
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCross-dehydrogenative coupling and oxidative-amination reactions of ethers and alcohols with aromatics and heteroaromatics
Mahesh K.
Lakshman
 *ab and
Prasanna K.
Vuram
a
*ab and
Prasanna K.
Vuram
a
aDepartment of Chemistry, The City College of New York, 160 Convent Avenue, New York 10031, USA. E-mail: mlakshman@ccny.cuny.edu
bThe Ph.D. Program in Chemistry, The Graduate Center of The City University of New York, New York 10016, USA
First published on 30th June 2017
Abstract
Cross-dehydrogenative coupling (CDC) is a process in which, typically, a C–C bond is formed at the expense of two C–H bonds, either catalyzed by metals or other organic compounds, or via uncatalyzed processes. In this perspective, we present various modes of C–H bond-activation at sp3 centers adjacent to ether oxygen atoms, followed by C–C bond formation with aromatic systems as well as with heteroaromatic systems. C–N bond-formation with NH-containing heteroaromatics, leading to hemiaminal ethers, is also an event that can occur analogously to C–C bond formation, but at the expense of C–H and N–H bonds. A large variety of hemiaminal ether-forming reactions have recently appeared in the literature and this perspective also includes this complementary chemistry. In addition, the participation of C–H bonds in alcohols in such processes is also described. Facile access to a wide range of compounds can be attained through these processes, rendering such reactions useful for synthetic applications via Csp3 bond activations.
A. Introduction
Reactions that lead to the formation of a C–C bond at the expense of two C–H bonds are now collectively categorized under the title cross-dehydrogenative coupling, abbreviated as CDC. Formally, an equivalent of H2 is lost in the transformation although in practice this typically does not occur, and an oxidant is generally employed for these reactions. Such CDC reactions have seen substantial growth and have now been used for sp3–sp3, sp3–sp2, sp3–sp, sp2–sp2, sp2–sp (Sonogashira type) and sp–sp (Glaser) bond formation. Importantly, for CDC reactions, there is no need for prefunctionalization of compounds before C–C or C–N bond formation can be performed. Numerous outstanding reviews have been published on such reactions and several have appeared since just 2010.1–16 In addition, several metal-centric reviews describing reactions effectuated by Fe,17,18 Pd,19,20 Cu,21 Rh,22 and Ru23 have also appeared in the literature. A general representation of a CDC reaction and the related oxidative amination is shown in Fig. 1. | ||
| Fig. 1 Pictorial representation of CDC and oxidative-amination reactions. | ||
Comparable to CDC reactions of aromatic compounds, an equivalent can be anticipated when heteroaryls are used. To complement prior reviews, herein, we focus specifically on the activation of Csp3–H bonds at α-positions to ether or alcohol oxygen atoms, and CDC-type reactions with aromatic and heteroaromatic systems. While with aromatic compounds the products result from α-arylation to the ether oxygen atom, reactions with heterocyclic systems can result in either C–C or C–N bond formation, depending on the heterocycle presented, conditions and/or catalysts used. Scheme 1 shows a schematic representation as well as metals/reagents used for the conversions that will be described herein. As far as possible, comparisons of the various approaches and mechanistic inquiries will be discussed. However, related CDC reactions at α-positions to nitrogen atoms, and CDC reactions of ethers with ketones, active methylene compounds, alkenes, alkynes and alkanes, as well as carbene insertion chemistry are not discussed herein, but they are all powerful reactions. Notably, the content presented does not overlap with, but is complementary to prior reviews on Csp3–H bond activation of ethers, and is a collection of reactions that the authors consider to be promising for synthetic applications and development.
 | ||
| Scheme 1 CDC reactions of aromatic and heteroaromatic compounds with ethers (Ar = aryl, Het = heteroaryl). Metals and reagents used for these reactions are shown in the orange color. | ||
B. Reactions of ethers with aromatic compounds
Reactions in this category that are described in the following sections involve the use of: catalytic salts of Fe and Cu in the absence or presence of DDQ, catalytic Pd(OAc)2/Cu(OTf)2/t-BuOOH, Ni catalysis, DDQ and Grignard reagents with or without PhI(OTFA)2 (PIFA), catalytic organocatalyst AZADOL and Grignard reagents, photochemical reactions with Ir and Pd–TiO2 catalysts. In each case an aryl ring is introduced α to the ether oxygen atom and, in one subgroup, a styrene unit.B.1. Using metal catalysts
 | ||
| Scheme 2 Reactions of isochroman with electron-rich aromatic systems. | ||
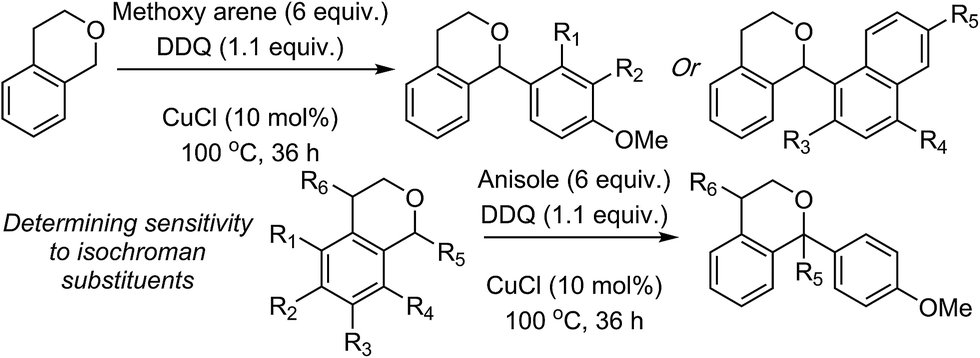 | ||
| Scheme 3 Reactions of isochroman with various methoxy arenes and those of various isochromans with anisole. | ||
The reaction was sensitive to substitution on the isochroman ring (Scheme 3). Isochromans with R1 and R6 as methyl groups reacted uneventfully. From a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture of 6- and 8-methylisochromans (R2 and R4 = Me), only the 8-methyl isomer yielded product. A methoxy substituent at the 6-position (R2 = OMe) prevented reactivity. On the other hand, 6-fluoroisochroman (R2 = F) reacted reasonably well. Placing a bromine atom at the 5-position (R1 = Br) diminished the product yield. Among substitutions at the 1- and 4-positions, good reactivity was observed with 4-methylisochroman (R6 = Me) whereas the 1-methyl isomer did not give product (R5 = Me).
:3 mixture of 6- and 8-methylisochromans (R2 and R4 = Me), only the 8-methyl isomer yielded product. A methoxy substituent at the 6-position (R2 = OMe) prevented reactivity. On the other hand, 6-fluoroisochroman (R2 = F) reacted reasonably well. Placing a bromine atom at the 5-position (R1 = Br) diminished the product yield. Among substitutions at the 1- and 4-positions, good reactivity was observed with 4-methylisochroman (R6 = Me) whereas the 1-methyl isomer did not give product (R5 = Me).
A proposed mechanism involves the cupration of anisole, followed by the formation of a radical, which reacts with an oxocarbenium ion formed through the oxidation of isochroman (Scheme 4). The formed CuI species is then reoxidized by the radical cation intermediate to CuII.
 | ||
| Scheme 4 A proposed mechanistic pathway for the arylation of isochroman by CuII. | ||
The reaction was then expanded to include other keto groups and esters at the R1 site (Scheme 5). All ketones reacted comparably but the aldehyde gave a lower yield. In general, yields from the ester derivatives were comparable amongst themselves and to that from the aldehyde, and the n-butyl ester gave the lowest yield.
 | ||
| Scheme 5 CDC reactions of resorcinol derivatives with THF. | ||
Application to 1- and 2-naphthols also proceeded in good yields, whereas reactions of a 4-chromenone and two xanthones were modest, and reaction times ranged from 6 to 24 h (Fig. 2). It appears that a mesomeric OMe donor group lowered the yield.
 | ||
| Fig. 2 CDC reactions of naphthols, a 4-chromenone and two xanthones with THF. | ||
Reactions of 1,4-dihydroxynaphthalenes were then evaluated with a view to accomplishing CDC as well as oxidation of the dihydroxynaphthalene to a 1,4-naphthoquinone (Scheme 6). Reaction with THF proceeded well but those of 1,3-dioxolane, 1,4-dioxane and dihydropyran were more modest. Notably, the product from 1,3-dioxolane had the naphthoquinone moiety appended at the C4 position, not flanked by the two oxygen atoms. Use of 5-hydroxy-1,4-naphthoquinone (R = OH) resulted in a 1:1 mixture of two regioisomers (R1 = OH, R2 = H and R1 = H, R2 = OH), also in a modest yield. Finally, CDC reactions with alcohols were evaluated and these are complementary to the reactions of ethers. The use of MeOH and EtOH proceeded uneventfully at 65 °C (60% yields of products, Fig. 3). However, under reflux, the reaction with EtOH gave 53% yield of a product that would otherwise arise from CDC with Et2O.
 | ||
| Scheme 6 CDC and oxidation leading to 1,4-naphthoquinones. | ||
 | ||
| Fig. 3 Reactions of alcohols with 1,4-dihydroxynaphthalene. | ||
Reactions with n-PrOH and n-BuOH also proceeded as anticipated (51% and 32% yields, respectively, Fig. 3). However, reaction with sec-BuOH gave a naphthoquinone ether derivative in 40% yield, and reaction with iPrOH did not proceed. Thus, it appears that 1° alcohols will react, some modestly, but increasing steric bulk at the β-position to the oxygen atom results in an ether, and alkyl substitution at the α-carbon prevents the reaction.
Mechanistically, in this reaction CuII is proposed to be responsible for the formation of t-BuO˙, which then causes the removal of a hydrogen atom from THF (not shown in Scheme 7), leading to an oxocarbenium ion. The phenol forms a PdII complex that reacts with the oxocarbenium ion from THF (Scheme 7).
 | ||
| Scheme 7 The Pd-mediated portion of the reaction of resorcinol with THF. | ||
Although an explanation for the need to have an electron-withdrawing group on the resorcinol is not mentioned, one must assume that increased acidity of the phenolic hydroxyl groups will favor the formation of the Pd-phenolates. In Scheme 7, both phenolic hydroxyl groups are influenced by the R group and either can form the Pd-phenolate. However, if carbonyl group involvement is necessary in stabilizing the Pd-phenolate, then the species shown in Scheme 7 would be reasonable.
Radical trap experiments indicated that these reactions could be radical processes. With TEMPO no product was formed, whereas with 1,1-diphenylethylene (DPE) product suppression was observed. The product shown in the box in Scheme 7 was formed in 43% yield and the THF–DPE adduct (also in the box) was formed in 35% yield.
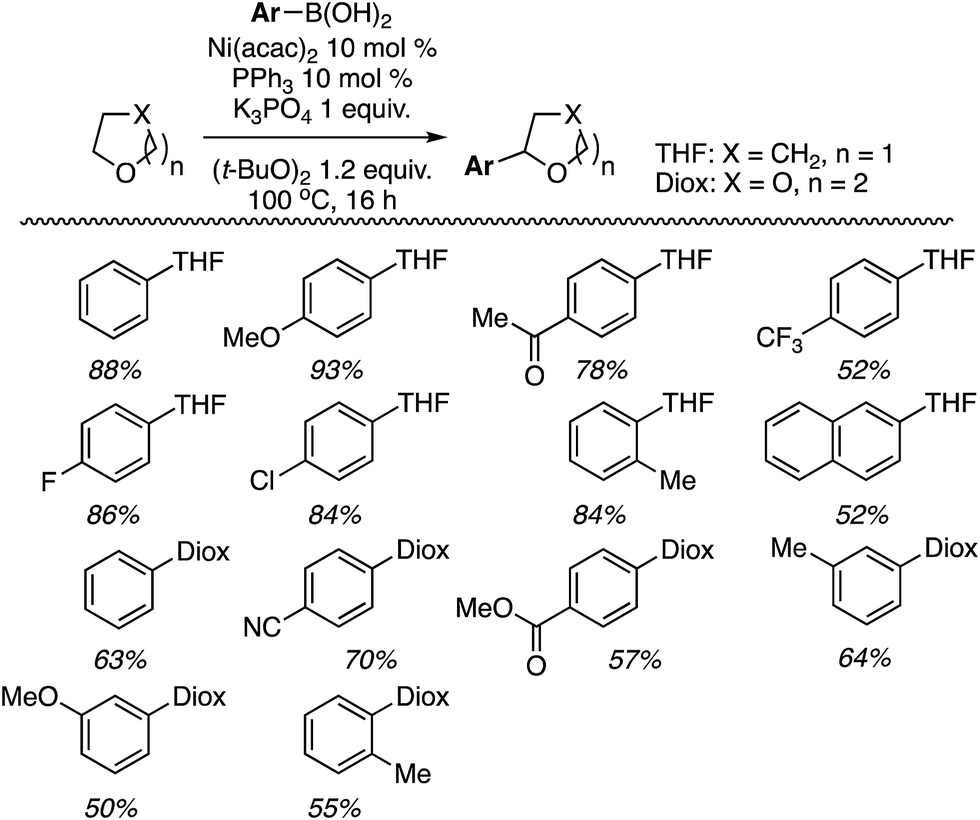 | ||
| Scheme 8 Reactions of aryl boronic acids with THF and THP, catalyzed by Ni(acac)2. | ||
Reactions of benzo[d]-1,3-dioxole, N,N-dimethylaniline, DMA and N-methylpyrrole with PhB(OH)2 were also investigated. Successful reactions were observed with all four and the products are shown in Fig. 4.
