
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A complex stereochemical relay approach to the antimalarial alkaloid ocimicide A1. Evidence for a structural revision†
Herman
Nikolayevskiy
 a,
Maung Kyaw
Moe Tun
a,
Paul R.
Rablen
b,
Choukri
Ben Mamoun
c and
Seth B.
Herzon
*ad
a,
Maung Kyaw
Moe Tun
a,
Paul R.
Rablen
b,
Choukri
Ben Mamoun
c and
Seth B.
Herzon
*ad
aDepartment of Chemistry, Yale University, New Haven, CT 06520, USA. E-mail: seth.herzon@yale.edu
bDepartment of Chemistry and Biochemistry, Swarthmore College, Swarthmore, PA 19081, USA
cDepartment of Internal Medicine, Yale School of Medicine, New Haven, CT 06520, USA
dDepartment of Pharmacology, Yale School of Medicine, New Haven, CT 06520, USA
First published on 4th May 2017
Abstract
Ocimicide A1 (1) and the semisynthetic derivative ocimicide A2 (2) are highly potent antimalarial agents efficacious against chloroquine-sensitive and -resistant Plasmodium falciparum strains with IC50 values in the nanomolar and picomolar range, respectively. Members of this family have demonstrated radical cure in rhesus monkeys, without detectable toxicity, but their structure–function relationships and mechanism of action are unknown. Herein we describe a twelve-step synthesis of an advanced N-acylated pentacyclic precursor to the proposed structure of 1 (11% overall yield). Instability and poor P. falciparum growth inhibition of the corresponding free donor–acceptor cyclopropylamine, and large discrepancies between reported and both experimental and DFT-calculated 13C chemical shifts and coupling constants, suggest that substantial revision of the proposed structures may be necessary.
The structures 1 and 3, designated ocimicide A1 and B1, respectively (Fig. 1), were disclosed in the patent literature1 as hexacyclic quinoline alkaloids found in the root bark of Ocimum sanctum. The semisynthetic derivatives ocimicide A2 (2) and ocimicide B2 (4) were prepared in one step from 1 and 3, respectively. The structures of 1–4 were determined by NMR, UV, IR, and HRMS analyses. Tabulated NMR spectroscopic data for 2 and 4 were provided, but no spectroscopic data for the natural isolates 1 and 3 were disclosed.1 All four compounds contain a tetrasubstituted aminocyclopropane and oxidized pyrrolidine rings, but the orientation of the quinoline rings in 1/2 and 3/4 are, surprisingly, different. The alkaloids 1–4 demonstrated potent activity against chloroquine (5)-sensitive and -resistant P. falciparum strains (IC50s = 26–35 nM, 0.7–1.1 nM for natural and semisynthetic derivatives, respectively).1 The compounds were highly selective in cell culture, and efficacious in both prophylactic and treatment assays in the P. berghei malaria murine model. In a separate patent, structurally-related isolates were reported to effect radical cure in rhesus monkeys, without detectable toxicity.2
 | ||
| Fig. 1 Structures of the alkaloids ocimicides A1 (1) and B1 (3), the semisynthetic derivatives ocimicides A2 (2) and B2 (4), and chloroquine (5). | ||
With resistance to the front-line antimalarial artemisinin increasing,3 there is a pressing need for the development of novel agents with unique modes of action.4 We initiated synthetic studies toward ocimicide A1 (1), with the goal of elucidating the structure–function relationships and mechanism of action of this new class of antimalarials.
Since E-ring substitution influences activity,1 we targeted late-stage construction of this ring from the pentacyclic intermediate 6 (Scheme 1). A tandem epoxide-opening–ring contraction of 8, followed by activation and invertive displacement of the alcohol 7, was used to install the aminocyclopropane5 and lactam substituents of 6. The epoxide 8 was simplified to the vinyl triflate 9 and the stannane 10. The vinyl triflate 9 was prepared in racemic form in four steps and 57% overall yield from 4-methoxypyridine by a sequence developed by Comins6 for closely-related substrates (see ESI†). The stannane 10 was prepared in one step and 70% yield by site-selective metalation7 of 2-cyano-6-methoxyquinoline8 with lithium tetramethylpiperidide, followed by the addition of trimethyltin chloride (see ESI†).
