
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the incompatibility of lithium–O2 battery technology with CO2†
Shiyu
Zhang‡
 a,
Matthew J.
Nava‡
a,
Gary K.
Chow
b,
Nazario
Lopez§
a,
Gang
Wu
c,
David R.
Britt
b,
Daniel G.
Nocera
*d and
Christopher C.
Cummins
*a
a,
Matthew J.
Nava‡
a,
Gary K.
Chow
b,
Nazario
Lopez§
a,
Gang
Wu
c,
David R.
Britt
b,
Daniel G.
Nocera
*d and
Christopher C.
Cummins
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139-4307, USA. E-mail: ccummins@mit.edu; Tel: +1 617 253 5332
bDepartment of Chemistry, University of California, Davis, One Shields Avenue, Davis, CA 95616, USA
cDepartment of Chemistry, Queen’s University, 90 Bader Lane, Kingston, Ontario K7L3N6, Canada
dDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138, USA
First published on 20th June 2017
Abstract
When solubilized in a hexacarboxamide cryptand anion receptor, the peroxide dianion reacts rapidly with CO2 in polar aprotic organic media to produce hydroperoxycarbonate (HOOCO2−) and peroxydicarbonate (−O2COOCO2−). Peroxydicarbonate is subject to thermal fragmentation into two equivalents of the highly reactive carbonate radical anion, which promotes hydrogen atom abstraction reactions responsible for the oxidative degradation of organic solvents. The activation and conversion of the peroxide dianion by CO2 is general. Exposure of solid lithium peroxide (Li2O2) to CO2 in polar aprotic organic media results in aggressive oxidation. These findings indicate that CO2 must not be introduced in conditions relevant to typical lithium–O2 cell configurations, as production of HOOCO2− and −O2COOCO2− during lithium–O2 cell cycling will lead to cell degradation via oxidation of organic electrolytes and other vulnerable cell components.
Introduction
The two-electron reduction of molecular oxygen to the peroxide dianion is an attractive cathode redox couple for developing rechargeable lithium–O2 batteries.1 Lithium carbonate (Li2CO3) formation is deleterious to battery performance because it passivates electrodes and causes a drastic reduction in the round trip efficiency of discharge–charge cycles.2,3 Carbonate formation is typically ascribed to oxidative degradation of organic electrolytes4–6 and carbon electrodes7 by superoxide8,9 and singlet oxygen.10 Although peroxide is often considered to be a strong oxidant in aqueous media, salts of its dianion (O22−) are poor oxidizers in organic media due to their extremely low solubility and so, for this reason, the possible role of peroxide in furnishing carbonate is underappreciated.11 The presence of carbonate-derived CO2 during the recharge cycle of lithium–O2 batteries2 prompted us to consider the possibility that carbonate formation may be a consequence of peroxide combination with carbon dioxide; this would likely confer increased solubility and yield powerful oxidizers. To address this topic, we utilized an anion-receptor solubilized form of the peroxide