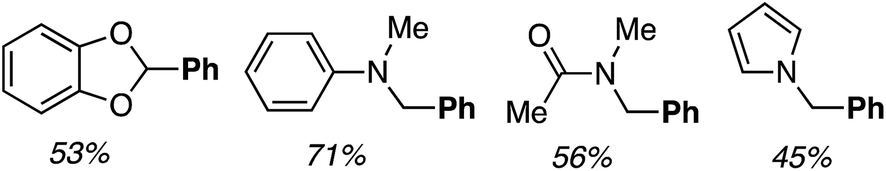 | ||
| Fig. 4 Products of the reactions of benzo[d]-1,3-dioxole, N,N-dimethylaniline, DMA and N-methylpyrrole with PhB(OH)2. | ||
Possible operative mechanisms proposed include hydrogen atom abstraction from THF by t-BuO˙, formed from the peroxide, leading to a THF radical. The THF radical can then form an oxocarbenium ion that reacts with the aryl boronic acid, leading to the product. Alternatively, with the Ni salt and peroxide, an aryl radical can be envisaged. Addition of the aryl radical to the oxocarbenium ion formed from THF will lead to a radical cation, which can then collapse to the product. A radical trapping experiment with 1.5 equiv. of TEMPO completely suppressed product formation. With 1,1-DPE as a radical trap, four products (shown in the boxes in Scheme 9) were obtained. Formation of 1,1,2-triphenylethene and 1,1,2-triphenylethane indicates that aryl radicals are likely involved, and the THF containing ethene and ethane products indicate that the THF radical is likely formed. Thus, the formation of aryl radicals in this reaction seems feasible and these can then react with an oxocarbenium ion produced from THF, leading to the product (Scheme 9).
 | ||
| Scheme 9 Four products obtained in radical trapping experiments with 1,1-DPE and a plausible mechanism. | ||
B.2. Metal-free reactions with organic oxidants
 | ||
| Scheme 10 Oxidation of ethers by DDQ followed by reactions with aryl Grignard reagents. | ||
Generally, good yields were obtained (77–97%) with some minor variations in conditions needed in individual cases. The yields seem independent of the nature of the alkyl ether unit and the Grignard reagent, but the thioether gave a lower (52%) yield. With 2,5-dihydroisobenzofuran a mixture of cis/trans diaryl products was obtained. The mechanism of the reaction is proposed to proceed via a single electron oxidation of the ether with DDQ, and subsequent formation of an oxocarbenium ion.
 | ||
| Fig. 5 Products obtained via the use of PIFA and DDQ. | ||
Notably, in this work,32 alkyl-, allyl- and vinylmagnesium iodides, p-toluenesulfonamides and sodium azide could be used as nucleophiles. The products shown in Fig. 6 were obtained in 73–90% yields. From a mechanistic standpoint, the reaction parallels that shown in Scheme 10, except that PIFA presumably reoxidizes the reduced DDQ-H.
 | ||
| Fig. 6 Alkyl-, allyl-, vinyl- and nitrogen-substituted isochromans. | ||
Reactions of isochroman with MeMgI and EtMgI, as well as with iPrMgBr and cyclopentyl-MgBr, proceeded in high yields, just as with DDQ/PIFA.32 This reaction allowed the introduction of other substituents, also in parallel to DDQ/PIFA reactions, and with comparable yields. Notably, products can also be formed with heterocycles, diethyl malonate and thiophenols (Fig. 7).
 | ||
| Fig. 7 Other products that can be produced with AZADOL/PIFA. | ||
In investigating the reaction mechanism, the use of hydroquinone monomethyl ether led to suppression of the reaction, indicating the presence of radical intermediates. Importantly, the authors present a reaction of benzyl alcohol within this set, in good yield. However, this is unlikely to be a CDC reaction, but rather oxidation of the alcohol to the aldehyde followed by addition of the Grignard reagent. These processes can be understood in the context of the plausible reaction mechanism shown in Scheme 11. Because an oxoammonium intermediate is likely formed from AZADOL in these reactions, such an intermediate can be expected to oxidize alcohols to carbonyl compounds.34
 | ||
| Scheme 11 A plausible reaction mechanism leading to an oxocarbenium ion for reaction with nucleophiles. | ||
B.3. Photochemical reactions
 | ||
| Scheme 12 IrIII(ppy)3-catalyzed benzylic arylation with p-dicyanobenzene. Ar = p-CN-Ph. | ||
The ensuing IrIV(ppy)3 species then undergoes reduction with a thiol catalyst, completing the metallic redox cycle. The thiyl radical then causes benzylic hydrogen atom abstraction from the ether. Radical–radical coupling and the expulsion of CN− (with overall loss of HCN) leads to diarylmethanes with one ring containing a cyano group. Both an added base (KH2PO4 and occasionally Na2CO3), plausibly for thiol deprotonation prior to its oxidation by IrIV, and an aldehyde were found to be necessary, using DMA as the solvent. The aldehyde is presumed to sequester the CN− anion produced in the electron-transfer step.
Several products were prepared in generally good (62–82%) yields. The reactions were tolerant of silyl and MEM ethers, as well as a free hydroxyl group. Heteroaryls and other substituents such as Cl and NMe2 were not deterrents. These data and the plausible mechanism are shown in Scheme 12.
Using the TBDMS ether of benzyl alcohol, several other aryl dinitriles and activated aryl nitriles were investigated (Fig. 8). The yields were in the 41–86% range and, notably, a sulfone was stable towards the radical–anionic intermediates formed. With unsymmetrical aryl dinitriles, as anticipated, regioisomeric products were observed. The ratio of regioisomeric products is also indicated in Fig. 8 with blue arrows.
 | ||
| Fig. 8 Other aryl dinitriles and activated nitriles used in the arylation of PhCH2OTBDMS, product yields as well as regioisomer ratios with unsymmetrical substrates are shown. | ||
In a single example, 2,5-dihydrofuran was subjected to the previously mentioned reaction conditions with p-dicyanobenzene. A single arylated dihydrofuran regioisomer was formed in an excellent 82% yield (Scheme 13).
 | ||
| Scheme 13 Arylation of 2,5-dihydrofuran with p-dicyanobenzene. | ||

Using 0.2 wt% Pd on TiO2, the reactions of benzene with Et2O, tetrahydropyan, THF and n-butyl methyl ether were evaluated. In every case selectivity was 99%; the products obtained and associated yields are shown in Fig. 9.
 | ||
| Fig. 9 Products obtained with 0.2 wt% Pd on TiO2 and yields. | ||
The authors investigated reactions of C6D6 with Et2O and C6H6 with Et2O-d10. In both reactions a kH/kD = 0.92–0.93 was observed. The authors, therefore, suggested that addition of the radical formed from Et2O to benzene, resulting in a cyclohexadienyl radical, then resulting in loss of a hydrogen atom and rearomatization of the ring.
B.4. Cu-Catalyzed reactions of 1,1-diarylethenes and styrenes
The preceding sections describe reactions of aromatic compounds with ethers. Reactions of aryl-substituted olefins, 1,1-diarylethenes and styrenes have also been studied.37 In combination with catalytic KI and an oxidant, CuI, Cu(OAc)2, Ni(acac)2, FeCl3, and CoBr2 were evaluated as metal catalysts for CDC reaction of 1,1-diphenylethene with THF. The oxidants tested were (t-BuO)2, t-BuOOH, m-CPBA and K2S2O8. From these, the combination of CuI, KI and (t-BuO)2 at 120 °C was optimal. Notably, the reaction proceeds in the absence of KI but in a lower yield. I2 in place of KI was ineffective, NaI gave a low yield and although n-Bu4N+I− was effective, it was less so in comparison to KI. The order of effectiveness is KI > n-Bu4N+I− > no iodide reagent ≫ NaI. Lowering the reaction temperature to 100 °C also caused a substantial lowering of the product yield. Reactions of various olefins with THF were evaluated and these results are shown in Scheme 14. | ||
| Scheme 14 Reactions of 1,1-diarylethenes and styrenes with THF. | ||
Next, several ethers were reacted with 1,1-DPE (one case with 1-methoxy-4-(1-phenylvinyl)benzene) and these products are shown in Fig. 10. Notably, with 1,3-dioxolane reaction at the C2 methylene, flanked by two oxygen atoms, was preferred. When presented with an ether containing 1° and 2° carbon atoms, reaction at the latter occurred with a significant preference.
 | ||
| Fig. 10 Reactions of other ethers with 1,1-diarylethenes. | ||
These reactions are radical processes and when a single equivalent of TEMPO was added to the reaction mixture, product formation was completely suppressed. The TEMPO–THF adduct was obtained in 53% yield. What is interesting about this work is that 1,1-DPE is, in fact, used as a probe for radical mechanisms.
C. Reactions of ethers with heteroaromatic compounds
As shown in Scheme 1, CDC reactions of ethers with heteroaromatics can result in either C–C bond-forming reactions or C–N bond formation, depending on the heteroaromatic system and reaction conditions utilized. With heteroaromatics that do not possess an NH unit, only C–C bond formation is anticipated. On the other hand, in the presence of an NH, either C–C or C–N bond formation can occur. Each category is discussed in the sections below.C.1. Formation of C–C bonds with heteroaromatic compounds that do not possess NH
Within this category of reactions, three sets of conditions have been utilized: (a) reactions of thiazoles and benzothiazoles catalyzed by Cu, (b) reactions of benzothiazoles, thiazoles and benzoxazoles, and one reaction of a benzimidazole catalyzed by Fe, (c) Co-catalyzed reactions of oxazoles and benzoxazoles and (d) reactions of benzothiazole and benzoxazole, and one reaction of benzimidazole without a metal catalyst. | ||
| Scheme 15 Cu(OTf)2/K2S2O8 promoted CDC reactions of heterocycles with ethers. | ||
Generally these reactions progressed in good yields. At least in the benzothiazole series, there may be an influence of the heteroaryl ring electronics on the product yields. For example, a methoxy group on the ring led to a better yield than the unsubstituted system. An electron-withdrawing fluorine atom caused a lowering of the yield and a strongly electron-withdrawing nitro group led to no product formation. Similar trends seem apparent in reactions of benzothiazoles with 1,3-dioxolane and 1,4-dioxane.
Mechanistically, this is plausibly a radical reaction because the reaction of 4,5-dimethylthiazole and THF was completely suppressed in the presence of TEMPO, and the TEMPO–THF adduct was detected. However, in the absence of K2S2O8 neither product nor the TEMPO–THF adduct was formed. This indicates that K2S2O8 is responsible for the formation of the radical from THF, possibly through thermal formation of SO4−˙.
There are two possible mechanistic manifolds shown in Scheme 16. Of these one, shown in solid arrows, was supported by DFT computational analysis. This mechanism may proceed via the addition of a THF radical to the imino carbon atom of the heteroaryl ring.
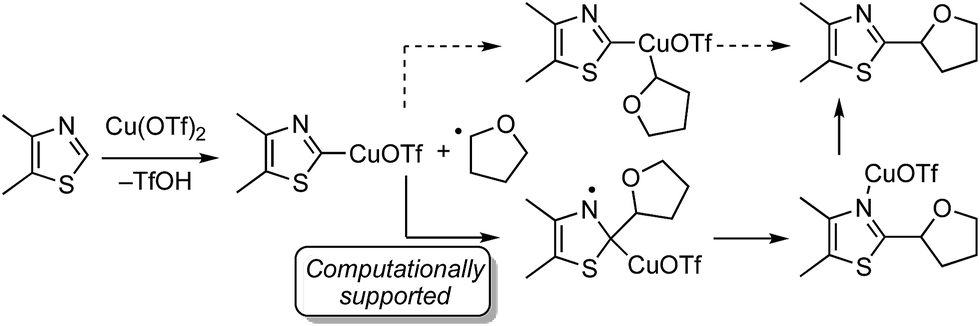 | ||
| Scheme 16 Two plausible reaction pathways. | ||
Notably, in contrast to the Cu(OTf)2-catalyzed reactions,38 where K2S2O8 was effective, with FeF2 this oxidant gave a 0% yield. DDQ, cumene hydroperoxide, dicumyl peroxide and (t-BuO)2 were all equally ineffective, whereas t-BuOOCOPh was modestly effective. Using the optimized combination several products were prepared (Scheme 17).
 | ||
| Scheme 17 FeF2/t-BuOOH promoted CDC reactions of heterocycles with ethers. | ||
Some side-by-side comparisons of the Cu(OTf)2- and FeF2-mediated reactions are feasible. In comparable cases, the reactions of thiazoles and benzothiazoles with the former catalyst combination gave higher product yields. Interestingly, with 1,3-dioxolane, the FeF2-based system gave higher yields. However, in contrast to the Cu-mediated process that gave only the C2 arylation product, the Fe catalyst gave isomeric mixtures of the C2 and C4 arylation products with this ether.
When presented with 1° and 2° carbon centers for potential reaction, as with 1,2-DME, both benzothiazole and 6-fluorobenzotriazole gave mixtures of products. However, reaction at the 2° center predominated (by ca. 2:1). Less acidic heteroaryls such as 1,2,3-triazoles and indoles, as well as ethers such as n-Bu2O and EtOH were unreactive.
This reaction also appears to proceed via radicals, with product suppression occurring in the presence of inhibitors such as TEMPO, BHT and DPE. With DPE, the same THF adduct as was observed in Scheme 7 was obtained in 10% yield. In a competition between THF and THF-d8, a significant kH/kD = 3.18 was observed, suggesting that hydrogen atom abstraction is likely to be rate determining. Computations showed that the formation of t-BuO˙ is stabilized by FeIII, and the reaction is proposed to proceed via an FeII–FeIII redox chemistry. The THF radical produced in the process forms an oxocarbenium ion. The hydroxide formed in the metal redox reaction is responsible for deprotonation of the heteroaryl (mechanism in Scheme 18).
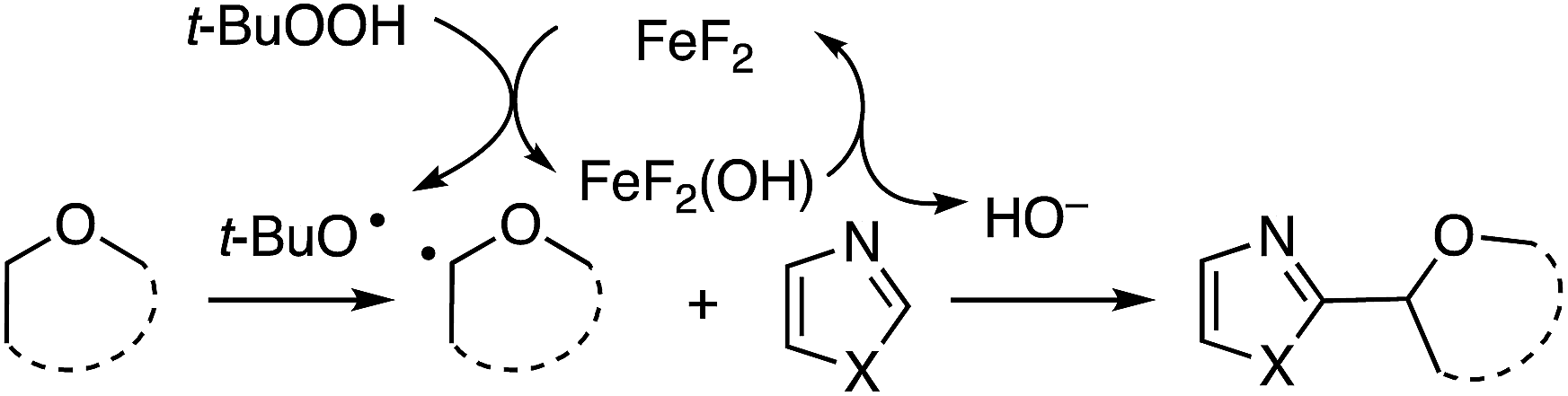 | ||
| Scheme 18 A plausible mechanism for FeF2/t-BuOOH mediated CDC reactions. | ||
 | ||
| Scheme 19 Reactions of oxazoles and benzoxazoles with 1,4-dioxane, catalyzed by Co2CO3. | ||
While reasonable product yields were generally obtained, a single electron-withdrawing group on the benzoxazole group appears to cause yield improvement. However, multiple electron depleting groups caused a lowering of the product yields, such as with the dichloro and fluoro bromo substrates. Other ethers were tested in these reactions and these data are summarized in Fig. 11.
 | ||
| Fig. 11 Products formed from reactions of oxazoles and benzoxazoles with other ethers. | ||
Some interesting factors emerge from Fig. 11. A single product was obtained from 1,2-diethoxyethane but, by contrast, two products resulted from 1,2-DME in a 1.9:1 ratio. A similar outcome was observed with n-butyl ethyl ether. With 1,3-dioxolane two products were obtained, with the C2 arylation product predominating. Finally, with 2-methyl THF two products were obtained, with the predominant reaction occurring at the 3° carbon atom. The authors have attempted to rationalize these findings on the basis of the C–H bond dissociation energies of the ethers. Interestingly, no product was obtained with tetrahydropyran.