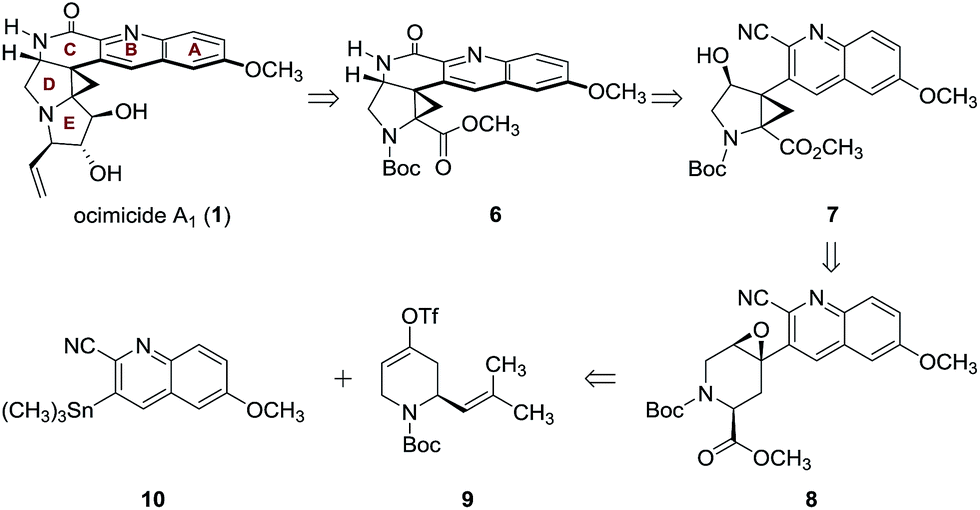 | ||
| Scheme 1 Retrosynthetic analysis of ocimicide A1 (1). | ||
Stille coupling of the vinyl triflate 9 and the stannane 10 [tetrakis(triphenylphosphine)palladium, copper iodide, cesium fluoride]9 provided the coupling product 11 in >99% yield (Scheme 2A). Site-selective oxidative cleavage of the exocyclic alkene within 11 [osmium tetroxide, then bis(acetoxy)iodobenzene],10 followed by in situ oxidation under Pinnick–Lindgren conditions11 provided the acid 12. The carboxylic acid function was employed in a carefully choreographed multistep relay to control the relative stereochemistry of the target. Treatment of 12 with N-bromosuccinimide and 4-dimethylaminopyridine provided the bromolactone 13 as a single regioisomer. The bromolactone 13 could be isolated, but in practice was treated directly with potassium carbonate in methanol to induce a ring-opening–ring-closing sequence, to form the epoxy ester 8 (45% from 11). The remaining mass balance was attributed to formation of a cyclic imidate resulting from addition of the alkoxide intermediate to the nitrile.
