P. Edwards and A. Walsh, Phys. Rev. Lett., 2011, 107, 1–5 CrossRef PubMed
- P. Jones, R. Lind and W. F. K. Wynne-Jones, Trans. Faraday Soc., 1954, 50, 972–979 RSC
- A. B. Velichenko, R. Amadelli, E. A. Baranova, D. V. Girenko and F. I. Danilov, J. Electroanal. Chem., 2002, 527, 56–64 CrossRef CAS
- M. J. Barmi and A. N. Nikoloski, Hydrometallurgy, 2012, 129–130, 59–66 CrossRef CAS
- M. Clancy, C. J. Bettles, A. Stuart and N. Birbilis, Hydrometallurgy, 2013, 131–132, 144–157 CrossRef CAS
- J. P. Carr and N. A. Hampson, Chem. Rev., 1972, 72, 679–703 CrossRef CAS
- J. P. Pohl and W. Schendler, J. Power Sources, 1981, 6, 245–250 CrossRef CAS
- N. K. Appandairajan and J. Gopalakrishnan, Proc. - Indian Acad. Sci., Anim. Sci., 1978, 87, 115–120 CAS
- V. Patil, Soft Nanosci. Lett., 2012, 2, 1–7 CrossRef
- E. Preisler, J. Appl. Electrochem., 1976, 6, 311–320 CrossRef CAS
- R. N. Deguzman, A. Awaluddin, Y. F. Shen, Z. R. Tian, S. L. Suib, S. Ching and C. L. Oyoung, Chem. Mater., 1995, 7, 1286–1292 CrossRef CAS
- B. Warnes, F. Aplan and G. Simkovich, Solid State Ionics, 1984, 12, 271–276 CrossRef CAS
- K. Kaneko and K. Inouye, J. Chem. Soc., Faraday Trans. 1, 1976, 72, 1258–1266 RSC
-
F. Basolo and R. G. Pearson, in Mechanisms of Inorganic Reactions, Wiley, New York, 1967 Search PubMed
- T. A. Betley, Y. Surendranath, M. V. Childress, G. E. Alliger, R. Fu, C. C. Cummins and D. G. Nocera, Philos. Trans. R. Soc., B, 2008, 363, 1293–1303 CrossRef CAS PubMed
-
J. F. Watts and J. Wolstenholme, An Introduction to Surface Analysis by XPS and AES, John Wiley & Sons, Ltd, Chichester, UK, 2003 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: CVs for unary metal oxides deposition, electrochemical stability at higher current densities for unary metal oxides at pH 2.5, EDS maps for CoMnOx and CoPbOx, STEM images and PXRD of CoMnOx and CoFePbOx, high-resolution XPS of Fe 2p for CoFePbOx, Pourbaix diagrams (of Mn, Co, Pb, and Fe), and elemental analysis. See DOI: 10.1039/c7sc01239j |
| This journal is © The Royal Society of Chemistry 2017 |