In mechanistic studies the reaction rate, as estimated by the product yield, for the reaction between 5-phenyloxazole and 1,4-dioxane was shown to be positively influenced by CoCO3. Further, the reaction is fully suppressed by 6 equiv. of TEMPO, but with 1 equiv. only a slight lowering of the product yield was observed. This led the authors to propose a possible radical mechanism. From a competition experiment between benzoxazole and C2 deutereobenzoxazole a KIE of 2.1 was obtained. Also, with C2 deutereobenzoxazole it was observed that very little loss of the isotope occurred in the absence of the catalyst (Scheme 20).
 | ||
| Scheme 20 Experiments involving monodeuterated benzoxazole. | ||
On the basis of the foregoing mechanistic evaluations a radical process has been proposed. This is summarized in Scheme 21. The immediately preceding section describes reactions of benzoxazoles with THF, 1,3-dioxolane and dioxane with yields in the 60–77% range.
 | ||
| Scheme 21 A mechanistic scheme for the Co-catalyzed CDC reactions of ethers with oxazoles (and benzoxazoles). | ||
 | ||
| Fig. 12 Products formed in metal-free CDC reactions with ethers. | ||
At least among the examples that are comparable to the metal-catalyzed processes, these reactions are highly competitive in terms of product yields. When presented with 1° and 2° carbon atoms, as in 1,2-DME, two regioisomeric products were observed. Among these, reaction at the 1° carbon is favored over the 2° (ratio of 1.25:1). This is a contrast to the FeF2-catalyzed reaction, where reaction at the 2° carbon atom of 1,2-DME predominated.39 Perhaps the only downside to the method is that minor, dimeric radical coupling products were observed.
A notable aspect of this chemistry is the CDC reaction of alcohols and benzothiazoles, which is complementary to that of ethers. MeOH, and 1° and 2° alcohols react under these reaction conditions to provide the α arylated alcohols in reasonable to high yields (Scheme 22).
 | ||
| Scheme 22 CDC reactions of alcohols with benzothiazole. | ||
 | ||
| Scheme 23 Photochemical α-heteroarylation of two ethers with benzothiazole. | ||
C.1.f.i. Pd-Mediated reactions. In 2013, Pd(OAc)2, PdCl2, (Ph3P)2PdCl2, (PhCN)PdCl2 and Pd(TFA)2 were investigated for catalyzing reactions of 4-methylquinoline-N-oxide with 1,4-dioxane.43 All of these salts enabled reactions in combination with aq. t-BuOOH, with Pd(OAc)2 being the most promising. Also analyzed were CuBr, CuBr2, NiCl2·6H2O, CoCl2and CuI, and again all, with the exception of CuI, showed product formation. As compared to t-BuOOH, dicumyl peroxide was less effective, whereas (t-BuO)2 and DDQ were ineffective in combination with Pd(OAc)2. Interestingly, the addition of water produced a dramatic yield increase, and this was further enhanced with stoichiometric n-Bu4N+Br−. Several quinoline-N-oxides, isoquinoline-N-oxide and a 2-substituted pyridine-N-oxide were evaluated for generality, and these data are shown in Scheme 24.
 | ||
| Scheme 24 CDC products obtained from quinoline, isoquinoline and 2-phenylpyridine N-oxides. | ||
Some interesting points emerge in reviewing the product data. With 4-methylquinoline-N-oxide, product yields were generally independent of the ether ring size and number of oxygen atoms. Quinoline- and 6-methylquinoline-N-oxide produced lower yields, possibly due to competing reactions at the C4 position as well as dialkylation at the C2 and C4 positions (observed by LC-MS). Electron-rich 6-methoxyquinoline gave an excellent product yield. 3-Bromoquinoline and isoquinoline N-oxides were reasonable substrates as well. Cyclic thioethers reacted efficiently and the method was also applicable to pyridine N-oxides, although the substrates contained a C2 phenyl substituent. 2-Methyl THF was suitable but produced tertiary/secondary carbon alkylation in a 1.2:1 ratio.
The reaction conditions could also be applied to a CDC reaction with EtOH (Scheme 25). This result should be considered in conjunction with CDC reactions of other alcohols (see section C.3.f.iii. below). However, the methodology was not applicable to the pyrazine, pyrimidine, imidazole and thiazole scaffolds.
 | ||
| Scheme 25 CDC reaction of 4-methylquinoline-N-oxide with EtOH. | ||
t-BuOOH is essential in this reaction, without which no reaction occurs. Thus, formation of t-BuO˙ is necessary for the abstraction of a hydrogen atom from the ether. A plausible mechanism is shown in Scheme 26.
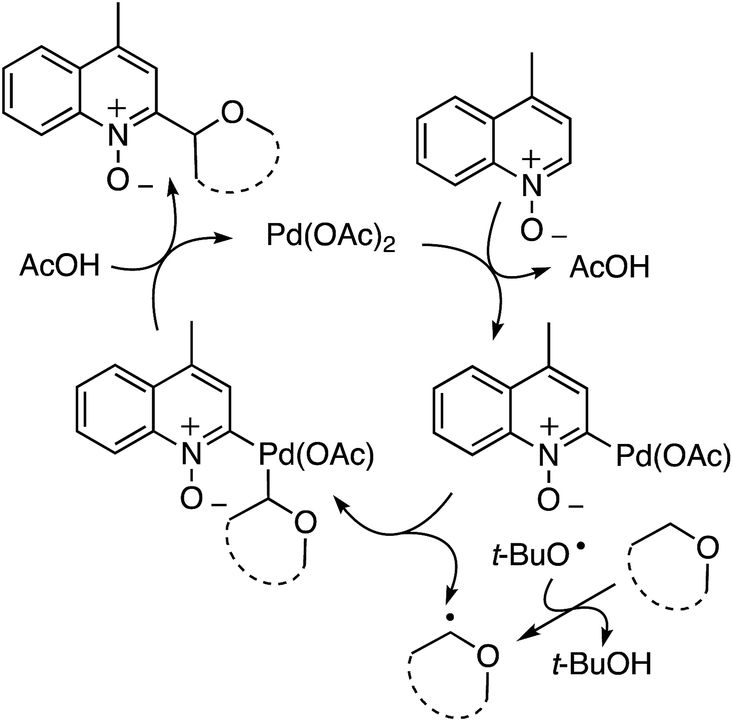 | ||
| Scheme 26 Plausible mechanism for the CDC reaction of quinoline N-oxides with ethers. | ||
C.1.f.ii. Cu-Mediated reactions. Because the Pd-mediated method described in C.1.f.i. was not directly applicable to unsubstituted pyridine N-oxides, a Cu-mediated approach for CDC reactions at the 2-position of pyridine N-oxides was sought.44 CuBr2, Cu(OAc)2, CuO, CuI, Cu2O, CuCO3 and CuSO4 were evaluated in combination with K2S2O8. The CuO/K2S2O8 combination, at 125 °C, gave a very promising reaction of pyridine-N-oxide with 1,4-dioxane. Lower temperatures gave lower yields, with no product formation occurring at 60 °C. FeCl3, NiBr2 and the [Ru(p-cymene)2Cl2]2 dimer were ineffective with K2S2O8 as the oxidant. Likewise, t-BuOOH, (t-BuO)2 and Ag2CO3 in combination with CuO were ineffective. The reaction did not progress in the absence of the copper catalyst and, notably, argon atmosphere instead of air is detrimental. Under argon, no product formation was observed. Several pyridine N-oxides were evaluated and the data are shown in Scheme 27.
 | ||
| Scheme 27 Cu-Catalyzed CDC reactions of pyridine N-oxides with cyclic ethers. | ||
These reactions were not amenable to the use of linear ethers and in two cases, with 3-methoxypyridine as well as 2-cyanopyridine-N-oxide, no product was observed. Interestingly, reaction of 1,3-dioxolane resulted in reaction at the C4 position, apparently, regioselectively.
This Cu-catalyzed reaction is prone to inhibition with TEMPO, indicating the involvement of a radical process. Thus, mechanistically, this reaction is similar to the Pd-mediated reaction in C.1.f.i. (Scheme 26), where t-BuO˙ mediated radical formation occurs from the ether, and cupration occurs at the α position to the heteroatom of the heteroaryl. Formation of the radical is likely initiated by SO4−˙, as in the Cu(OTf)2/K2S2O8 promoted CDC reactions of ethers with thiazoles (Scheme 16). Finally, C–C bond formation results in the products, with expulsion of CuI that is reoxidized.
C.1.f.iii. Metal-free reactions. In 2015, a metal-free Minisci-type reaction was reported.45 The precedent for this is a 2009 report on the alkylation of pyridine N-oxides with cycloalkanes and a solitary example of alkylation with 1,4-dioxane.46 In the earlier work,46 the reaction of pyridine-N-oxide with 1,4-dioxane and (t-BuO)2 at 135 °C gave a 2.2
:1 mixture of 2,6-dialkylated and 2,4,6-trialkylated pyridine-N-oxide in 61% yield (Scheme 28).
 | ||
| Scheme 28 CDC reaction of pyridine-N-oxide with dioxane using (t-BuO)2. | ||
Reaction conditions were evaluated for the CDC reaction of pyridine-N-oxide with THF at 145 °C. H2O2, m-CPBA, (t-BuO)2 and K2S2O8 as oxidants gave no reaction. However, 2 equiv. of cumene hydroperoxide or t-BuOOH showed product formation, with yield increasing when the amount of t-BuOOH was increased to 4 equiv. However, the addition of K2CO3 gave a dramatic increase in yield. Na2CO3 and Cs2CO3 did not perform as well, indicating that the selection of carbonate base is critical. Other bases such as t-BuOK, KOAc and DBU were also not as effective as K2CO3. The use of CuI, Pd(OAc)2, FeCl3 or n-Bu4N+Br− as additives lowered the product yield, and n-Bu4N+I− suppressed the reaction. Fig. 13 shows various products prepared from pyridine N-oxides and THF, using 4 equiv. of t-BuOOH and 1 equiv. of K2CO3, at 145 °C.
 | ||
| Fig. 13 Products from metal-free CDC reactions of pyridine N-oxides with THF. | ||
The yields in these reactions were generally good, in the 44–73% range. Interestingly, with 2-methylpyridine-N-oxide no product formation was observed. With 3-bromopyridine-N-oxide, two regioisomeric products were observed in a 2.2:1 ratio, with the predominant reaction occurring adjacent to the bromine atom.
The reaction scope was then expanded and these data are shown in Fig. 14. A low yield was obtained with 2,3-dihydrobenzofuran and t-butyl methyl ether was also compatible. With 1,3-dioxolane, regioisomeric products were observed with the C4 isomer predominating over the C2. Finally, several of the products were then reduced to the free C2 alkylated pyridines using Zn/aq. NH4Cl in THF at 40 °C (Scheme 29).
 | ||
| Fig. 14 Expanding the scope of the metal-free CDC reactions of pyridine N-oxides. | ||
 | ||
| Scheme 29 Reduction of some of the CDC products to the free pyridines. | ||
This reaction must be a radical process, where the decomposition of t-BuOOH occurs to t-BuO˙ and HO˙. Then hydrogen atom abstraction from THF results in a THF radical that adds at the C2 position of the pyridine N-oxide. Finally, loss of a hydrogen radical and rearomatization results in the CDC products. In support of this radical reaction mechanism, the addition of TEMPO completely suppressed product formation.
C.1.f.iv. Photochemical CDC reactions of azaheteroaromatics via Ir catalysis. In order to eliminate the need for metallic catalysts, as well as the stoichiometric or higher quantities of peroxides and the relative high temperatures needed for Minisci-type reactions, a phototoredox system was developed for the generation of radicals α to the ether oxygen atoms followed by addition to azaheteroaryls.47
As with the photochemical CDC reactions of dicyanobenzenes with ethers described in B.3.a. above, photoexcitation of an IrIII catalyst with a fluorescent, house-hold light bulb produces a long-lived IrIII* species that is a good reducing agent, capable of converting S2O82− to SO4−˙. The latter can then become involved in hydrogen atom abstraction from an ether. The radical so derived can add to a protonated electron-deficient azaarene to produce a radical cation. Loss of a proton and electron transfer to the IrIV species that is produced in the formation of SO4−˙ regenerates the catalyst and leads to product formation. These mechanistic details are shown in Scheme 30.
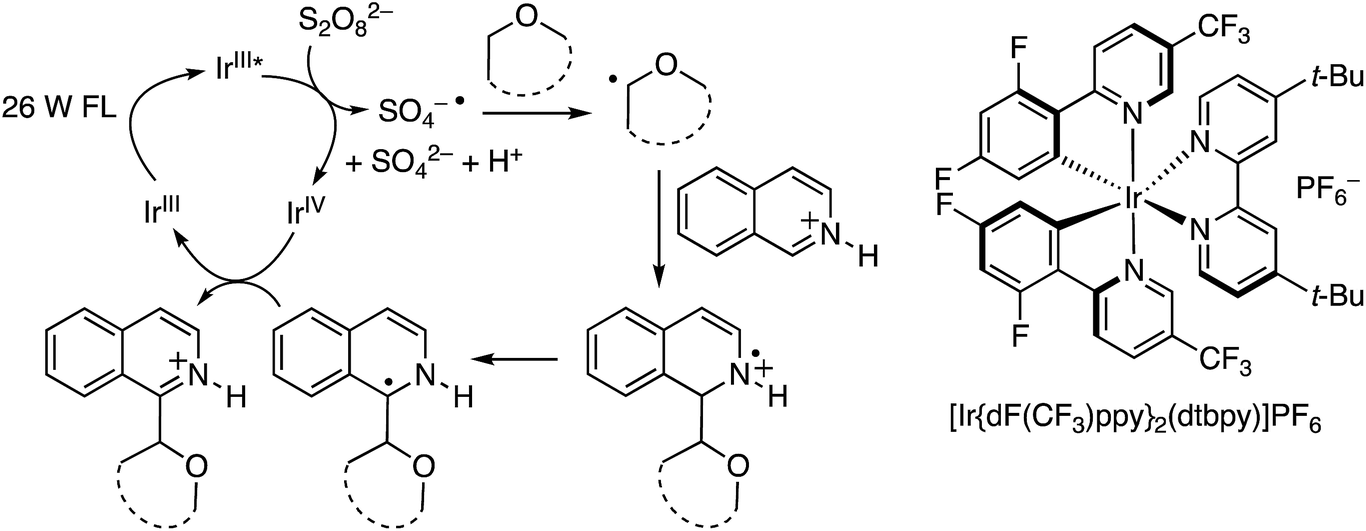 | ||
| Scheme 30 Photochemical IrIII mediated Minisci-type process. | ||
For this approach ammonium, potassium and sodium persulfates were assessed in combination with three Ir salts and one Ru salt. The combination of [Ir{df(CF3)ppy}2(dtbpy)]PF6, Na2S2O8 and TFA in MeCN/H2O, proved optimal in evaluation reactions of isoquinoline and dihydropyran. Water is likely necessary for solubility of the persulfate salt, which in turn makes for effective irradiation. With this combination various azaheteroaryls were reacted with tetrahydropyran (THP) and the products are summarized in Fig. 15. Reactions occur at the most electron-deficient sites in the heteroaryls and, notably, chlorine and bromine atoms are well tolerated. When the C2 position of quinoline is blocked, reaction occurs at the C4 position. As was proposed, the reactions were generally tested on electron-deficient heteroaryls.
 | ||
| Fig. 15 Products from the photochemical Ir-mediated CDC reactions with THP. | ||
Because the oxidative-dimerization of products is possible, in the isoquinoline cases EtCN was used as the solvent. This change, which ameliorated the problem, has been ascribed to the lower polarity of EtCN in comparison to MeCN, which could influence the electron-transfer rates in the reactions.