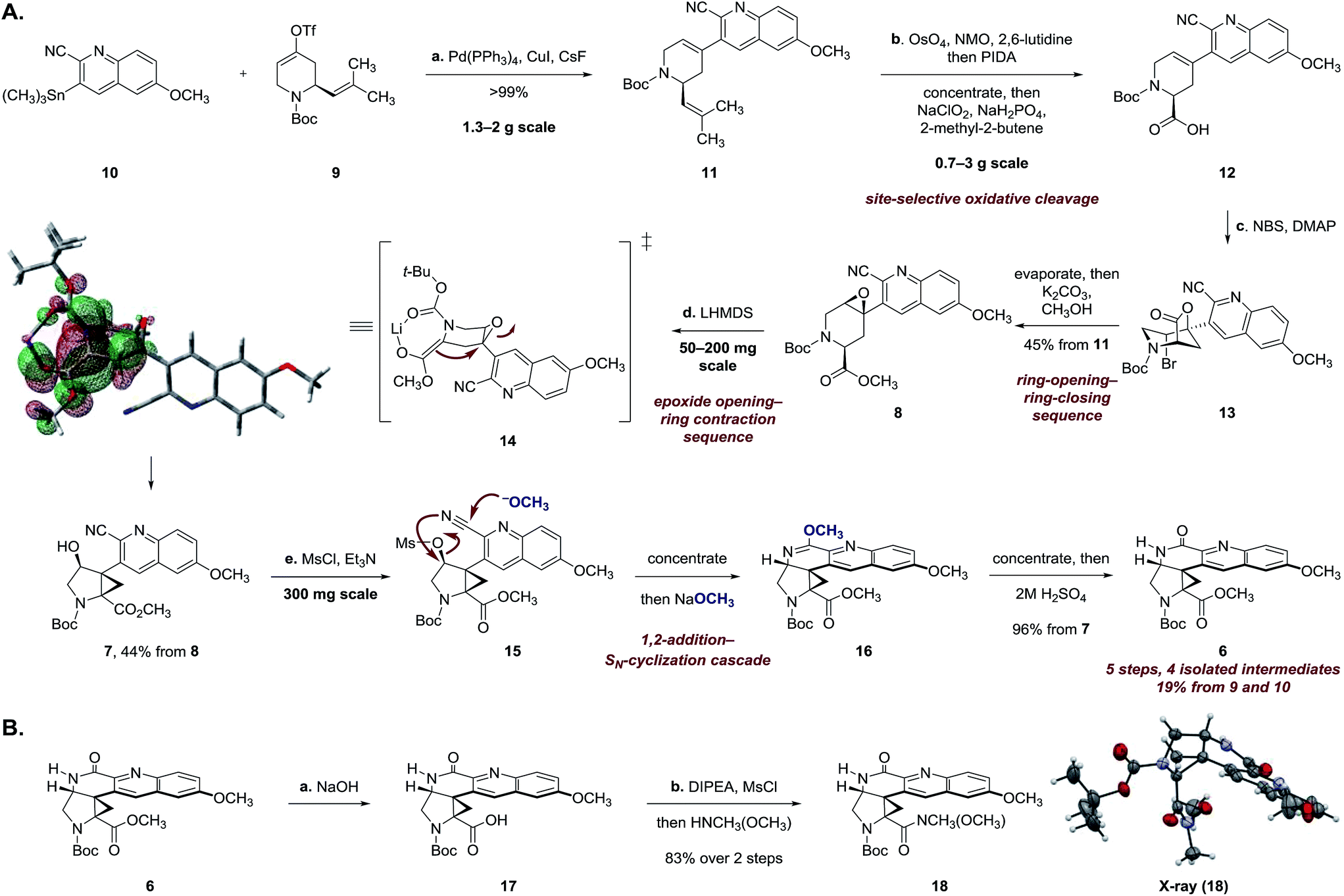 | ||
Scheme 2 (A) Synthesis of the lactam 6. Reaction conditions: (a) Pd(PPh3)4, CsF, CuI, DMF, 24 °C, >99%; (b) 2,6-lutidine, N-methylmorpholine N-oxide, OsO4, CH3COCH3–H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 24 °C, then bis(acetoxyiodo)benzene, 24 °C, evaporate, then NaClO2, NaH2PO4, 2-methyl-2-butene, THF–H2O–t-BuOH (4:3:1), 24 °C; (c) 4-dimethylaminopyridine, N-bromosuccinimide, CH2Cl2, 24 °C, evaporate, then K2CO3, CH3OH, 24 °C, 45% from 11; (d) lithium bis(trimethylsilyl)amide, PhCH3, 103 °C, 44%; (e) methanesulfonyl chloride, triethylamine, CH2Cl2, 0 → 24 °C, then NaOCH3, CH3OH, 24 °C; evaporate, then NaOCH3, CH3OH, 65 °C; evaporate, then 2 M H2SO4, THF, 24 °C, 96% from 7. The HOMO of the enolate 14 was calculated using M062X/6-31+G(2df,p) with simulated tert-butanol as solvent and a lithium counterion. (B) Synthesis of the Weinreb amide 18. Reaction conditions: (a) NaOH, CH3OH, 80 °C; (b) methanesulfonyl chloride, N,N-di-iso-propylethylamine, THF, 0 °C, then N,O-dimethylhydroxylamine, 83% over 2 steps. :1), 24 °C, then bis(acetoxyiodo)benzene, 24 °C, evaporate, then NaClO2, NaH2PO4, 2-methyl-2-butene, THF–H2O–t-BuOH (4:3:1), 24 °C; (c) 4-dimethylaminopyridine, N-bromosuccinimide, CH2Cl2, 24 °C, evaporate, then K2CO3, CH3OH, 24 °C, 45% from 11; (d) lithium bis(trimethylsilyl)amide, PhCH3, 103 °C, 44%; (e) methanesulfonyl chloride, triethylamine, CH2Cl2, 0 → 24 °C, then NaOCH3, CH3OH, 24 °C; evaporate, then NaOCH3, CH3OH, 65 °C; evaporate, then 2 M H2SO4, THF, 24 °C, 96% from 7. The HOMO of the enolate 14 was calculated using M062X/6-31+G(2df,p) with simulated tert-butanol as solvent and a lithium counterion. (B) Synthesis of the Weinreb amide 18. Reaction conditions: (a) NaOH, CH3OH, 80 °C; (b) methanesulfonyl chloride, N,N-di-iso-propylethylamine, THF, 0 °C, then N,O-dimethylhydroxylamine, 83% over 2 steps. | ||
Extensive experimentation was required to develop conditions to effect the epoxide-opening–ring contraction sequence (Scheme 2A).12 Ultimately, we found that treatment of 8 with lithium hexamethyldisilazide (1.10 equiv.) in toluene at 103 °C provided the cyclopropyl alcohol 7 in 44% yield (+10% unreacted 8). The temperature profile of this step was critical; although deprotonation occurred at 24 °C (as evidenced by deuterium incorporation experiments), heating to 103 °C for short amounts of time (2 h) was essential to obtain the product 7 in workable yields. Higher temperatures and longer reaction times led to degradation.