dianion12 to elucidate the molecular level details of its reaction with carbon dioxide. As reported herein, we observed the formation of strongly oxidizing peroxy(di)-carbonate intermediates and studied their reaction with organic solvents to produce carbonate. In a complementary line of investigation, we showed that carbon dioxide activation of insoluble Li2O2 similarly engenders solvent oxidation with the concomitant production of carbonate. Our findings shed light on the identity and behavior of the hot oxidants generated upon the facile and quantitative combination of O22− with CO2via direct spectroscopic detection and exploratory reaction chemistry.Results and discussion
Reaction of O22− with CO2 using an anion receptor
Despite the drastic and deleterious effect that CO2 has upon the performance of a cycling lithium–oxygen battery, our understanding of the chemical entities responsible for this effect is poor and based primarily upon computational studies or observation of terminal reaction products.2,8 To examine the effect of CO2 on the oxidative power of peroxide, an anion receptor complex13 of the peroxide dianion, [O2⊂mBDCA-5t-H6]2− (1, Fig. 1),12 was employed as a soluble source of peroxide dianion. The anion receptor mBDCA-5t-H6 encapsulates the peroxide dianion via six N–H⋯O hydrogen bonds. Since its discovery, this cryptate has enabled exploration of the reactivity of the peroxide dianion with small molecules in polar organic media without the complicating influence of acidic protons.12,14 Despite being a simple molecule, the peroxide dianion has yielded rich and previously unknown chemistry, including metal-free oxidation of carbon monoxide (CO) generating carbonate, which is encapsulated by the anion receptor as [CO3⊂mBDCA-5t-H6]2− (2, Fig. 1).14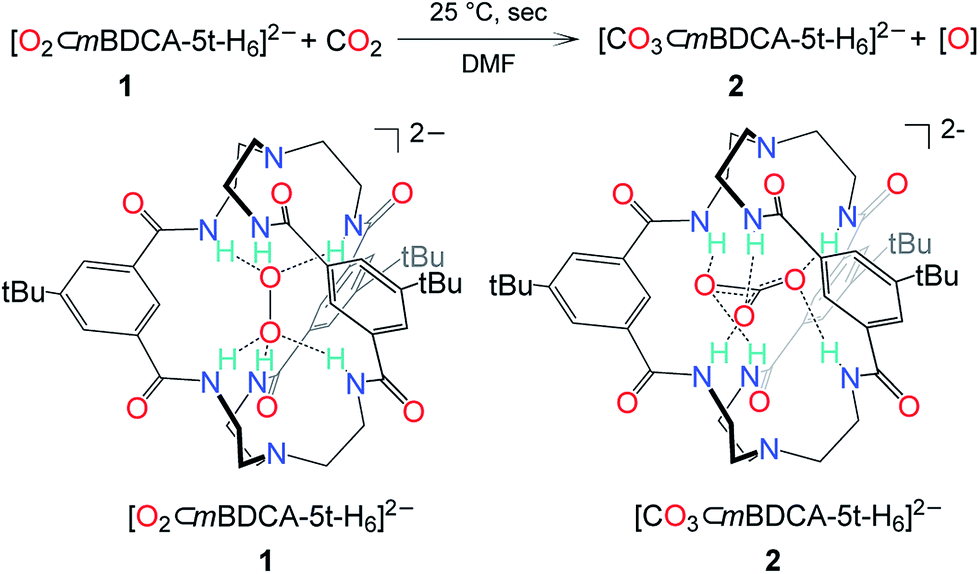 | ||
| Fig. 1 The reaction scheme of peroxide cryptate 1 with CO2 and a line drawing of [O2⊂mBDCA-5t-H6]2− and [CO3⊂mBDCA-5t-H6]2−. | ||
While the conversion of 1 to 2 under CO (1 atm, 40 °C) takes two hours to go to completion, exposing a dimethylformamide-d7 (DMF-d7) solution of 1 to CO2 (1 atm, 25 °C) resulted in the essentially instantaneous formation of carbonate cryptate [CO3⊂mBDCA-5t-H6]2− as indicated by 1H NMR spectroscopy. Formation of O2 gas was not observed by gas chromatography (GC) analysis of the reactor headspace gases,15 suggesting the possibility of oxygen incorporation into the solvent molecules. To probe the fate of the “missing oxygen atom” according to the equation at the top of Fig. 1, the reaction of CO2 and 1 was next performed in the presence of oxygen atom acceptors. While 1 on its own is unreactive towards PPh3 and methoxythioanisole at 25 °C, exposing a mixture of 1 and an organic oxygen-atom acceptor to CO2 (1 atm, 25 °C) resulted in the rapid formation of triphenylphosphine oxide (90%, Fig. 2) or 1-(methylsulfinyl)-4-methoxybenzene (61%, Fig. 2), respectively.