Thus, several ethers were used in CDC reactions with isoquinoline and the yields as well as the products are shown in Scheme 31. Excellent yields were obtained with cyclic and acyclic ethers. With oxetane the reaction was conducted in MeCN (without water), without TFA but with added n-Bu4N+Cl−. The ammonium salt was used to solubilize the persulfate salt. The reactions of 1,3-dioxolane and 1,2-DME gave regioisomeric products. C2 arylation was favored with the former and reaction at the 2° carbon atom was favored with the latter. In the case of (tetrahydrofuran-2-yl)methyl acetate, reaction occurred remote from the side chain but a mixture of diastereomers was obtained. Finally, reaction with 2-methyl THF gave a regioisomeric mixture of products, with the predominant reaction occurring at the 2° carbon atom. In this case, diastereomeric products ensued. Reaction at the 3° carbon atom led to a minor isomer.
 | ||
| Scheme 31 Photochemical CDC reactions of various ethers with isoquinoline (ratios of isomeric products are indicated with blue arrows). | ||
C.1.f.v. PIFA/NaN3 mediated reaction of 2,7-dichloroquinoline with 1,4-dioxane. During the course of investigating CDC reactions of alkanes with 4,7-dichloroquinoline, mediated by PIFA and NaN3, a single reaction of 1,4-dioxane was reported (Scheme 32).48 The reaction with alkanes is anticipated to be a radical process due to product suppression with radical inhibitors. Also, a large KIE value of 7.6 was observed in a reaction with cyclohexane/cyclohexane-d12. A possible reaction mechanism, shown in Scheme 32, involves the formation of PhI(N3)OCOCF3 (or PhI(N3)2), which produces N3˙ thermally. This species can produce radicals from the (alkane or) ether. Protonation of the nitrogen heterocycle by the formed CF3CO2H, followed by radical addition, will then result in a nitrogen-centered radical cation. Oxidation of the radical cation and deprotonation will then ultimately result in the alkylated product.
 | ||
| Scheme 32 A possible mechanism for the CDC reaction of 4,7-dichloroquinoline with 1,4-dioxane, mediated by PIFA/NaN3. | ||
C.1.f.vi. N-Hydroxysuccinimide/(NH4)2S2O8 promoted reactions of azaheteroaryls. While this perspective was under review, as a follow up to the photochemical α-heteroarylation of ethers using (NH4)2S2O8,42 the use of N-hydroxysuccinimide (NHS) to promote CDC reactions of nitrogenated heterocycles and ethers was reported.49 During initial assessments on the efficacy of additives in reactions of isoquinoline with THF, 13 reagents were tested. These were CH3CONH2 (on the basis of prior results42), acetone, CF3CONH2, CH3SO2NH2, PhCONH2, CH3CONMe2, iPr2NEt, pyrrolidine, quinuclidine, 3-quinuclidinol, CH3CONHOH and NIS. As in their earlier work,42 the “amine moiety” in the amide is essential for successful reaction. All other amides, with the exception of CH3CONMe2, were ineffective, as were the amines. NIS displayed outstanding reactivity under a wide range of conditions tested (60–88% yields as determined by 1H NMR). Most notably, the use of 20 versus 10 equiv. of THF showed only a marginal lowering of the yield. Even with 5 equiv. of THF the yield was 60–67%.
Using the identified reaction conditions, several reactions were conducted (products shown in Scheme 33). Substituted isoquinolines were reacted with 10 equiv. of THF (yields from reactions with 5 equiv. of THF are shown in parentheses) and high regioselectivity for the C1 position was observed. The reaction of quinoline occurred at the C2 and C4 positions, in a reaction time of 72 h. Reaction of 4-cyanopyridine gave the mono and bis alkylated products in a 10:1 ratio. 3-Cyanopyridine gave only the mono alkylation product. Reactions of the benzothiazoles and benzimidazoles were conducted with 20 equiv. of THF. While the reactions of benzothiazoles proceeded well, those of benzimidazoles were less successful. Benzothiazole and the N-Cbz-protected analogue gave traces of product, and deprotection of the Cbz group was observed with the latter. N-Methylbenzimidazole reacted reasonably well as did a carboline (Scheme 33).
 | ||
| Scheme 33 Reactions of isoquinolines, quinolines, pyridines, benzothiazoles, benzimidazoles and a carboline with THF. | ||
Reactions of isoquinoline with several ethers were tested (Scheme 34). Notably, many functionalized ethers were evaluated, which augments the potential further utilities of the products. Two regioisomers are possible with unsymmetrical dioxo ethers. With 1,3-dioxane the C4 arylated product was slightly favored over the C2 isomer, whereas with 1,3-dioxolane the C2 isomer was substantially favored in comparison to the C4. This selectivity is greater than that observed in the Ir-mediated reaction (C.1.f.iv., Scheme 31).47 The use of 2,2-dimethyl-1,3-dioxolane resulted in deprotection of the acetonide under those conditions, giving an ethylene glycol alkylation product. With 2-methyl THF the less-hindered C5 position was favored. In comparison, the selectivity was slightly better in the Ir-mediated reaction (Scheme 31).47 On the other hand with a CH2OH or CH2OAc substituent at the C2 position of THF, there was complete selectivity for the C5 position, and this was observed in the Ir-catalyzed reaction as well (Scheme 31).47 The diastereoselectivity in the three THF cases was ca. 2:1. A crown ether could be efficiently utilized as also other THF substrates containing hydroxyl groups.
 | ||
| Scheme 34 Reactions of other ethers with isoquinoline, mediated by NHS (sites and ratios of regioisomeric products are indicated with blue arrows, diastereomeric ratios are indicated by dr and the yields indicated with an asterisk were obtained with 5 equiv. of the ether). | ||
From a mechanistic standpoint, the reaction of isoquinoline and THF was suppressed by TEMPO. In a competitive KIE determination with THF/THF-d8, a value of 3.0 was observed, and the KIE from two parallel experiments was 2.2. From extensive experimentation, it is believed that oxygen centered radicals are not involved but that amines such as quinuclidine and 3-quninuclidinol are oxidized to nitrogen-centered radical cations. Similar nitrogen-centered radicals may be possible from amide additives and NHS. A possible mechanism is shown in Scheme 35.
 | ||
| Scheme 35 A plausible mechanism for the (NH4)2S2O8/amine mediated CDC reaction of THF with isoquinoline. | ||
C.1.f.vii. Pd-Catalyzed CDC reactions of azaheteroaromatic compounds with alcohols. In Scheme 25, a single example of a CDC reaction of EtOH with quinoline-N-oxide is shown, involving the use of Pd(OAc)2/t-BuOOH/n-Bu4N+Br−, in EtOH and H2O.43 Related to this, a general approach to CDC reactions of alcohols with quinolines, isoquinolines, phenanthridine and quinazoline has been reported.50 Sc(OTf)3, FeCl3, InCl3, RhCl3, PdCl2 and PdBr2 were investigated as catalysts for the reaction of 4-methylquinoline with EtOH, with dicumyl peroxide as oxidant. With the Pd and Ru catalysts, the effect of added phosphines and a phosphine oxide was also assessed.
In these assays it was discovered that the use of 5 mol% of PdCl2 in conjunction with 5 mol% of (±)-BINAP, and 3.5 equiv. of dicumyl peroxide, in alcohol solvent at 120 °C in the presence of air, gave a good yield of the C2 alkylation product. On this basis, generality assessments were conducted and these data are shown in Fig. 16. Yields in the reactions of EtOH with 4-methylquinoline, isoquinoline, 3-methylisoquinoline, 3-carbomethoxyisoquinoline and phenanthridine were good to high. The reaction of isoquinoline with n-PrOH gave a modest yield, which seems to improve with decreased electron density on the aryl moiety. Consistent with this, n-BuOH also gave a low yield with quinoline. In the reactions of quinoline and 6-methoxyquinoline with EtOH, a mixture of isomeric C2 and C4 alkylation products, as well as C2, C4-bis alkylation products were observed. A lower overall yield of products was obtained with the electron-rich 6-methoxyquinoline.
 | ||
| Fig. 16 CDC reactions of alcohols with azaaromatics catalyzed by PdCl2/(±)-BINAP. | ||
In order to evaluate whether isoquinoline-N-oxide is formed in the reaction, then leading to product, a reaction of isoquinoline-N-oxide and EtOH was conducted in the absence of the oxidant. No product was observed and, in the presence of dicumyl peroxide, 18% yield of the CDC product was obtained. Also, another query that was evaluated was whether oxidation of the alcohol to the aldehyde occurs followed by addition of the heterocycle. However, in a reaction between isoquinoline and n-butanal no product formation was observed in the absence of the peroxide, and when peroxide was present a 35% yield of the product was obtained.
Mechanistically, it is possible that thermal decomposition of the peroxide generates a radical that then abstracts a hydrogen atom from the alcohol, α to the oxygen atom. This new radical can participate in a Minisci-type reaction, leading to the product.
C.1.f.viii. Au-Catalyzed CDC reactions of azaheteroaromatic compounds with alcohols. In 2012, immediately following the report on a methodology for Pd-catalyzed reactions of alcohols and heteroaromatics (C.1.f.vii.),50 Au catalysis was studied for similar conversions.51 Six gold catalysts shown in Scheme 36 were evaluated in combination with t-BuOOH.
 | ||
| Scheme 36 Reaction of 4-methylquinoline with EtOH using six Au catalysts. | ||
While all of the catalysts gave product, 5 mol% of HAuCl4·4H2O in combination with 4.5 equiv. of t-BuOOH proved to be most promising (Scheme 36). The use of FeCl3 or CuI gave no product formation, but with NiCl2 23% product formation was observed. Other oxidants such as m-CPBA, TEMPO and NaClO gave only traces or no product. A reduction in the amount of t-BuOOH or the catalyst led to reduced product yield.
Using these optimized conditions, several heteroaryls were acylated and these are shown in Fig. 17. Notably, an increase in the alcohol chain length from methyl to ethyl caused the yield to be lowered, but further increase did not diminish the yield further. These reactions are highly regioselective.
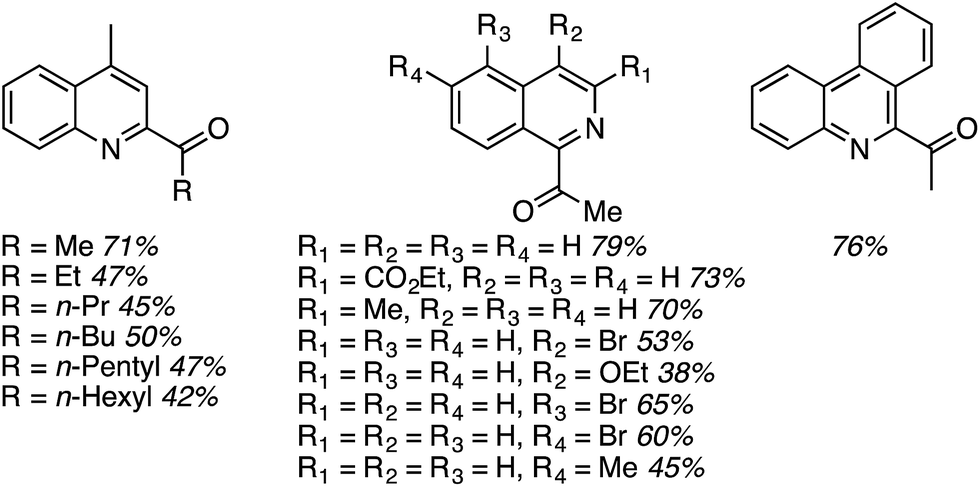 | ||
| Fig. 17 Gold-catalyzed acylation of heteroaromatics. | ||
The reaction of 4-methylquinoline with EtOH was followed by ESI-MS, which led to the following data (Scheme 37). At 3 h, only 4-(1-methylquinolin-2-yl)ethan-2-ol was observed, but at 5 h both this alcohol and the oxidized 4-(1-methylquinolin-2-yl)ethan-2-one were seen. Finally, at 24 h, only the ketone was observed. Thus, the alcohol is formed en route to the final acyl product.
 | ||
| Scheme 37 Monitoring of the reaction of 4-methylquinoline and EtOH by ESI-MS. | ||
Given the above observations, the next question was about potential reaction intermediates and a mechanism. The reaction was fully suppressed in the presence of radical inhibitors: TEMPO and BHT. Therefore, the reaction likely originates from the formation of a radical from the alcohol that then adds to the heteroaryl group (Scheme 38). Although this type of reaction can be postulated to occur in the absence of the metal, no reaction occurred in the absence of Au. The authors propose coordination of Au to the heteroaryl group, as well a role of Au in the final oxidation.
 | ||
| Scheme 38 A possible mechanism for the Au-catalyzed acylation reaction. | ||
C.2. Formation of C–C bonds with heteroaromatic compounds that possess NH
In this section C–C reactions of heterocycles that possess an NH unit are discussed. Notably, indole is the heterocycle that has been the focus of studies, and the various reactions that have been evaluated are described in the following sections.C.2.a.i. Fe-Catalyzed CDC reactions of indoles and ether ring cleavage. One of the early reports on the reaction of heterocycles with ethers is the iron-catalyzed reactions of indoles with ethers.52 From the various iron-species tested (Fe2(CO)9, Fe(OAc)2, Fe(acac)2, Fe(acac)3, FeCl2, FeBr2, FeI2 and FeCl3), FeCl2 proved optimal, in combination with (t-BuO)2. t-BuOOH and t-BuOOCOPh in combination with FeCl2 were inferior. No reaction occurred in the absence of FeCl2.