Density functional theory calculations [M062X/6-31+G(2df,p)] suggest that the enolate is required to adopt a boat-like conformation to effect invertive opening of the epoxide (see 14). This conformation orients the quinoline ring in a pseudoaxial position, leading to destabilizing non-bonded interactions in the transition state. The calculated activation energy of 27.9 kcal mol−1 for this step is in agreement with the experimentally-determined reaction temperature (103 °C).
The synthesis of 6 was completed by a high-yielding cascade sequence. Exposure of the cyclopropyl alcohol 7 to methanesulfonyl chloride and triethylamine provided the mesylate 15. Following concentration of the reaction mixture, the unpurified mesylate 15 was dissolved in anhydrous methanol and treated with sodium methoxide to effect 1,2-addition of methoxide to the nitrile and invertive displacement of the mesylate (15 → 16). Concentration of the reaction mixture, followed by dilution with sulfuric acid, resulted in smooth hydrolysis of the methyl imidate 16 to provide the key pentacycle 6 (96% from 7). The facile addition of methoxide to 15 is likely a reflection of electrophilic activation of the nitrile substituent by the quinoline ring. By this route, the racemic lactam 6 was obtained in only five steps, four isolated intermediates, and 19% yield from the Stille coupling partners 9 and 10 (twelve steps and 11% yield overall from commercial reagents).
The structural assignment of 6 was confirmed by X-ray analysis of the crystalline Weinreb amide 18,13 which was prepared by saponification, activation of the resulting acid 17 with methanesulfonyl chloride,14 and addition of N-methoxy-N-methylamine (Scheme 2B; 83% overall). While 6, 18, and related N-acylated intermediates were amenable to purification and handling, the corresponding free amines were unstable and underwent rapid decomposition. For example, attempted neutralization of the trifluoroacetate salt 19, formed by exposure of 6 to trifluoroacetic acid, led to extensive decomposition (Scheme 3). Although the complexity of the decomposition mixtures precluded characterization, we believe that the conjugation of the secondary lactam through the electron-deficient quinoline ring effectively renders the cyclopropane within 19 a donor–acceptor system.15 Decomposition may occur by amine-initiated ring-opening.