 | ||
| Fig. 2 Addition of CO2 to 1 in the presence of an oxygen-atom acceptor. | ||
Aiming to establish the chemical identity of the oxidant(s) generated upon exposure of peroxide cryptate 1 to CO2, we followed the reaction by variable temperature 13C NMR spectroscopy. A strong new signal at δ = 156.9 ppm, together with one minor species resonating at δ = 157.4 ppm, was observed at −50 °C (Fig. 3). We first considered peroxycarbonate (−OOCO2−,Fig. 3) and hydroperoxycarbonate (HOOCO2−, Fig. 3) as candidates to correspond to the observed 13C NMR signals, since hydroperoxycarbonate is known to be active for sulfide oxidation.16,17 The salt [PPN][HOO13CO2] (PPN = bis(triphenylphosphine)iminium), which was generated in situ from H2O2 and bicarbonate [PPN][H13CO3] (δ = 160.0 ppm),18–20 showed a single 13C resonance at δ = 157.5 ppm, confirming the identity of the minor intermediate as HOOCO2−.
 | ||
| Fig. 3 Variable temperature 13C NMR (left) and 17O NMR (right) analysis of the reaction between 13CO2 and 1. | ||
Moreover, 13C Gauge-Independent Atomic Orbital (GIAO) NMR calculations of the chemical shifts of potential candidates were performed.15 From a range of potential chemical species (Fig. 4), symmetric peroxydicarbonate (−O2COOCO2−) emerged as the most plausible assignment for the major product at δ = 156.9 ppm, having the best match between the observed and calculated 13C NMR chemical shift.15 In an effort to independently generate −O2COOCO2−, an experiment was carried out in which excess 13CO2 was added to a frozen mixture of potassium tert-butoxide and bis(trimethylsilyl) peroxide giving rise to a single new 13C NMR resonance at δ = 155.5 ppm (−40 °C), tentatively supporting our identification of the major 1 + CO2 product as symmetric peroxydicarbonate. Differences in the medium and reaction conditions may account for the observed chemical shift difference (155.5 ppm here versus 156.9 ppm, above). Similarly, superoxide (O2˙−) has been documented to absorb two equivalents of CO2, generating unsymmetrical peroxydicarbonate (Fig. 4) as a precipitate.21 In our hands, the low solubility of this unsymmetrical peroxydicarbonate material precluded its characterization by solution 13C NMR studies under conditions we employed successfully for in situ characterization of −O2COOCO2− and HOOCO2−. This establishes that different oxidants are generated upon addition of CO2 to superoxide as compared with the peroxide dianion (Fig. 4).
 | ||
| Fig. 4 Possible intermediates during the conversion of 1 and CO2 to 2 (top) and formation of symmetric and unsymmetric peroxydicarbonate (bottom). | ||
Further support for the formation of HOOCO2− and −O2COOCO2− upon interaction of CO2 with peroxide sources was provided by variable temperature 17O NMR spectroscopy. Due to the fast relaxation times of 17O nuclei, observation of the 17O resonance for mid-size molecules such as 1-17O2 and 2-CO17O2 was expected to be challenging in solution.22 Indeed, 17O NMR measurements of independently prepared peroxide cryptate 1-17O2 and carbonate cryptate 2-CO17O2 (70%, 17O-enriched) showed no resonances between δ = −1100 and +1800 ppm (H2O used as a reference, δ = 0 ppm) in DMF. However, solid-state 17O NMR measurements for 1-17O2 and 2-CO17O2 were successful, as reported previously in the case of 2-CO17O2,14 and in the present work for 1-17O2, providing the benchmark 17O NMR chemical shifts (δ = 260 ppm for 1-17O2 and 170 ppm for 2-CO17O2) (Fig. 5, Table 1). As seen in Fig. 3, 70% 17O-enriched samples of HOOCO2− and −O2COOCO2− generated in DMF solution at −78 °C from the reaction of 1-17O2 and 13CO2 resulted in a broad 17O NMR resonance at δ = 275.3 ppm, assigned as overlapping