Several indoles were tested for reactions with ethers, under neat reaction conditions at 80 °C. Products generally arise through the reaction of two indole moieties with the ether. The ethers used were THF, 2-methyl THF (which gave a 1:1 mixture of regioisomers), 2,3-dihydroisobenzofuran, isochroman, benzyl methyl ether and diethyl ether. The use of 2-ethoxy ethyl acetate gave two natural products, vibrindole A (37% yield) and strepindole (17% yield), in a single chemical transformation. The 1,1-bis-indolylmethane products from these reactions are summarized in Fig. 18.
 | ||
| Fig. 18 1,1-Bis-indolylmethane derivatives prepared and their yields. | ||
During the course of previous investigations, it was discovered that reactions of 2,5-dihydroisobenzofuran were highly dependent on the nature of the indole. As can be seen from the data in Fig. 19, with an increased electron-depleting nature of the indole substituent, the extent of monoalkylation increased. These results implied that introduction of the second indole is via a Friedel–Crafts process. On the basis of this observation, several unsymmetrical 1,1-bisindolylmethanes were synthesized from 2,5-dihydroisobenzofuran and isochroman, in generally good to high yields (Fig. 19).
 | ||
| Fig. 19 Ratio of products as a function of the indole substituent (top) and unsymmetrical compounds synthesized (bottom). | ||
Studies were conducted to gain mechanistic insight. For this, the monoalkylation product from the reaction of 5-nitroindole and 2,5-dihydroisobenzofuran was prepared. This product was used in a competitive reaction with indole and 5-carbomethoxyindole. The overall product yield was 70% and, notably, the product from indole predominated over that from 5-carbomethoxyindole by 6:1. This also indicated that the second step is akin to a Friedel–Crafts process, where the electron-deficient nucleophile is expected to be slower reacting. In support of this, a competitive reaction between indole and 5-carbomethoxyindole with THF gave a nearly 1:1 yield of the two symmetrical bisindolyl products, indicating that the first mono-alkylation step is independent of the indole ring electronics.
Reaction with 1,3-dideuterioindole and THF led to no deuterium incorporation in the product. Furthermore, a reaction of indole and THF was completely prevented by a radical inhibitor, TEMPO. Thus, a proposed plausible mechanism is shown in Scheme 39.
 | ||
| Scheme 39 A plausible mechanism for the formation of 1,1-bis-indolylmethanes. | ||
C.2.a.ii. Fe-Catalyzed cascade cyclization CDC reaction leading to oxindoles. An interesting C–C bond-forming CDC reaction is, in fact, a cascade-type process where a radical generated α to an ether oxygen atom adds to an olefin of an N-arylacrylamide, eventually resulting in cyclization to oxindoles.53 From among a variety of iron catalysts, FeCl2, FeCl3, FeBr3, Fe(acac)2 and CuCl2, FeCl3 was optimal in combination with a ligand and t-BuOOH. DABCO, TMEDA, DMAP, 4-aminopyridine and DBU were tested as ligands, from which DBU was selected.
Initially, a series of ethers, an amine and a thioether were reacted with N-methyl-N-phenylmethylacrylamide (Scheme 40, blue dots represent the point of C–C bond formation). Reactions with THF generally needed the addition of 5 mol% of CuCl2.
 | ||
| Scheme 40 Synthesis of oxindoles from N-methyl-N-phenylmethylacrylamide. | ||
A range of oxindoles, summarized in Fig. 20, was then synthesized via this approach. Notably, in two cases regioisomeric products were obtained and the reaction failed when no substituent was present on the nitrogen atom or when the nitrogen atom bore an acetyl group.
 | ||
| Fig. 20 Various oxindoles synthesized via the cascade CDC cyclization. | ||
Mechanistically, this is also a radical reaction, like the previous example above, which is a recurring theme in almost all other C–H bond activations of ethers. In support of this, there was no observable kinetic isotope effect in: (a) a competitive reaction between the amide and the pentadeutero amide, (b) an intramolecular reaction with an ortho-deuterated amide, and (c) reaction with THF-d8.
Additionally, when TEMPO was added to a reaction with Et2O, product formation was completely suppressed, and formation of the Et2O–TEMPO adduct occurred in a high 91% yield. Thus, in a FeII–FIII redox cycle, t-BuO˙ is formed, which enables the formation of a radical from the ether. Addition of this radical to the olefin of the acrylamide is followed by reaction at the ortho-position of the aromatic ring. Loss of a hydrogen radical in the rearomatization step then results in the oxindoles. A portion of the mechanism is shown in Scheme 41.
 | ||
| Scheme 41 Mechanism for the formation of oxindoles. | ||
:3 to 2:3 and, hence, were uninteresting.
The Ni catalysts, however, showed more promise and a summary of the catalyst/additive analysis is shown in Table 2. Notably, in each case C2 and C3 alkylation products were observed. However, the additives had an important influence on these ratios.
|
|
||||
|---|---|---|---|---|
| Entry | Ni salt | Additive | Solvent | Yield, C2:C3 ratio |
| a Catalyst = 10 mol%, additive (when used) = 10 mol%, (t-BuO)2 = 2 equiv., reaction temperature = 120 °C. b Reaction was conducted at 130 °C. c Reaction was conducted under argon. d Reaction with 1.2 equiv. of (t-BuO)2. | ||||
| 1 | Ni(acac)2 | None | None | 18%, 3:2 |
| 2 | Ni(acac)2 | None | None | 35%, 2:1b |
| 3 | Ni(acac)2 | None | 1,4-Dioxane | 63%, 3:2 |
| 4 | Ni(acac)2 | None | MeCN | Not available |
| 5 | Ni(acac)2 | None | DMF | Not available |
| 6 | Ni(acac)2 | PPh3 | 1,4-Dioxane | 71%, 3:2 |
| 7 | Ni(acac)2 | Zn(OTf)2 | 1,4-Dioxane | 68%, 1:19 |
| 8 | Ni(acac)2 | Zn(OTf)2 | 1,4-Dioxane | 76%, 1:19c |
| 9 | Ni(acac)2 | Zn(OTf)2 | 1,4-Dioxane | 51%, 1:19d |
| 10 | Ni(OAc)2 | PPh3 | 1,4-Dioxane | 26%, 2:1 |
| 11 | NiCl2 | PPh3 | 1,4-Dioxane | 16%, 2:1 |
| 12 | Ni(OH)2 | PPh3 | 1,4-Dioxane | 44%, 5:1 |
| 13 | NiF2 | PPh3 | 1,4-Dioxane | 72%, 9:1 |
| 14 | NiF2 | Zn(OTf)2 | 1,4-Dioxane | 47%, 4:3 |
| 15 | NiF2 | None | 1,4-Dioxane | 49%, 9:1 |

With Ni(acac)2 and PPh3, C2 alkylation was slightly preferred over that at C3 (entry 6). However, with Zn(OTf)2 C3 alkylation was preferred over C2 by 19 to 1 (entry 7), and the yield improved with the use of an argon atmosphere, while the ratio remained constant. Notably, reducing the amount of oxidant only reduced the overall yield, not the C2/C3 alkylation ratio. Thus, the conditions in entry 8 were chosen for CDC reactions at the C3 and the products prepared are shown in Fig. 21.
 | ||
| Fig. 21 C3 alkylation of indole with 1,4-dioxane using Ni(acac)2/Zn(OTf)2. | ||
As can be seen from Fig. 21, the reaction is quite general to a wide range of indoles, and even when a C2 substituent is absent the reaction occurs at the C3 position with very good selectivity. Only with 5-methylindole was a 5:1 ratio of C3/C2 alkylation observed. The reaction conditions are also well suited for 2-substituted and 1,2-disubstituted indoles.
The conditions in Table 2 also provided insight into a possible catalytic system for C2 alkylation, i.e., the NiF2/PPh3 combination (entry 13). Using this combination, complementary CDC reactions at the C2 position could be conducted with a range of indoles (Fig. 22). Among the substrates tested was one example of a 1-substituted and two examples of 2-substituted indoles, as well as one example of benzofuran.
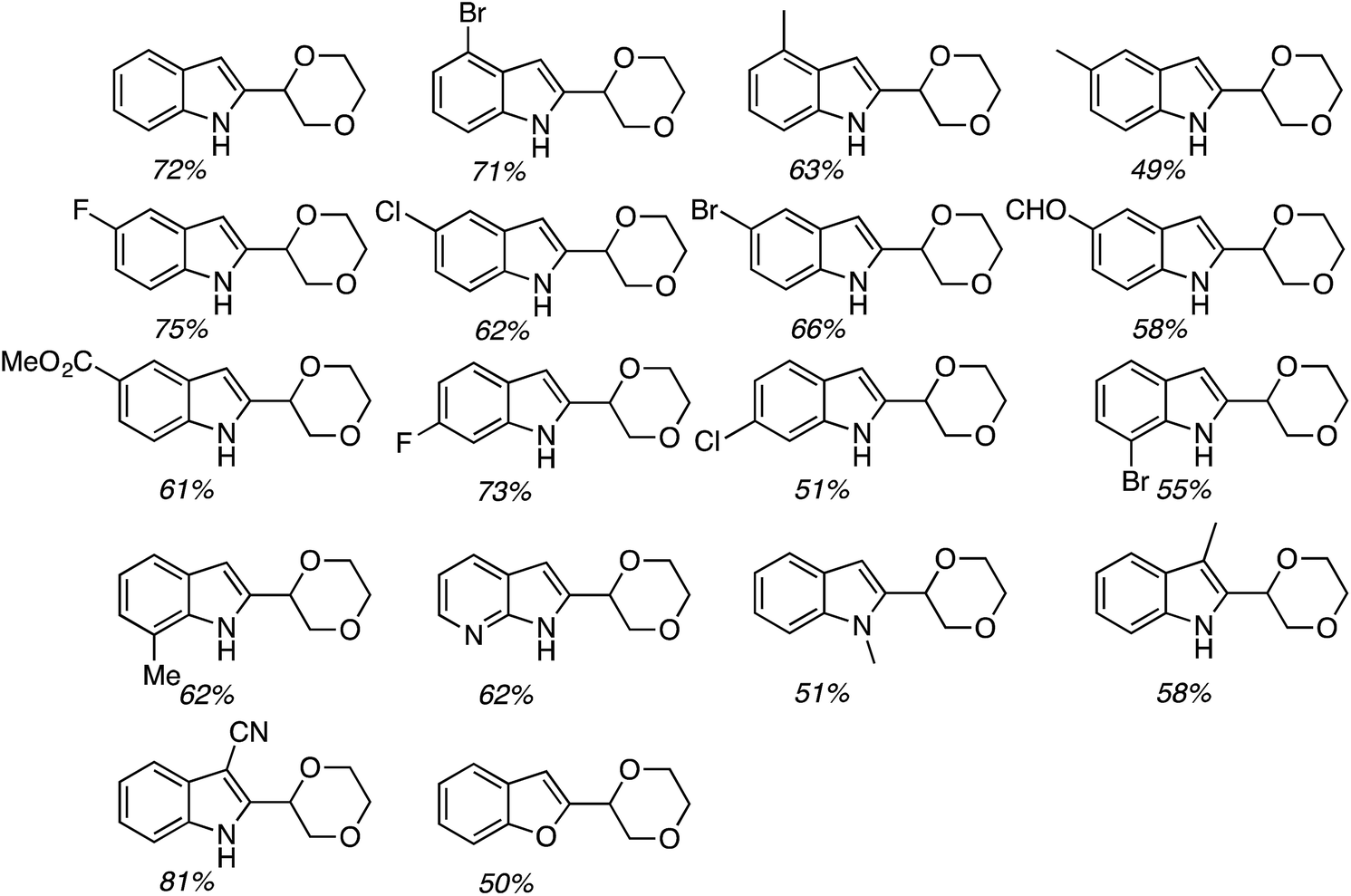 | ||
| Fig. 22 C2 alkylation of indoles and a benzofuran with 1,4-dioxane using NiF2/PPh3. | ||
All of the indoles tested underwent satisfactory reactions and those with electron-donating groups generally gave lower yields than those with electron-withdrawing substituents. Other substrates were tested in reactions with indole under the two sets of conditions identified and the results are summarized in Table 3. Notably, with DMF N,N-dimethyl-1H-indole-1-carboxamide was formed as the product.





Reactions of indole and 1,4-dioxane under each set of reaction conditions were inhibited upon addition of TEMPO, and in both cases the 1,4-dioxane–TEMPO adduct was isolated in 16–18% yield. Formation of the product was not observed in either case, indicating that radical processes are likely occurring. Further, two indole substrates shown in Fig. 23 were reacted with 1:1 1,4-dioxane/1,4-dioxane-d8, under the two separate, optimized conditions. The kH/kD values observed are shown below each structure, indicating that C–H bond breakage in the ether, possibly through a radical mechanism, may be involved in the rate-limiting step.
 | ||
| Fig. 23 Two indoles used in competitive reactions with 1,4-dioxane/1,4-dioxane-d8. | ||
C.2.c.i. Metal-free CDC reaction of indoles with isochroman and other ethers. In 2015, reactions of isochromans and indoles were investigated in the presence of oxidants, but in the absence of metal catalysts.55 The oxidants evaluated were O2, t-BuOOH, (t-BuO)2, DDQ, (PhCOO)2 and (t-BuO)2. From these, t-BuOOH and (t-BuO)2 emerged as promising, yielding in addition to the desired CDC product, small amounts of isochroman-1-one. The use of an argon atmosphere depressed the yield slightly as compared to air. Using a set of optimized conditions that involved 1.2 equiv. of (t-BuO)2 under air atmosphere at 120 °C, several indoles were reacted with isochroman. The products and yields are shown in Fig. 24.
 | ||
| Fig. 24 Uncatalyzed CDC reactions of indoles with isochroman. | ||
The yields were generally good, however, the presence of an N-methyl group appears to be one feature that is yield lowering. A methyl group elsewhere is not as problematic, an aldehyde is also tolerated reasonably well, and a phenolic hydroxyl group did not pose difficulties. Other ethers were tested and the products of these reactions are shown in Fig. 25. Here, chroman and isochroman-3-one were not suitable substrates for these CDC reactions.
 | ||
| Fig. 25 Other ethers tested in the metal-free CDC reactions with indoles. | ||
A neat reaction of isochroman with (t-BuO)2 in the absence of indole resulted in a good yield of isochroman-1-one. However, isochroman-1-one is not an intermediate to the product because no product was formed from it and indole under the reaction conditions. Also, no reaction occurred in the absence of the oxidant, and the reaction was suppressed by the addition of TEMPO. Clearly, this reaction appears to be a radical process as well and a plausible mechanism is shown in Scheme 42.
 | ||
| Scheme 42 A plausible mechanism for the CDC reaction of indole with isochroman. | ||
C.2.c.ii. Metal-free CDC reactions of indoles with 1,4-dioxane and other ethers. In 2016, metal-free CDC reactions of mainly 3-carboxylic esters, aldehyde and nitrile derivatives of indole, and a few 2-carboxylic esters of indole with 1,4-dioxane were reported, with only an oxidant.56 In a screening of conditions, t-BuOOH, K2S2O8 and (PhCOO)2 yielded no product at 140 °C. On the other hand, 1.5 equiv. of (t-BuO)2 or t-BuOOCOPh resulted in product formation, with the former being superior. The amount of (t-BuO)2 was critical and even 2.0 equiv. caused a lowered product recovery. Additives such as Pd(OAc)2, CuI and KI did not alter the yield that was obtained with just the peroxide. On the other hand, n-Bu4N+I− caused a substantial decrease in yield.