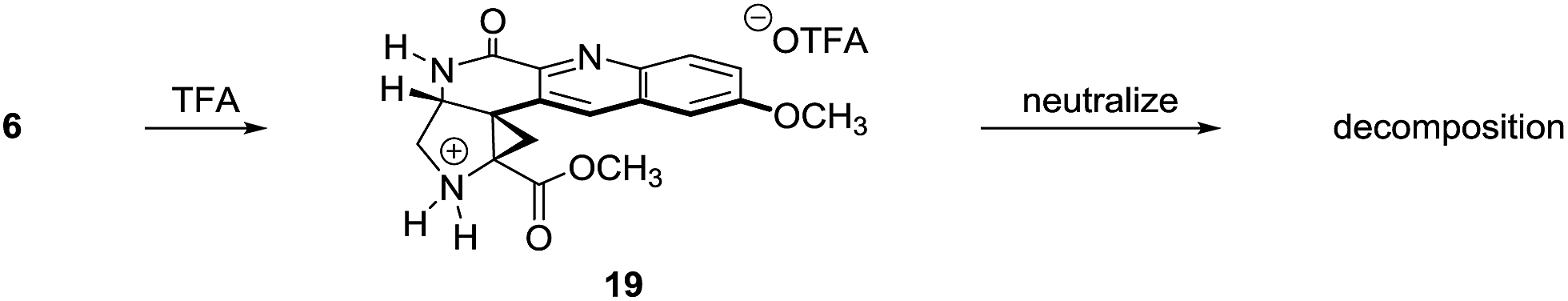 | ||
| Scheme 3 Synthesis of the trifluoroacetate salt 19. | ||
The instability of our synthetic intermediates prompted us to reexamine the original structural assignment using density functional theory.16 As spectroscopic data for 1 were not disclosed,1 we focused on the dimethyl ether derivative ocimicide A2 (2), for which tabulated spectroscopic shifts were presented. Following the protocol of Hoye and co-workers,17 32 structures (corresponding to all possible diastereomers at nitrogen 15 and carbons 12, 13, 14 and 17 of 2,18Fig. 2) were generated. These structures were imported into BOSS19 and each was separately subjected to a conformational search. Conformers within 5.02 kcal mol−1 of the lowest energy isomer (8–30 conformers for each diastereomer) were advanced to density functional theory geometry optimization [gas phase, B3LYP/6-31+G(d,p)]. Geometry-optimized conformers were confirmed as real local-minima by the absence of imaginary frequencies. The chemical shifts of the optimized conformers were then calculated using a modified WC04 functional20 and 6-31G(d) basis set in methanol (an initial screen of several functional and basis set combinations indicated that this level of theory provided an acceptable compromise of computational time and accuracy). Finally, the 13C chemical shifts were Boltzmann-averaged to generate the heat maps shown in Tables S1 and S2.† The geometry optimization and NMR calculation methods selected were benchmarked against 19-(E)-hunteracine (20),21 the structure of which has been unequivocally established by X-ray analysis.22 The mean absolute error (MAE) for 20 was 2.8 ppm [absolute error (AE) = 0.2–5.7 ppm], which was within the error typically obtained from similar levels of theory under optimized conditions.23
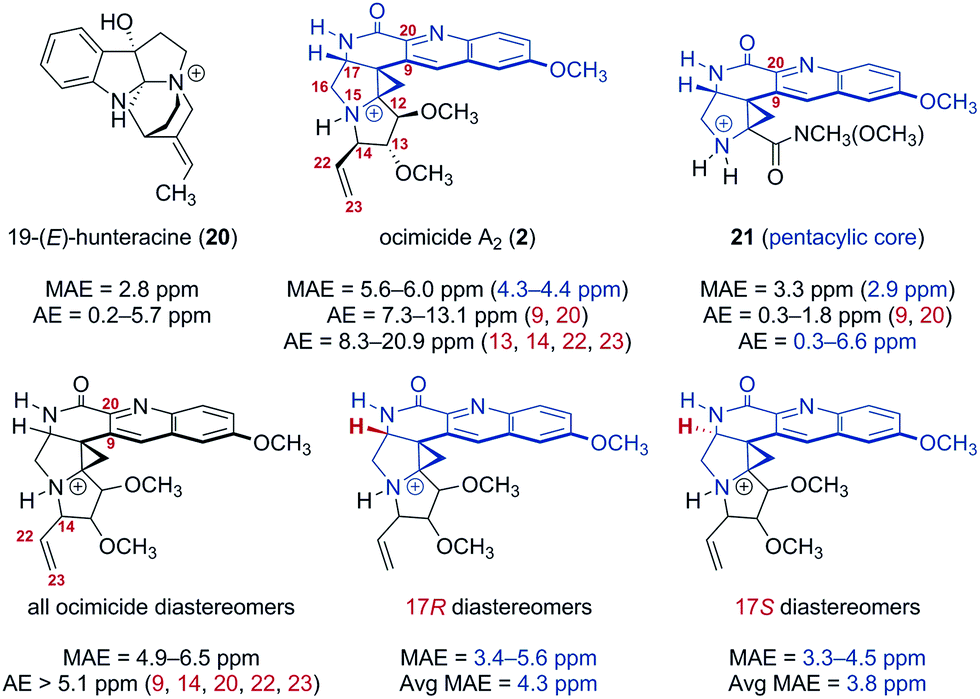 | ||
| Fig. 2 Mean absolute error (MAE) and absolute error (AE) of calculated 13C chemical shifts for the reference compound 19-(E)-hunteracine (20), ocimicide A2 (2; numbered as in patent1), the trifluoroacetate salt 21, and all ocimicide diastereomers (varied configuration at carbons 12, 13, 14, 17, and nitrogen 15). Geometries were optimized using B3LYP/6-31+G(d,p). 13C NMR chemical shifts were calculated using modified WC04/6-31G(d). Data highlighted in blue corresponds to the pentacyclic core. | ||