signals of HOOCO2− and −O2COOCO2−. Upon gradual warming of the sample to −10 °C, the intensity of the signal decayed; the signal ultimately resolved into two peaks with equal intensities at δ = 278.7 and 264.0 ppm, distinct from those observed for 1-17O2 and 2-CO17O2. The two peaks observed are attributed to HOOCO2− which contains two chemically inequivalent 17O atoms (δ = 278.7 ppm for HOOCO2− and 264.0 ppm for HOOCO2−), in contrast to the situation for −O2COOCO2− in which the peroxy oxygen atoms are related by symmetry. The appearance of the relatively sharp 17O NMR signals assigned to HOOCO2− coupled with the concurrent observation of monodeprotonated cryptand ([mBDCA-5t-H5]−)14 by 1H NMR spectroscopy strongly suggests that HOOCO2− is not strongly sequestered inside the anion receptor. The observed 17O NMR chemical shifts are in accordance with expectations arising from 17O NMR absolute shielding calculations and compare well with data for benchmark organic compounds containing the peroxy functional group.23
 | ||
| Fig. 5 Experimental (black trace) and simulated (red trace) solid-state 17O NMR spectra of (a) 1-17O2, (b) 2-CO17O2, and (c) product resulting from the treatment of solid 1-17O2 with CO2. All solid-state 17O NMR experiments were performed on a Bruker Avance-600 (14.1 T) spectrometer under static conditions. A Hahn echo sequence was used for recording the static spectra to eliminate the acoustic ringing from the probe. A 4 mm Bruker MAS probe was used without sample spinning. The effective 90° pulse was of a duration of 1.7 μs. High power 1H decoupling (70 kHz) was applied in all static experiments. A liquid H2O sample was used for both RF power calibration and 17O chemical shift referencing (δ = 0 ppm). | ||
| Compound | δ iso /ppm | δ 11/ppm | δ 22/ppm | δ 33/ppm | C Q/MHz | η Q/MHz | |
|---|---|---|---|---|---|---|---|
| a The uncertainties in the experimental data are: δiso ± 2 ppm; δii ± 10 ppm; CQ ± 0.2 MHz; ηQ ± 0.1. b See ref. 24. c See ref. 25. | |||||||
| 1 | Exp | 260 | 335 | 335 | 110 | −16.6 | 0.0 |
| ADF | 308 | 388 | 388 | 148 | −17.5 | 0.000 | |
| 2 | Exp | 170 | 266 | 194 | 50 | 7.5 | 0.7 |
| ADF | 223 | 335 | 222 | 112 | 7.03 | 0.95 | |
| O22− | ADF | 221 | 398 | 398 | −13.3 | −18.66 | 0.000 |
| H2O2 | Expb | 180 | — | — | — | −16.31 | 0.687 |
| ADF | 182 | 383 | 211 | −48 | −16.81 | 0.969 | |
| Li2O2 | Expc | 227 | 352 | 352 | −23 | −18.66 | 0.00 |
The mechanism of CO2/peroxide driven oxidation
Having thereby established the identity of the active oxidants generated from the combination of O22− and CO2 as HOOCO2− and −O2COOCO2−, we next turned our attention to the mechanism of CO2/peroxide driven oxidation. The reaction of 18O-labeled 1 and CO2 was performed in the presence of an oxidizable substrate. Exposure of a mixture of 1-18O2 and PPh3 to CO2 furnished 18OPPh3 as the oxidized product based on GCMS analysis.15 The obtained 18O isotope labeling data precluded the possibility of O–O bond cleavage prior to the oxygen atom transfer (OAT) reaction, as such a process would yield isotopic scrambling and result in a mixture of 16OPPh3 and 18OPPh3. Therefore, H18O2CO2 – with its peroxy unit intact as it was derived from the peroxide dianion – is implicated as the active species for the OAT conversion of PPh3 to OPPh3 (Fig. 6, OAT pathway). In contrast, addition of CO2 to a solution of 1-18O2 in the presence of the hydrogen atom donor 9,10-dihydroanthracene (DHA) led to a statistical mixture of anthraquinone products with 16O and 18O incorporation.15 The observed isotope scrambling was likely due to a sequence of H-atom abstraction/radical recombination reactions. By analogy to the behavior of organic peroxydicarbonates,26 symmetrical peroxydicarbonate would be expected to undergo O–O bond homolysis generating two equivalents of the reactive carbonate radical CO3˙− (Fig. 6, hydrogen atom transfer (HAT) pathway).27 Quantum chemical calculations indicate that homolytic cleavage of the O–O bond in −O2COOCO2− is only mildly endergonic (reaction free energy +14 kcal mol−1). This species thus has an unusually weak O–O bond. | ||