On the basis of the screening exercise, several indoles and 1,4-dioxane were reacted using 1.5 equiv. of (t-BuO)2 at 140 °C, under a nitrogen atmosphere. These results are shown and are shown in Fig. 26.
 | ||
| Fig. 26 Uncatalyzed CDC reactions of 3- and 2-substituted indoles with 1,4-dioxane. | ||
Generally the product yields were good, and as with reactions of isochroman described in Section C.2.c.i.,55 substrates with a methyl group on the nitrogen atom and a formyl group gave lowered yields. CDC reactions with the 2-carboxylic esters gave lower yields as compared to the 3-carboxylic analogues. These data can be seen in Fig. 26.
Subsequently CDC reactions of methyl 1H-indole-3-carboxylate with other ethers and four cycloalkanes were evaluated. These results are summarized in Fig. 27. Notably, cycloalkanes also underwent CDC reactions in modest to good yields. Just as with CDC reactions of the isomeric indole carboxylic esters with 1,4-dioxane, an indole-2-carboxylic ester underwent reaction with cyclohexane with a yield lower than the 3-carboxylic ester isomer. Notably, both 1,3-dioxane and 1,3-dioxolane underwent reactions at the C4 position, flanked by one oxygen atom, and not at the C2 atom. This is comparable to the Ni-catalyzed reactions described in C.2.b. (see Table 3).54
 | ||
| Fig. 27 CDC reactions of indole carboxylic esters with other ethers and cycloalkanes. | ||
Mechanistically, this too should be a radical process, just as the reactions of isochroman described in C.2.c.i.,55 proceeding via a similar pathway (Scheme 42). KIE measurement (Scheme 43) gave a kH/kD value of 3.3. The use of TEMPO resulted in only a trace amount of product, and the 1,4-dioxane–TEMPO adduct was isolated in 60% yield. Thus, C–H bond cleavage by t-BuO˙ is likely involved in the rate-limiting step.
 | ||
| Scheme 43 KIE measurement in the reaction of methyl 1H-indole-3-carboxylate with 1,4-dioxane and 1,4-dioxane-d8. | ||
C.2.c.iii. Addition to a vinylogous imine. An interesting reaction involving indoles was reported after this manuscript was reviewed. This is the addition of an ether-derived radical to a vinylogous imine derived from indole.57 Whereas this is not formally a CDC type reaction, it is worth considering this transformation within the context of C–C bond formation with indoles. The concept relies on the elimination of a p-Ts group from a 3-indolylmethane derivative, followed by the addition of a radical derived from an ether (see scheme above Table 4). Because the reaction requires an elimination, various bases (K2CO3, Cs2CO3, NaOH and Et3N) were tested. Optimal results were obtained with 1 equiv. of K2CO3 in (t-BuO)2, at 120 °C (2 M substrate concentration in the peroxide).
|
|
||||
|---|---|---|---|---|
| Entry | R, R1, and R2 | Ether |
anti:syn |
Yield |
| 1 | R = H, R1 = Me, R2 = Ph | 1,4-Dioxane | 1.9:1 |
67% |
| 2 | R = H, R1 = R2 = Ph | 1,4-Dioxane | 1.5:1 |
71% |
| 3 | R = R1 = Me, R2 = Ph | 1,4-Dioxane | 1.9:1 |
64% |
| 4 | R = Cl, R1 = Me, R2 = Ph | 1,4-Dioxane | 1.8:1 |
69% |
| 5 | R = H, R1 = Me, R2 = 4-Me-Ph | 1,4-Dioxane | 2.0:1 |
62% |
| 6 | R = H, R1 = Me, R2 = Ph | Tetrahydropyran | 1.3:1 |
61% |
| 7 | R = H, R1 = R2 = Ph | Tetrahydropyran | 1.7:1 |
80% |
| 8 | R = R1 = Me, R2 = Ph | Tetrahydropyran | 1.7:1 |
65% |
| 9 | R = Cl, R1 = Me, R2 = Ph | Tetrahydropyran | 1.5:1 |
59% |
| 10 | R = H, R1 = Me, R2 = 4-Me-Ph | Tetrahydropyran | 1.7:1 |
70% |
| 11 | R = H, R1 = Me, R2 = 2 F-Ph | Tetrahydropyran | 1.3:1 |
61% |
| 12 | R = R1 = H, R2 = Ph | Tetrahydropyran | 1.2:1 |
45% |
| 13 | R = H, R1 = Me, R2 = Ph | THF | 2.0:1 |
64% |
| 14 | R = H, R1 = R2 = Ph | THF | 1.6:1 |
63% |
| 15 | R = H, R1 = Me, R2 = 4-Me-Ph | THF | 2.0:1 |
48% |
| 16 | R = H, R1 = CO2Et, R2 = Ph | THF | 1.3:1 |
60% |

In every case the anti isomer was the major product. The yield with an indole that had no substituent at the 2-position was lower than 50% (entry 12). With 1,2-DME three products, that are shown in Fig. 28, were obtained in a 45% yield. Reaction at the 2° carbon atom resulted in two diastereomers, and the third product was formed by reaction at the 1° carbon atom. The ratio of these products was 1:1:1.3, indicating a preference for reaction at the 2° carbon center. However, with this acyclic ether the anti/syn preference was equal.
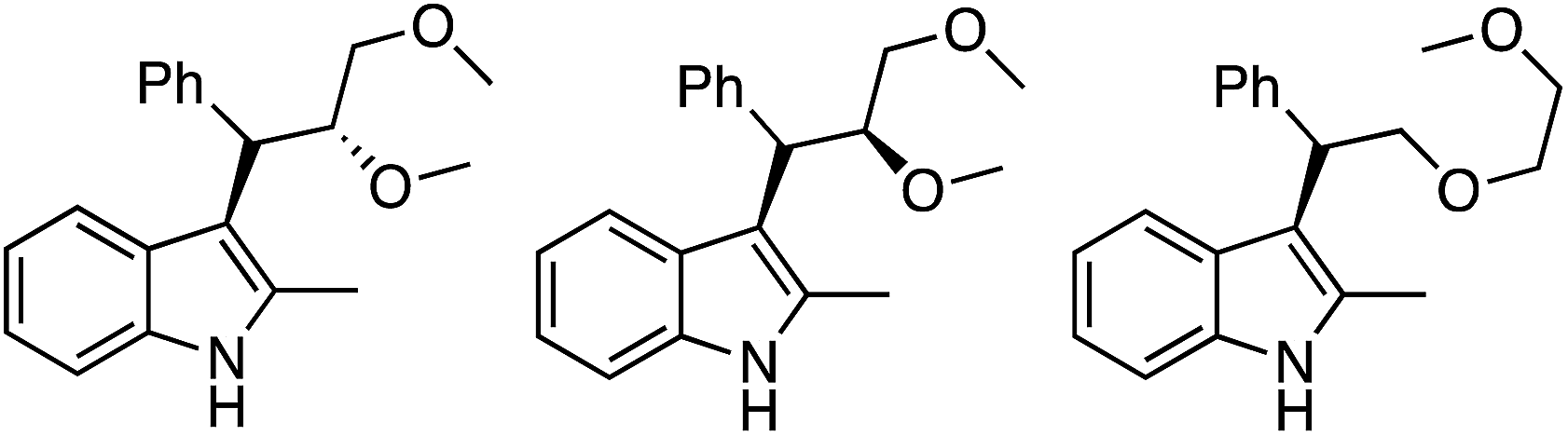 | ||
| Fig. 28 Three products obtained from the reaction of indole with 1,2-DME. | ||
From a mechanistic standpoint, this is a radical reaction with t-BuO˙, produced from the peroxide, generating a radical at the α-position to the ether oxygen. This ether radical reacts with the vinylogous imine formed from the indole and K2CO3 (see scheme above Table 4). The ensuing nitrogen-centered radical is ultimately converted to the product.
C.3. Formation of C–N bonds with heteroaromatic compounds
Several reports have appeared on the oxidative-amination reactions of ethers with heteroaromatic compounds, resulting in the formation of a C–N bond. Products from these reactions are hemiaminal ethers. This area involves a serendipitous result involving Rh-catalysis and oxidative amination reactions with Fe, Cu and Ru catalysts. In addition, reactions not involving metals, such as via the use of catalytic n-Bu4N+I−, and uncatalyzed processes have also emerged recently. Related to the reactions of ethers with heteroaryls, recently, uncatalyzed reactions of alcohols with heteroaryls have also been reported, also as a cascade process. All of these are described in the following topics.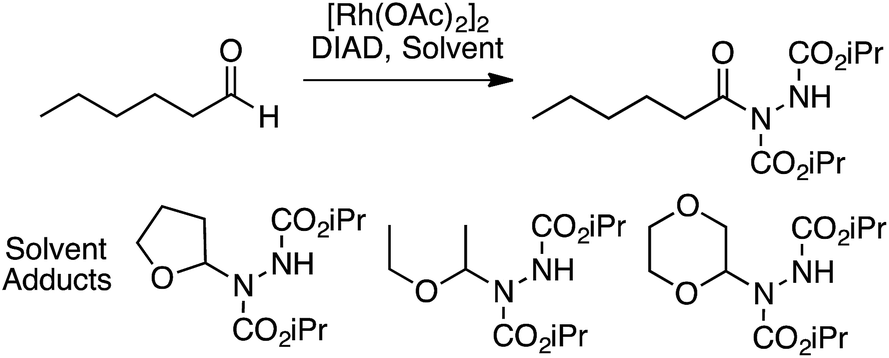 | ||
| Scheme 44 Rh-Mediated reaction of ethers with diisopropyl azodicarboxylate. | ||
C.3.b.i. Fe-Catalyzed oxidative aminations of imidazoles and benzimidazoles. Generally easy to use, earth abundant, Fe-based catalysts are obvious choices as catalytic systems. In 2010, a series of Fe salts were screened, in combination with t-BuOOH for effecting C–N bond-forming reactions between imidazole and THF.59 From among FeCl2, FeBr2, FeCl3, Fe(OAc)2, Fe(CO)9, Fe(acac)3 and FeCl3·6H2O, 2.5 mol% of FeCl3·6H2O and 3 equiv. of t-BuOOH gave high product yields in DCE at 80 °C. Subsequently, four different conditions were used for the reactions of several imidazoles, one pyrazole and 1,2,4-triazole with THF (conditions 1: 10 equiv. of THF, 3 equiv. of t-BuOOH, DCE, 80 °C; conditions 2: 5 equiv. of t-BuOOH, 4 Å MS, EtOAc, 80 °C; conditions 3: THF as solvent, 3 equiv. of t-BuOOH, 80 °C; conditions 4: THF as solvent, 5 equiv. of t-BuOOH, 4 Å MS, 80 °C). These conditions were then compared and the data are shown in Fig. 29 (although the communication reports NMR yields as well, only isolated yields are shown as this is more relevant to synthetic utilities).
 | ||
| Fig. 29 Fe-Catalyzed reactions of imidazoles, benzimidazole, pyrazole and 1,2,4-triazole with THF. Conditions 1, 2 and 4 are shown in parentheses along with yields. | ||
Next, benzimidazoles were studied in generality evaluations with other ethers. Reactions of imidazoles, a pyrazole and 1,2,4-triazole with THF were evaluated but not with other ethers. Reactions of benzimidazole were conducted with 10 equiv. of the ether, 5 equiv. of t-BuOOH and 4 Å MS, in EtOAc as the solvent, at 80 °C. The only exception was with 2-methyl THF, where it was used as the solvent. Results from the reactions of symmetrical and unsymmetrical ethers are shown in Fig. 30.
 | ||
| Fig. 30 Products from oxidative-amination reactions of benzimidazole and ethers. BIm = 1H-benzimidazol-1-yl. | ||
Good product yields were observed in most cases (Fig. 30 only lists the isolated yields and not the NMR yields). With 2-methyl THF exclusive regioselectivity was observed at the less-hindered carbon (erroneously termed regiospecificity), with the formation of diastereomers in a 1:1 ratio. With unsymmetrical butyl ethyl ether and 1,2-diethoxyethane, regioisomeric products were formed in 1:1 ratios. An interesting electronic effect comes to light in the reaction of 2-ethoxyethyl acetate. Here, exclusive formation of a single product was observed. When comparing this to the reaction of 1,2-diethoxyethane, the acetoxy group prevents reaction at the carbon atoms of the ethylene glycol moiety.
Because imidazolium compounds are highly important as both carbene precursors as well as ionic liquids, the Fe-catalyzed synthesis of three imidazolium salts was investigated. The product arising from the reaction of THF with imidazole was alkylated with MeI, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2Br and PhCH2Br. Only NMR yields are reported and they are 98, 96 and 95%, respectively.
CHCH2Br and PhCH2Br. Only NMR yields are reported and they are 98, 96 and 95%, respectively.
A competitive reaction of THF with imidazole, 3,5-dimethylpyrazole and 1,2,4-triazole was conducted in order to gain insight into the relationship of azole nucleophilicities and their pKa values. Three C–N products were isolated in a 1:0.77:0.8 ratio, leading the authors to conclude that conjugate base nucleophilicity rather than azole acidity correlates with azole reactivity. As a repeating theme, the addition of TEMPO completely suppressed product formation, and a competitive reaction of THF and THF-d8 with imidazole gave a kH/kD value of 4.0:1, indicating again that ether C–H bond scission is likely involved in the rate-limiting step.