Large deviations, particularly in proximity to the lactam (carbons 9 and 20; AE = 7.3–13.1 ppm) and pyrrolidine rings (carbons 13, 14, 22, and 23; AE = 8.3–20.9 ppm), were observed between the calculated and reported113C chemical shifts for ocimicide A2 (2) (MAE = 5.6–6.0 ppm). While 17 of the 30 diastereomers investigated achieved better agreement (MAE = 4.9–5.5 ppm), no diastereomer could, within acceptable error, replicate reported values at carbons 14, 22, or 23 (AE > 6.6 ppm).
For comparison, the trifluoroacetate salt 21 was synthesized (via treatment of 18 with trifluoroacetic acid) and computationally subjected to analogous geometry optimization and NMR calculation methods.24 A focused analysis of pentacyclic core MAE values for 2, 21, and alternate ocimicide diastereomers revealed that while 21 was comparable in error to the benchmark 20 (2.9 ppm vs. 2.8 ppm), ocimicide diastereomers (including 2) had, on average, higher MAEs (4.3 ppm for 17R; 3.8 ppm for 17S). Moreover, calculated AE values for carbons 9 and 20 were significantly lower for 21 than any ocimicide diastereomer (0.3–1.8 ppm vs. 5.1–13.8 ppm). These data suggest our computational methods achieved an acceptable level of accuracy in this system.
The rigidity of the azabicyclo[3.1.0] ring system present within 6 and 2 suggests that a reasonable comparison of coupling constants may be made between the methine proton of carbon 17 and the methylene protons of carbon 16. For this three-proton spin system, the J values observed in 6 (5.2 and 0 Hz) poorly match those reported for 2 (9.2 and 4.4 Hz). For comparison, coupling constants were calculated for all diastereomers with the reported 17R and the alternate 17S configuration. Interestingly, while J values for the reported diastereomers (3.3–4.1 Hz and 0.1–0.3 Hz) closely resemble the experimental J values of 6, the reported data for 2 is better approximated by the alternate diastereomer (7.6–8.1 Hz and 5.7–6.7 Hz). While not definitive, the computational studies described herein suggest that a structural revision of the ocimicides is required.