| Fig. 6 Proposed mechanistic pathways for CO2-mediated solvent decomposition in lithium–O2 batteries. OER is an “oxygen evolving reaction”, HAT is a “hydrogen atom transfer” oxidative process involving hydrogen atom abstraction by carbonate radical anion, and OAT is “oxygen atom transfer” to a substrate, S. | ||
Homolytic cleavage of the O–O bond and generation of CO3˙− appears to be favorable for two reasons: (i) repulsion of the negative charge due to poor solvation in organic solvents resulting in coulombic explosion28 and (ii) resonance stabilization of the unpaired electron of the carbonate radical anion over the carbonate π system. Carbonate radicals have been generated previously via laser photolysis of aqueous persulfate in the presence of bicarbonate.29 Carbonate radicals have been implicated in guanine oxidation27 and are also believed to be formed upon treatment of peroxynitrite (ONOO−) with CO2, and in that case generate nitrogen dioxide as a byproduct.27,30,31 Furthermore, in the manganese-catalyzed oxidation of amino acids by H2O2, the formation of reactive oxygen species only occurred when HCO3− buffer was used.32,33 Carbonate radicals generated in lithium–oxygen batteries can then engage in HAT reactions with solvents containing weak C–H bonds, driven by the high O–H bond strength (BDE ≅ 107 kcal mol−1) of the bicarbonate that is formed.27 Accordingly, we suggest that for stability under lithium–O2 cell cycling conditions, an organic solvent/electrolyte should have no C–H bonds of BDE ≅ 107 kcal mol−1 or less.
To experimentally confirm the generation of CO3˙−, 1 was treated with CO2 in the presence of the spin trap 5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide (BMPO), and the reaction was monitored by EPR spectroscopy.34 Upon addition of the CO2, signals for the hydroxyl adduct [BMPO–OH]˙ together with small quantities (ca. 5%) of an unidentified spin-trap adduct suspected to be [BMPO–OCO2]˙− were observed within seconds (Fig. 7). Formation of [BMPO–OH]˙ is proposed to occur via a rapid reaction between the chemically generated CO3˙− and BMPO, initially yielding [BMPO–OCO2]˙−, followed by decarboxylation and protonation. The proton source under these conditions could be the anion receptor mBDCA-5t-H6 (Fig. 7C). This sequence is directly along the lines proposed for the related spin trap DMPO under exposure to carbonate radicals.35,36 On longer timescales, [BMPO–OH]˙ was further oxidized to [BMPO–O]˙ and other unidentified decomposition products.34
 | ||
| Fig. 7 (a) X-Band EPR spectra of: pristine BMPO in DMF (red), BMPO + 1 without adding CO2 in DMF (purple), and exposure of BMPO + 1 to CO2 in DMF (black). (b) Simulation of the EPR spectra of BMPO + 1 + CO2 in DMF by linear combination of the contribution from: [BMPO–OCO2]˙− (green), [BMPO–OH]˙ (red), and [BMPO–O]˙ (yellow). (c) Formation of [BMPO–O]˙ from [BMPO–OH]˙ and an oxidant “[O]”. | ||
Activation of solid Li2O2 with CO2 in aprotic organic media