On the basis of Gif chemistry,60 it is plausible to infer that FeIII is converted to FeIIvia reaction with t-BuOOH, releasing t-BuOO˙. Either this species or t-BuO˙ formed in the reoxidation of FeII to FeIII with the peroxy radical can cause hydrogen atom abstraction from the ether. Reduction of the FeIII intermediate with the newly formed ether-based radical will then result in an oxocarbenium ion and the reformation of FeII (Scheme 45).
 | ||
| Scheme 45 A potential reaction mechanism involving an FeIII–FeII redox cycle. | ||
C.3.b.ii. Fe-Catalyzed oxidative aminations of benzimidazoles and benzotriazoles. Complementary FeCl3-centric CDC reactions of diarylmethanes (not described in this review) with benzimidazole and benzotriazole were reported in 2012.61 DDQ, Ag2CO3, O2, (t-BuO)2, (PhCOO)2 and t-BuOOH were analyzed as oxidants, in solvents such as DCE, DMSO, PhCl, MeCN and PhNO2. The best reaction occurred with 20 mol% of FeCl3 in neat t-BuOOH at 100 °C. Reactions of 1,4-dioxane and THF with benzimidazole, 2-methylbenzimidazole and benzotriazole are reported, using 20 mol% of FeCl3 in 5 equiv. of t-BuOOH at 100 °C. Products from these reactions are shown in Fig. 31. A mechanism shown in Scheme 45 can be envisioned here as well because this reaction involves FeCl3/t-BuOOH. A slightly better product yield was observed in the reaction of benzimidazole and THF in section C.3.b.i.59
 | ||
| Fig. 31 Reactions of benzimidazole, 2-methylbenzimidazole and benzotriazole with 1,4-dioxane and THF. | ||
C.3.b.iii. Fe-Catalyzed oxidative aminations of 1,2,3,4-tetrazoles. Along similar lines to the Fe-catalyzed processes described in the preceding two sections, in 2015 5-aryltetrazoles were subjected to oxidative-amination reactions with ethers.62 Initial tests of conditions were performed with 5-phenyltetrazole and THF. FeCl3·6H2O, FeCl3, FeBr3, Fe(acac)3, FeCl2 and Fe(OAc)2 were tested in various combinations with t-BuOOH as well as one reaction each with FeCl3·6H2O and H2O2 or (t-BuO)2. The optimal combination was 5 mol% of FeCl3·6H2O and 5 equiv. of t-BuOOH, at 90 °C in EtOAc. While modest to good product yields were obtained in reactions of several 5-aryltetrazoles and THF, reactions with precursors containing a nitro group and a phenolic hydroxyl were not productive. Results from these reactions are shown in Fig. 32.
 | ||
| Fig. 32 FeCl3·6H2O/t-BuOOH mediated reactions of 5-aryltetrazoles with THF. | ||
Next, 5-phenyl-1H-tetrazole was reacted with a series of ethers (Fig. 33). In these, reactions with THF, THP, 1,4-dioxane, 1,2-DME and di-n-butyl ether proceeded as anticipated. However, some unusual products were obtained with others. With 1,3-dioxolane, the desired product was not observed, but rather the N-t-butyl and ethoxy methyl products were obtained. The N-t-butyl tetrazole was also obtained with Et2O, and with 3,4-dihydro-2H-pyran the 1,2-addition product, which is identical to that obtained from the reaction of THP, was obtained.
 | ||
| Fig. 33 Reactions of 5-phenyl-1H-tetrazole with other ethers (EtOAc was used as the reaction solvent except with 1,4-dioxane, where DCE was used). | ||
As with all previous reactions and, specifically, the Fe/t-BuOOH mediated reactions described previously, TEMPO inhibited formation of the product in a reaction of 5-phenyl-1H-tetrazole and THF. Also, competitive reaction of this tetrazole with THF and THF-d8 gave a kH/kD value of 2.7:1 (compared to the value of 4.0 obtained in the reaction of imidazole in C.3.b.i.).59
C.3.c.i. Cu-Catalyzed oxidative aminations of indoles. In sections C.2.b. and C.2.c. CDC reactions leading to C–C bond formation at the C2 and C3 positions of indole have been discussed.54–56 In 2015,63 Cu-catalyzed C–N bond formation between indole and ethers, was disclosed. This study also included reactions of carbazoles, which is in effect a benzo-annulated indole.
Optimizations were carried out with methyl 1H-indole-3-carboxylate and THF using a variety of Cu salts such as CuCl2, CuBr2, Cu(OAc)2, CuBr, CuO and CuCl2 in combination with pyridine-based or amine ligands, and (t-BuO)2 at 140 °C. From these, the combination of 10 mol% each of CuCl2 and 2,2′-bipyridine (bipy) and 3 equiv. of (t-BuO)2, at 140 °C provided efficient reaction, and this combination was used for analysis of the reaction scope (Scheme 46).
 | ||
| Scheme 46 CuCl2/(t-BuO)2 mediated C–N bond formation of indoles and a benzimidazole. | ||
A variety of indoles with electron-withdrawing substituents at the 3-position reacted with generally good yields. The placement of ester substituents at the 2-position resulted in significantly lower yields, just as in the uncatalyzed CDC reactions of indoles leading to C–C bond formation, described in C.2.c.ii.56 A formyl group was tolerated but gave a poor yield, 7-azaindole also reacted successfully, albeit modestly, and 2-methylbenzimidazole gave a good product yield. Notably, the use of air gave a slightly higher product yield as compared to a nitrogen atmosphere, as exemplified in the reaction of methyl 1H-indole-3-carboxylate with THF (95% under an air atmosphere versus 87% under a nitrogen atmosphere).
The same reaction conditions were utilized for reactions of carbazoles with THF. While generally good to excellent product yields were obtained, there is possibly a steric effect because 2-methylcarbazole gave the lowest (43%) yield of product (Fig. 34).
 | ||
| Fig. 34 Reactions of carbazoles with THF. | ||
While the preceding reactions involved only THF, a series of ethers were then tested for reactivity with methyl 1H-indole-3-carboxylate, ethyl 1H-indole-2-carboxylate and carbazole. Generally, the yields ranged from low to good (data in Fig. 35).
 | ||
| Fig. 35 Oxidative-amination reactions of two indoles and two carbazoles with ethers. | ||
In mechanistic investigations, the reaction of carbazole with THF was almost completely suppressed upon addition of TEMPO, resulting in only a trace of product and a reasonable yield of the THF–TEMPO adduct (30%). In a competitive reaction of THF/THF-d8 a KIE value of 4.0 was observed, indicating that the reactions are potentially radical processes and that cleavage of the C–H bond in the ether is likely involved in the rate-limiting step. Copper catalysis, thus, offers complementary reactivity to Ni(acac)2/Zn(OTf)2 and NiF2/PPh3, as well as uncatalyzed CDC reactions of indoles.54–56
C.3.c.ii. Cu-Catalyzed reactions of isoquinolines leading to N-benzylisoquinolin-1(2H)-ones. The Minisci reactions described in C.1.f. involve C–C bond-forming reactions of quinolines and isoquinolines with ethers and alcohols. Recently, an interesting transformation of isoquinolines leading to N-benzylisoquinolin-1(2H)-ones was demonstrated (Scheme 47).64 For this, 20 mol% of Cu0, CuI (CuOAc, CuSCN, CuI, CuCl, CuBr and Cu2O) and CuBr2 were tested in combination with 2 equiv. of (t-BuO)2 at 120 °C. Among these, only CuBr gave a yield >20%, all of the others gave trace to 17% of product. Other oxidants, t-BuOOH, K2S2O8 and dicumyl peroxide, were evaluated in combination with CuBr, but in each case only traces of the product were observed. An increase in the amount of either CuBr or (t-BuO)2 led to yield improvements and, finally, with 1 equiv. CuBr and 4 equiv. of (t-BuO)2 at 100 °C a good 78% yield of N-benzylisoquinolin-1-(2H)-one was attained (Scheme 47).
 | ||
| Scheme 47 Synthesis of N-benzylisoquinolin-1(2H)-one from isoquinoline and toluene. | ||
A variety of substituted toluenes were reacted with isoquinoline (7 examples), and toluene was reacted with 4-, 5- and 6-bromoisoquinolines, 8-chloroisoquinoline, phthalazine and quinoline. In each case the corresponding N-alkyl amide product was obtained (yields ranged from 37–91%). Notably, in a reaction of isoquinoline with p-methylanisole two products were obtained: one from reaction at the benzylic methyl group and the other from reaction at the methoxy carbon (Scheme 48).
 | ||
| Scheme 48 The two products from the reaction of isoquinoline with p-methylanisole. | ||
On the basis of the foregoing, and of particular relevance to this perspective, five other ethers were tested, and these data are shown in Fig. 36. Reactions with anisole, 1,3-dimethoxybenzene, p-bromoanisole and 1,4-dioxane proceeded well. Reaction with 1,2-DME gave, as anticipated, products from reaction at both the 1° and 2° carbon atoms, and reaction at the 2° carbon atom led to the major isomer.
 | ||
| Fig. 36 Products from the reactions of other ethers with isoquinoline. | ||
When reactions of o- and p-bromoanisole were conducted at 120 °C, 2-(t-butoxymethyl)isoquinolin-1(2H)-one (shown in the dotted box) was obtained. With o-bromoanisole, this compound was 25% of the products formed whereas with p-bromoanisole it was 50% of the products formed. Formation of this product has been termed “competing exchange reaction”64 by the authors.
This methodology has been extended to the synthesis of natural product scaffolds (Scheme 49). The reaction of isoquinoline with o-bromoanisole using CuBr and (t-BuO)2 gave the corresponding N-(2-bromobenzyl)isoquinolin-1(2H)-one. This could be converted, either through Pd-mediated C–C bond formation or through a radical reaction, to the two scaffolds shown in Scheme 49.
 | ||
| Scheme 49 Utility of the N-benzylation reaction for access to natural product structures. | ||
In order to understand the mechanism of this transformation, a reaction of benzyl bromide and isoquinoline was conducted under these reaction conditions. Only a trace of N-benzylisoquinol-1(2H)-one was formed. On the other hand, a reaction of toluene and isoquinoline, that gave 78% product yield, was fully suppressed in the presence of BHT. This indicates that the reactions are likely radical processes. The use of ESI/MS (positive mode) to monitor the reaction indicated three cationic intermediates, as shown in Fig. 37.
 | ||
| Fig. 37 Cationic intermediates, identified using ESI-MS, in the reaction of isoquinoline with toluene. | ||
On the basis of these observations, it is plausible to deduce that the oxygen atom of the amide is obtained from (t-BuO)2. Thus, a mechanism for the reaction of isoquinoline with toluene has been proposed. This is adapted in Scheme 50 for the reaction of isoquinoline with anisole. One point for consideration is that the CuII/CuI redox cycling could occur when PhOCH2˙ is formed, leading to an oxocarbenium ion. This could then alkylate the nitrogen atom. The rest of the pathway could then involve ionic intermediates.
 | ||
| Scheme 50 Plausible mechanism for the reaction of isoquinoline with anisole. | ||
In the course of investigating N-directed C–H bond activations of nucleosides, we discovered that 1H-benzotriazol-1-yl 4-methylbenzenesulfonate (BtOTs) underwent Ru-catalyzed C–H bond activation and C–N bond formation with NMP, giving a 2.1:1 ratio of isomeric N1 and N2 alkylated benzotriazoles, in a good 69% yield (Scheme 51). The use of 1H-benzotriazole (BtH) gave the same two products but in a lower 33% yield, and in a 5.6:1 ratio. This led to the screening of several Ru species and conditions for effecting the reaction between NMP and BtH (Scheme 51). The use of RuCl3·3H2O and t-BuOOH in nonane, in DCE at 80 °C, gave a 66% yield of the N1 and N2 alkylation products in a 4.1:1 ratio.
 | ||
| Scheme 51 Reactions of BtOTs and BtH with NMP. | ||
These conditions were then tested for reactions of BtH and 5,6-dimethyl BtH with NMP and ethers (4 Å MS were used in the reactions of ethers on the basis of an incomplete reaction with THF that was observed in the absence of sieves). The results of these experiments are collated in Table 5. In every case N1 and N2 products were formed and these were readily separated using chromatography on silica gel, with the N1 isomers generally being more polar. The ratio of the two isomeric products varied and more about this follows later. When comparing the reactions of benzotriazole in this Ru-catalyzed and the previously described Fe-catalyzed reactions (C.3.b.ii. and Fig. 31), the former produced a better yield in a reaction with THF. With 1,4-dioxane, however, a better yield was obtained under the Fe-catalyzed conditions, although both were around the 50% mark.
| Entry | Azole | Reactant | Time | Yield | N1 isomer | N2 isomer | N1/N2 ratio |
|---|---|---|---|---|---|---|---|
| a Reactions were performed with 0.5 M BtH or 5,6-dimethyl BtH in DCE, with 5 mol% of Ru catalyst, 10 equiv. of NMP or ether and 3 equiv. of t-BuOOH in nonane, at 80 °C. Reactions with ethers were conducted with 4 Å MS (0.2 g). b Reaction remained incomplete and yields are not based upon the recovered benzotriazole. c Reaction temperature was 110–120 °C. d Data for the second entry was obtained with 20 mol% of the Ru catalyst. | |||||||
| 1 |

|
7 h | 66% | 53% | 13% | 4.1:1 |
|
| 2 |
|
6 h | 84% | 64% | 20% | 3.2:1 |
|
| 3b |
|

|
20 h | 65% | 57% | 8% | 7.1:1 |
| 4b,c |

|
24 h | 46% | 29% | 17% | 1.7:1 |
|
| 5b,c,d | 24 h | 45% | 27% | 18% | 1.5:1 |
||
| 6 |

|
16 h | 73% | 46% | 27% | 1.7:1 |
|
| 7 |
|
8 h | 57% | 53% | 4% | 13.2:1 |
|
| 8 |

|
7 h | 75% | 55% | 20% | 2.7:1 |
|
| 9 |

|
6 h | 68% | 47% | 21% | 2.2:1 |
|
| 10 |
|

|
24 h | 46% | 31% | 15% | 7.1:1 |
| 11b,c |

|
24 h | 30% | 23% | 7% | 3.3:1 |
|
| 12 |

|
3.5 h | 68% | 40% | 28% | 1.4:1 |
|
| 13 |

|
8 h | 62% | 59% | 3% | 19.7:1 |
|
The conditions were also applicable to reactions of THF with benzimidazole, imidazole and 1,2,3- as well as 1,2,4-triazoles (Fig. 38). Reaction with benzimidazole gave a high yield, in the same range as that obtained in the two FeCl3·6H2O catalyzed reactions.59,61 The reaction of imidazole and 1,2,4-triazole under Fe-catalysis gave better yields.59 With 1,2,4-triazole, in both methodologies only the N1 isomer was detected. 1,2,3-Triazole also reacted via the N1 atom although a trace amount of the N2 isomer could have been produced (as observed using HRMS analysis). See sections C.3.b.i. and C.3.b.ii., Fig. 29 and 31 for comparisons.
 | ||
| Fig. 38 Reactions of other azoles with THF under the Ru-catalyzed conditions. | ||
Major differences from other methods were noted and novel observations were made in the mechanistic studies of this Ru-catalyzed process. In a reaction of BtH and THF, the addition of 3 equiv. of TEMPO produced the N1 and N2 hemiaminals in 48% and 24% yield, respectively. Although this appears to be a suppression of the reaction, two other relatively elusive products, shown in Fig. 39, were obtained in 15% yield, bringing the total yield of all products to 87% (compared to the 84% in entry 2 of Table 5). Relative stereochemical assignments were made on the basis of NMR experiments. The formation of such products offers a cautionary note. That is, suppression (rather than prevention) of product formation may not necessarily indicate capture of the radical species. The diversion of products to other materials that could escape identification could occur.
 | ||
| Fig. 39 Unusual products formed in the radical-trapping experiment with TEMPO. | ||
Several control experiments were conducted, and it was shown that: (a) THF and TEMPO react in the presence of t-BuOOH, (b) in the absence of the Ru catalyst, BtH and THF react to give product, although the reaction remains incomplete, (c) TEMPO prevents the reaction of BtH with THF in the absence of the Ru catalyst (note: this is a departure from the lack of inhibition by TEMPO in the presence of the catalyst, described earlier) and (d) a reaction of BtH, THF and TEMPO in the presence of the Ru catalyst, but in the absence of t-BuOOH also gave no product. These results indicate that a THF radical is produced through reaction with the peroxide, and in the absence of the metal the radical is inefficiently captured by BtH.