Finally, to probe the antimalarial activity of our synthetic intermediates, we examined the growth inhibitory potential of several compounds against the P. falciparum 3D7 isolate (Table 1). While the bromolactone 13, the epoxy ester 8, and the cyclopropyl alcohol 7 inhibited parasitic growth by 20–28% at 100 nM, more advanced synthetic intermediates were significantly less active. The pentacyclic lactam 6 completely failed to inhibit parasitic growth, while the amine 19 (Scheme 3) demonstrated only 15% inhibition at 500 nM. These data and in particular the low activity of 6 and 19 provide some circumstantial support for a structural revision of the metabolites.
| Compound | % inhibition, 100 nM | % inhibition, 500 nM |
|---|---|---|
| 13 | 26 | 40 |
| 8 | 20 | 30 |
| 7 | 28 | 41 |
| 6 | 0 | 0 |
| 19 | 8 | 15 |
Conclusions
In summary, we have described an expedient synthesis of the lactam 6, an advanced pentacyclic intermediate that contains the tetrasubstituted aminocyclopropane of the ocimicide alkaloids. Our synthesis of 6 proceeds in five steps, four isolated intermediates, and 19% yield from the vinyl triflate 9 and the stannane 10 and twelve steps and 11% yield overall from commercial reagents. Notable features of the synthesis include a complex multistep stereochemical relay to establish the relative configuration of the target and the formation of a tetrasubstituted cyclopropylamine via an epoxide-opening–ring contraction sequence. Our NMR calculations suggest that the published structures of the ocimicides may require revision. Given the reported success of these alkaloids to achieve radical cure of malaria in rhesus monkeys,2 efforts to fully elucidate their structures are ongoing.25 These studies highlight the ability of modern computational methods to guide experimental synthesis.Acknowledgements
Financial support from the National Institutes of Health (R01GM110506) and Yale University is gratefully acknowledged. We thank Dr Julian Tirado-Rives and Mr Steven Swick for assistance with the DFT calculations, Drs Chris Incarvito and Brandon Mercado for X-ray analysis, and Dr Apra Garg and Mr Will Zhao for assessing the antimalarial activity of our compounds.Notes and references
- S. Zhu, US 20100087469 A1, 2010.
- S. Zhu, US 20130023552 A1, 2013.
- (a) H. Noedl, Y. Se, K. Schaecher, B. L. Smith, D. Socheat, M. M. Fukuda and C. Artemisinin, Resistance in Cambodia 1 Study, N. Engl. J. Med., 2008, 359, 2619–2620 CrossRef CAS PubMed; (b) A. M. Dondorp, F. Nosten, P. Yi, D. Das, A. P. Phyo, J. Tarning, K. M. Lwin, F. Ariey, W. Hanpithakpong, S. J. Lee, P. Ringwald, K. Silamut, M. Imwong, K. Chotivanich, P. Lim, T. Herdman, S. S. An, S. Yeung, P. Singhasivanon, N. P. Day, N. Lindegardh, D. Socheat and N. J. White, N. Engl. J. Med., 2009, 361, 455–467 CrossRef CAS PubMed.
- The Gates Foundation invested $64 million in the development of a biosynthetic manufacturing process for artemisinin ( M. Peplow, Nature, 2016, 530, 389–390 CrossRef CAS PubMed ) but an inexpensive synthesis of the target potentially amenable to commercial manufacturing has been disclosed: C. Zhu and S. P. Cook, J. Am. Chem. Soc., 2012, 134, 13577–13579 CrossRef PubMed.
- For a review of aminocyclopropane synthesis, see: H. Wang, X. K. Zhou and Y. J. Mao, Heterocycles, 2014, 89, 1767–1800 CrossRef CAS.
- (a) D. L. Comins, H. Hong and J. M. Salvador, J. Org. Chem., 1991, 56, 7197–7199 CrossRef CAS; (b) D. L. Comins and A. Dehghani, Tetrahedron Lett., 1992, 33, 6299–6302 CrossRef CAS.
- J. A. Mccauley, N. J. Liverton, M. T. Rudd, K. F. Gilbert, M. Ferrara, V. Summa and B. Crescenzi, WO/2012/040040, 2012.