To examine the effect of CO2 on the oxidative power of Li2O2 under conditions relevant to the charging of lithium–air cells, commercially available solid Li2O2 was exposed to CO2 (1 atm, 25 °C, 48 h) in 1,2-dimethoxyethane (DME). In contrast to the results from the control experiments carried out similarly but in the absence of CO2, substantial amounts of methyl methoxyacetate were identified among the products of DME oxidation (Fig. 8). Approximately 51% of the Li2O2 was consumed, and quantitative conversion of the consumed Li2O2 to Li2CO3 (based upon lithium) was observed by 13C NMR spectroscopy and total inorganic carbonate (TIC) analysis.15 The consumed peroxide must generate an oxidizing equivalent; 74% was identified as evolved O2 and 15% as methyl methoxyacetate (Fig. 8), with the remainder unidentified. We also introduced solid Li2O2 into neat DMSO under a CO2 atmosphere (1 atm, 25 °C, 48 h), given the reported use of DMSO in lithium–O2 cells.37 More than 90% of the Li2O2 consumed participated in the conversion of DMSO to DMSO2 (Fig. 8). Viewed in the context of cycling lithium–O2 cells, the rate of CO2-induced solvent decomposition in bona fide lithium–air cells is perhaps lower than that observed in the current study due to the difference in CO2 partial pressures. Nonetheless, considering the low cycling rate and long cycling time of a typical lithium–air battery,38 our findings highlight that extensive oxidative degradation of the electrolyte in a cell will occur during cell cycling even when a small amount of CO2 is introduced or otherwise generated in the system.39 During cell cycling, CO2 is generated at the surface of lithium peroxide-impregnated carbon electrodes,39 leading us to speculate that the proposed chemistry (Fig. 8) should be expected to occur on a polarized electrode/electrolyte interface as well. It should be noted that while commercial Li2O2 was used in the present study, it is conceivable that the varied morphologies of electrochemically generated Li2O2 may react with CO2 at different rates. Due to the preponderance of conditions which result in varied Li2O2 crystallinity, size and surface structure,40,41 commercial Li2O2 was chosen as an ideal benchmark reactant with CO2. | ||
| Fig. 8 CO2-mediated oxidation of organic solvents by Li2O2. Addition of excess CO2 to solid Li2O2 in the organic solvent 1,2-dimethoxyethane (DME) generates an oxidizing equivalent “O”, which converts to O2 (74%) and methyl methoxyacetate (15%) with the remainder unidentified. A similar reaction performed in dimethylsulfoxide (DMSO) generated dimethylsulfone (DMSO2) in a 90% yield. | ||
A recent publication reported that when present in a charging lithium–air cell (>3.5 V vs. Li+/Li), the secondary amine 2,2,6,6-tetramethyl-4-piperidone (4-oxo-TEMP) was converted to the oxyl amine radical 4-oxo-TEMPO, as confirmed by EPR spectroscopy. 4-oxo-TEMP has been used in the past as a trap for singlet oxygen, leading the authors to propose that singlet oxygen was responsible for the observed conversion.10 An alternative explanation for the production of 4-oxo-TEMPO involves the oxidants being generated by activating Li2O2 with CO2, considering that the onset potential of CO2 formation in a typical lithium–air cell is also 3.5 V.42 Accordingly, we found that exposing Li2O2 to CO2 (1 atm, 25 °C, 24 h) in the same solvent and electrolyte as described in the literature, but without the application of an electrode potential, also resulted in the formation of 4-oxo-TEMPO. Of the oxidizing equivalents generated during the transformation of Li2O2 to Li2CO3, ca. 15% were incorporated into the 4-oxo-TEMPO reaction product based on EPR spin quantification. These experiments suggest that the oxidation of 4-oxo-TEMP is most likely due to CO2/peroxide-derived oxidants as opposed to singlet oxygen formation or at the very least that 4-oxo-TEMP is not a selective probe for singlet oxygen in Li–O2 cells under conditions of CO2 availability.
Conclusions
While previous studies on lithium–O2 batteries have attributed the low cycling number and capacity fading to singlet oxygen10 and superoxide,4–7 it is now clear that CO2/peroxide-derived oxidants are responsible for carbonate formation by way of the active oxidants HOOCO2− and CO3˙−via−O2COOCO2−. Since prototypical lithium–air cells (ether electrolyte, carbon cathode) lose 5–7% of their capacity to parasitic CO2 formation per complete cycle39 and have a typical cycling number of ca. 50, the resulting CO2/peroxide dianion-derived oxidants were expected to cause organic electrolyte degradation. This oxidative degradation may occur both during discharge through reaction of the peroxide dianion with CO2 and during recharge through electrochemical oxidation of carbonate initially generating the carbonate radical (CO3˙−). It has been established that recharging a lithium–O2 battery regenerates CO2 from Li2CO3, however identification of the mechanism and product(s) of electrochemical Li2CO3 degradation have been unclear.2 Our studies provide evidence for a mechanistic pathway by which carbonate radical anions, when generated, engage in C–H abstraction from the solvent (C–H bond ≅ 107 kcal mol−1, or less)27 and lead to solvent degradation and reformation of CO2 (Fig. 6). The regenerated CO2 sets in motion a decomposition cycle, therefore if even a small percentage of the total Li2O2 is converted to CO2, extensive oxidative degradation of the electrolyte in a cell will occur over the course of many cycles. If CO2 cannot be excluded from these systems then it is critical that the electrolyte and other cell components are invulnerable to reactive CO2/peroxide-derived oxidants if the full potential of rechargeable lithium–O2 battery systems is to be realized.Acknowledgements
This publication is based on work funded by the Robert Bosch Company and the National Science Foundation under CHE-1305124. The EPR spectroscopy in this study was funded by the National Science Foundation under CHE-1305124. We thank Carl Brozek, Ioana Knopf, Wesley Transue and Chong Liu for assistance with the instrumentation. We thank Prof. Yogesh Surendranath (MIT) for helpful discussion.References
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2011, 11, 172 CrossRef.
- S. R. Gowda, A. Brunet, G. M. Wallraff and B. D. McCloskey, J. Phys. Chem. Lett., 2013, 4, 276–279 CrossRef CAS PubMed.
- H.-K. Lim, H.-D. Lim, K.-Y. Park, D.-H. Seo, H. Gwon, J. Hong, W. A. Goddard, H. Kim and K. Kang, J. Am. Chem. Soc., 2013, 135, 9733–9742 CrossRef CAS PubMed.
- S. A. Freunberger, Y. Chen, Z. Peng, J. M. Griffin, L. J. Hardwick, F. Bardé, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 8040–8047 CrossRef CAS PubMed.
- Y. Chen, S. A. Freunberger, Z. Peng, F. Bardé and P. G. Bruce, J. Am. Chem. Soc., 2012, 134, 7952–7957 CrossRef CAS PubMed.
- D. G. Kwabi, T. P. Batcho, C. V. Amanchukwu, N. Ortiz-Vitoriano, P. Hammond, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2014, 5, 2850–2856 CrossRef CAS PubMed.
- M. M. O. Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2013, 135, 494–500 CrossRef PubMed.
- S. Yang, P. He and H. Zhou, Energy Environ. Sci., 2016, 9, 1650–1654 CAS.
- X. Yao, Q. Dong, Q. Cheng and D. Wang, Angew. Chem., Int. Ed., 2016, 55, 11344–11353 CrossRef CAS PubMed.
- J. Wandt, P. Jakes, J. Granwehr, H. A. Gasteiger and R.-A. Eichel, Angew. Chem., Int. Ed., 2016, 55, 6892–6895 CrossRef CAS PubMed.
- C. W. Jones and J. H. Clark, Introduction to the preparation and properties of hydrogen peroxide, R. Soc. Chem., 1999, 1–36 Search PubMed.
- N. Lopez, D. J. Graham, R. McGuire, G. E. Alliger, Y. Shao-Horn, C. C. Cummins and D. G. Nocera, Science, 2012, 335, 450–453 CrossRef CAS PubMed.
- The CAS registry number of the anion receptor is: 1360563-21-8.
- M. Nava, N. Lopez, P. Muller, G. Wu, D. G. Nocera and C. C. Cummins, J. Am. Chem. Soc., 2015, 137, 14562–14565 CrossRef CAS PubMed.
- See ESI.†.
- D. A. Bennett, H. Yao and D. E. Richardson, Inorg. Chem., 2001, 40, 2996–3001 CrossRef CAS PubMed.
- D. E. Richardson, H. Yao, K. M. Frank and D. A. Bennett, J. Am. Chem. Soc., 2000, 122, 1729–1739 CrossRef CAS.
- D. P. Jones and W. P. Griffith, J. Chem. Soc., Dalton Trans., 1980, 2526–2532 RSC.
- J. Flangan, D. P. Jones, W. P. Griffith, A. C. Skapski and A. P. West, J. Chem. Soc., Chem. Commun., 1986, 20–21 RSC.