- 2-Cyano-6-methoxyquinoline is commercially available or can be prepared in one step from 6-methoxyquinoline. See: D. L. Boger, C. E. Brotherton, J. S. Panek and D. Yohannes, J. Org. Chem., 1984, 49, 4056–4058 CrossRef CAS.
- S. P. H. Mee, V. Lee and J. E. Baldwin, Angew. Chem., Int. Ed. Engl., 2004, 43, 1132–1136 CrossRef CAS PubMed.
- K. C. Nicolaou, V. A. Adsool and C. R. H. Hale, Org. Lett., 2010, 12, 1552–1555 CrossRef CAS PubMed.
- (a) B. O. Lindgren and T. Nilsson, Acta Chem. Scand., 1973, 27, 888–890 CrossRef CAS; (b) B. S. Bal, W. E. Childers and H. W. Pinnick, Tetrahedron, 1981, 37, 2091–2096 CrossRef CAS.
- For related ring-contractions of α-lithioamines, see: P. Beak, S. Wu, E. K. Yum and Y. M. Jun, J. Org. Chem., 1994, 59, 276–277 CrossRef CAS.
- S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CrossRef CAS.
- J. C. S. Woo, E. Fenster and G. R. Dake, J. Org. Chem., 2004, 69, 8984–8986 CrossRef CAS PubMed.
- H.-U. Reissig and E. Hirsch, Angew. Chem., Int. Ed. Engl., 1980, 19, 813–814 CrossRef.
- For a review of computational approaches to NMR chemical shift calculations, see: M. W. Lodewyk and D. J. Tantillo, J. Nat. Prod., 2011, 74, 1339–1343 CrossRef CAS PubMed.
- P. H. Willoughby, M. J. Jansma and T. R. Hoye, Nat. Protoc., 2014, 9, 643–660 CrossRef CAS PubMed.
- The numbering used herein corresponds to that presented in ref. 1.
- W. L. Jorgensen and J. Tirado–Rives, J. Comput. Chem., 2005, 26, 1689–1700 CrossRef CAS PubMed.
- The modified WC04 functional was invoked using WC04 internal option (IOp) statements and the B3LYP functional. The unmodified WC04 functional was originally described in: K. W. Wiitala, T. R. Hoye and C. J. Cramer, J. Chem. Theory Comput., 2006, 2, 1085–1092 CrossRef CAS PubMed . IOp statements were obtained from: G. K. Pierens, Comput. Chem., 2014, 35, 1388–1394 CrossRef PubMed.
- Z. E. D. Torres, E. R. Silveira, L. F. R. E. Silva, E. S. Lima, M. C. de Vasconcellos, D. E. D. Uchoa, R. Braz Filho and A. M. Pohlit, Molecules, 2013, 18, 6281–6297 CrossRef PubMed.
- R. H. Burnell, A. Chapelle, M. F. Khalil and P. H. Bird, J. Chem. Soc. D, 1970, 772–773 RSC.
- (a) K. W. Wiitala, T. R. Hoye and C. J. Cramer, J. Chem. Theory Comput., 2006, 2, 1085–1092 CrossRef CAS PubMed; (b) M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef CAS PubMed.
- A conformational search of 21 was not conducted prior to geometry optimization. Instead, the Weinreb amide conformer found in the X-ray crystal structure of 18 was utilized. This may account for the large deviations observed near the Weinreb amide carbonyl (AE = 6.6–8.9 ppm).
- Natural samples of the ocimicides are no longer available ( S. Zhu, personal communication, 2014). We cultivated three strains of Ocimum sanctum (Krishna, Rama, and Kapoor; purchased from Horizon Herbs) at the Yale Farm in New Haven, CT. Using the isolation protocol reported in ref. 1, we were unable to detect 1 or 3 in the dried root bark extract (UPLC/MS analysis).
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary schemes, figures, and tables, general experimental remarks, synthetic procedures, catalogues of experimental and calculated nuclear magnetic resonance spectra. CCDC 1536046. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01127j |
| This journal is © The Royal Society of Chemistry 2017 |