- M. G. Bonini, S. A. Gabel, K. Ranguelova, K. Stadler, E. F. DeRose, R. E. London and R. P. Mason, J. Biol. Chem., 2009, 284, 14618–14627 CrossRef CAS PubMed.
- J. L. Roberts, T. S. Calderwood and D. T. Sawyer, J. Am. Chem. Soc., 1984, 106, 4667–4670 CrossRef CAS.
- I. P. Gerothanassis, in Encyclopedia of Magnetic Resonance, 2011, pp. 1–15 Search PubMed.
- G. Cerioni and F. Mocci, 17O NMR Spectroscopy of Organic Compounds Containing the –O–O– group, John Wiley & Sons, Ltd, 2009 Search PubMed.
- J. Lu, X. Kong, V. Terskikh and G. Wu, J. Phys. Chem. A, 2015, 119, 8133–8138 CrossRef CAS PubMed.
- M. Leskes, N. E. Drewett, L. J. Hardwick, P. G. Bruce, G. R. Goward and C. P. Grey, Angew. Chem., Int. Ed., 2012, 51, 8560–8563 CrossRef CAS PubMed.
- W. A. Strong, Ind. Eng. Chem., 1964, 56, 33–38 CrossRef CAS.
- N. B. Surmeli, K. L. Nadia, A.-F. Miller and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 17174–17185 CrossRef CAS PubMed.
- D. A. Armstrong, W. L. Waltz and A. Rauk, Can. J. Chem., 2006, 84, 1614–1619 CrossRef CAS.
- R. E. Huie, C. L. Clifton and P. Neta, Radiat. Phys. Chem., 1991, 38, 477–481 CrossRef CAS.
- S. V. Lymar and J. K. Hurst, J. Am. Chem. Soc., 1995, 117, 8867–8868 CrossRef CAS.
- M. G. Bonini, R. Radi, F.-S. Gerardo, A. M. D. C. Ferreira and O. Augusto, J. Biol. Chem., 1999, 274, 10802–10806 CrossRef CAS PubMed.
- B. S. Berlett, P. B. Chock, M. B. Yim and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 389–393 CrossRef CAS.
- M. B. Yim, B. S. Berlett, P. B. Chock and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 394–398 CrossRef CAS.
- H. Zhao, J. Joseph, H. Zhang, H. Karoui and B. Kalyanaraman, Free Radical Biol. Med., 2001, 31, 599–606 CrossRef CAS PubMed.
- D. B. Medinas, G. Cerchiaro, D. F. Trindade and O. Augusto, IUBMB Life, 2007, 59, 255–262 CrossRef CAS PubMed.
- F. A. Villamena, E. J. Locigno, A. Rockenbauer, C. M. Hadad and J. L. Zweier, J. Phys. Chem. A, 2007, 111, 384–391 CrossRef CAS PubMed.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- B. D. Mccloskey, A. Speidel, R. Scheffler, D. C. Miller, V. Viswanathan, J. S. Hummelshoj, J. K. Norskov and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS PubMed.
- S. Lau and L. A. Archer, Nano Lett., 2015, 15, 5995–6002 CrossRef CAS PubMed.
- R. R. Mitchell, B. M. Gallant, Y. Shao-Horn and C. V. Thompson, J. Phys. Chem. Lett., 2013, 4, 1060–1064 CrossRef CAS PubMed.
- Y.-C. Lu, E. J. Crumlin, T. J. Carney, L. Baggetto, G. M. Veith, N. J. Dudney, Z. Liu and Y. Shao-Horn, J. Phys. Chem. C, 2013, 117, 25948–25954 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental, crystallographic and spectroscopic data. CCDC 1512972. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01230f |
| ‡ These authors contributed equally. |
| § Current address: Centro de Investigaciones Químicas, IICBA, Universidad Autónoma del Estado de Morelos, Avenida Universidad 1001, Colonia Chamilpa, C.P. 62209, Cuernavaca, Morelos, Mexico. |
| This journal is © The Royal Society of Chemistry 2017 |