However, in the presence of the metal an alternate course of events unfolds, where either a long-lived radical is not produced or the rapid formation of an oxocarbenium ion occurs. Peroxide is needed for the reaction, plausibly to oxidize the metal. Finally, reaction of BtH with 1:1 THF/THF-d8 gave a KIE of 5.2–6.1 as assessed using NMR and HRMS measurements on both the N1 and N2 alkylated products. This KIE value is larger that those obtained in the FeCl3 catalyzed reaction of imidazole (4.0, C.3.b.i.)59 and the Cu-catalyzed reaction of carbazole (4.0:1, C.3.c.i.).63 Thus, in all these processes breakage of a C–H bond in the ether may be involved in the rate-limiting step. A plausible mechanism for the Ru-catalyzed reaction is shown in Scheme 52.
 | ||
| Scheme 52 A plausible mechanism for the Ru-catalyzed reaction. | ||
Perhaps the most important observation, not made prior to this study, is that the N2-alkylated benzotriazoles undergo rearrangement to the N1-isomers. This was first observed when a product obtained from isochroman was reanalyzed after 6 months at 2–5 °C. In this case, the pure N2-alkylated benzotriazole was completely isomerized to the N1 product. While the THF-derived product was relatively stable upon storage, the fact that this too can undergo rearrangement was proven through an NMR tube experiment, where heating the N2 isomer as a neat material caused almost complete isomerization to the N1 product.
This isomerization most likely occurs through disconnection of the C–N bond of the hemiaminal ether, producing an oxocarbenium ion, followed by reattachment of the azole via the N1 atom (Scheme 53). The fact that the isochroman product isomerized quite easily is consistent with the fact that it can produce a more stabilized oxocarbenium ion than the THF product.
 | ||
| Scheme 53 Isomerization of the N2-alkylated benzotriazoles to the N1 isomers, and a possible mechanism. | ||
C.3.e.i. Reactions of phthalimide. Investigation of PhI(OAc)2 with or without I2, m-CPBA/PhI or H2O2 with either PhI or n-Bu4N+I−, and t-BuOOH either independently or in combination with FeCl3·6H2O or CuBr, all proved to be ineffective for the reaction of phthalimide with THF at 75 °C (sealed tube).66 On the other hand, n-Bu4N+F− and anhydrous t-BuOOH gave good reaction progress with the best outcome occurring with 5 equiv. of the peroxide and 20 mol% of the ammonium salt. Phthalimides, succinimide and benzotriazole were tested and these data are shown in Scheme 54. Also tested under the conditions were N-benzoylbenzamide and p-toluenesulfonamide (data not shown), giving moderate product yields of 59% and 42%, respectively.
 | ||
| Scheme 54 Reactions of THF with phthalimides, succinimide and benzotriazole. | ||
While most of the phthalimides and succinimide reacted well, the nitro substituent proved to be problematic, and not only was the reaction slow but also the product yield was low. Notably, a good yield was obtained with benzotriazole. In comparison with the Fe- and Ru-catalyzed reactions of BtH with THF (see discussions in sections C.3.b.ii. and C.3.d., Fig. 31 and Table 5), this yield was comparable to the Ru-catalyzed process.59,65 In contrast to the Fe-mediated reaction,59 where only the N1 isomer was reported, this result is similar to the Ru-mediated chemistry,65 and the N1 to N2 alkylation ratios in the two methods are also comparable (3.2:1 in the Ru-based chemistry and 3.4:1 here).
Other ethers were tested for reactivity with phthalimide and these data are summarized in Fig. 40. With THP and 1,3-dioxolane the product yields were modest to good, whereas better yields were obtained with the aliphatic ethers (an excellent yield was observed with Et2O). With 2-methyl THF, regioisomeric products were obtained, and the reaction favored the 2° center over the 3° carbon atom. With 1,2-DME, the preference is for reaction at the 2° center over the 1° carbon atom. The reaction with MTBE presented an interesting scenario. Two products were observed in this case and, notably, the hemiaminal was formed to a slightly greater extent and was isolable!
 | ||
| Fig. 40 Products from the reactions of phthalimide with other ethers. Phthal = phthalimido. | ||
As anticipated, the proposed mechanism (shown in Scheme 55) involves a radical species formed from the ether. However, the radical inhibition experiment was conducted with 1 equiv. each of phthalimide and TEMPO, resulting in a 92% product yield and 88% yield of the THF–TEMPO adduct. A KIE value of 15.1 was obtained from the reaction of phthalimide with THF/THF-d8, which is the largest observed in these types of reactions.
 | ||
| Scheme 55 Hypoiodite or iodite intermediates formed in n-Bu4N+I−-catalyzed reactions of ethers with imides. | ||
Mechanistically, the alkylammonium iodide can be oxidized to a hypoiodite (I+) or iodite (I3+). Hydrogen atom abstraction from THF results in a radical that can be further oxidized to an oxocarbenium ion, with loss of hydroxide. Reoxidation of iodide occurs with the peroxide, and the released hydroxide can cause deprotonation of the imide. Product formation occurs through capture of the oxocarbenium ion by the imide anion (Scheme 55 shows a truncated catalytic cycle).
Because the n-Bu4N+I− system has been used in the other amination reactions described below, the mechanistic considerations of Scheme 55 will not be repeated in each case.
C.3.e.ii. Reactions of saccharin. n-Bu4N+I− (10 mol%) in combination with 70% t-BuOOH in water (4 equiv.) at 120 °C was effective for the reaction of saccharin with a number of anisole derivatives.67 In initial screening experiments with anisole, the use of (t-BuO)2 and H2O2 as oxidizing agents also gave conversions, albeit in low yields. However, K2S2O8 was ineffective and no reaction occurred with t-BuOOH in the absence of n-Bu4N+I−. Interestingly, a positive reaction was also observed with n-Bu4N+AcO−, n-Bu4N+Br− and n-Bu4N+Cl−. In fact, the ammonium chloride gave a good 79% product yield, but this is bettered by the iodide. The several products synthesized are summarized in Fig. 41. The reaction progressed well with several anisole derivatives. Also interestingly, with p-methylanisole the reaction occurred at the methyl group rather than at the methoxy carbon. Thioanisole also reacted well but 1-methoxynaphthalene was unreactive. Therefore, a question is whether this chemistry is only applicable to monoaryl systems.
 | ||
| Fig. 41 Products derived from saccharin and other isothiazolone 1,1-dioxides. | ||
These conditions were also applied to the reactions of cyclic and acyclic ethers. The products and yields are shown in Fig. 42. With 1,2-DME, a slightly greater preference for reaction at the 2° carbon atom was observed over that for the 1° center. This reactivity is comparable to the reaction of phthalimide with 1,2-DME (see C.3.e.i.).66
 | ||
| Fig. 42 Products from the reactions of saccharin with other ethers and yields. Sacc = saccharin. | ||
As can be anticipated this is a radical process, with complete inhibition of reactivity in the presence of TEMPO and BHT. With the latter radical trap, an 86% yield of a product from the reaction of saccharin with BHT was obtained (Fig. 43). A competitive reaction of THF/THF-d8 and saccharin returned a KIE value of 4.0, which contrasts to the value of 15.1 observed for the reaction of phthalimide (C.3.e.i.).66
 | ||
| Fig. 43 A saccharin–BHT adduct obtained in a radical inhibition experiment. | ||
C.3.e.iii. Reactions of tetrazoles, benzimidazoles, benzotriazole, triazoles and pyrazoles. The chemistry described in this section is complementary to the FeCl3·6H2O/t-BuOOH mediated reactions of 1,2,3,4-tetrazoles described in C.3.b.iii. (also see Fig. 32).62 In a preliminary screening exercise, the reaction of 1,4-dioxane and 5-phenyl-2H-tetrazole with both CuBr and notably 20 mol% of FeCl3·6H2O in combination with 5 equiv. of t-BuOOH at 80 °C, in a sealed tube, gave trace amounts of the product.68 As described in Section C.3.b.iii., 5 mol% of FeCl3·6H2O and 5 equiv. of t-BuOOH in EtOAc at 90 °C gave a successful reaction between the same two reactants.62 Therefore, there appear to be subtle factors at play in these reactions.
In the present case,68 H2O2, O2, NaOCl, K2S2O8 or oxone in combination with n-Bu4N+I− were ineffective. Also, the use of other tetrabutylammonium salts (chloride, bromide), NaI and I2 gave only traces of product. The use of 20 mol% of n-Bu4N+I− and 5 equiv. of t-BuOOH (70% aqueous solution) at 80 °C, in a sealed tube, gave good conversion to the hemiaminal ether. Absence of the ammonium salt resulted in no product. Interestingly, an O2 atmosphere lowered the yield in comparison to an inert nitrogen atmosphere, and the latter was comparable to air. Using these conditions several 5-aryl-2H-tetrazoles were reacted with 1,4-dioxane (Fig. 44). The yield obtained from 5-phenyl-2H-tetrazole via this protocol was lower than that with FeCl3·6H2O/t-BuOOH in EtOAc.62 A phenolic hydroxyl and a 2-furanyl group were incompatible in this procedure.
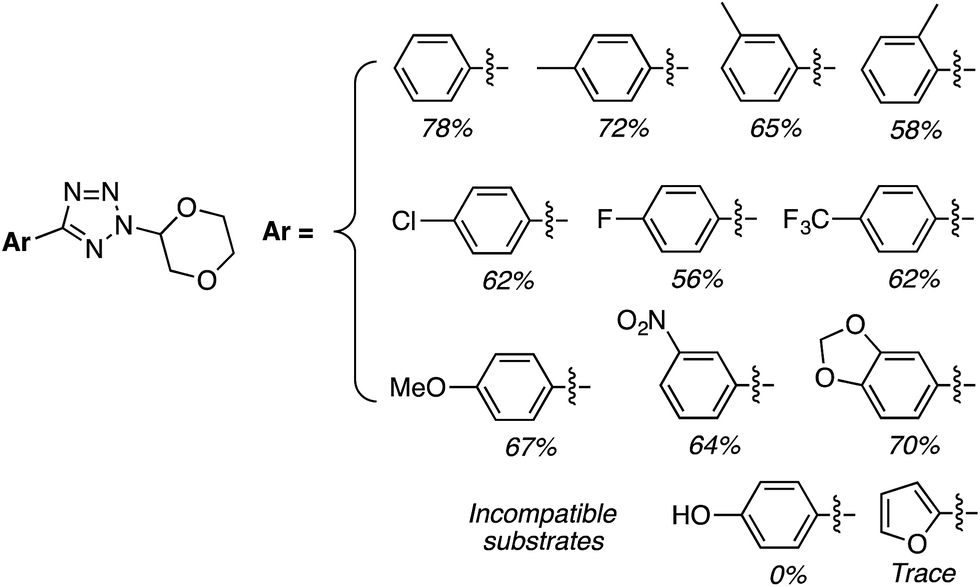 | ||
| Fig. 44 Products obtained in reactions of 5-aryl-2H-tetrazoles with 1,4-dioxane. | ||
Other alkyl ethers were evaluated in reactions with 5-phenyl-2H-tetrazole. There are notable comparisons to be made to the FeCl3·6H2O/t-BuOOH reactions (C.3.b.iii.).62 Where comparisons are possible, Fig. 45 shows yields from both protocols, where the yields in parentheses are from the Fe-catalyzed process.
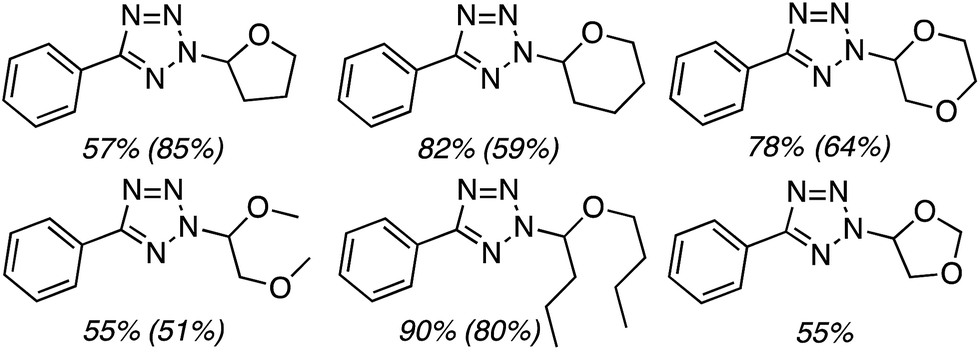 | ||
| Fig. 45 Products from the reactions of 5-phenyl-2H-tetrazole with other ethers. | ||
In this procedure, better yields were obtained with THP, 1,4-dioxane and di-n-butyl ether, but THF gave a lower product yield. 1,2-DME gave a comparable yield and showed preference for reaction at the 2° C atom. Interestingly, 1,3-dioxolane, which gave unusual products in the Fe-catalyzed process, gave a reasonable yield of the expected product via this protocol. Et2O gave no product here, whereas an N-t-Bu product was obtained with the Fe-catalyst. Benzyl methyl ether was not compatible with these reaction conditions.
Because 2-azido-1,4-dioxane is not available for CuAAC reactions with alkynes,69 six 4-aryl-NH-1,2,3-triazoles were investigated in reactions with 1,4-dioxane (Fig. 46). These were generally modest yielding, with none exceeding the 50% mark.
 | ||
| Fig. 46 Products from the reactions of 4-aryl-1,2,3-triazoles with 1,4-dioxane. | ||
The reaction of 5-phenyl-2H-tetrazole with dioxane/dioxane-d8 gave a kH/kD value of 12.8. This large value, though smaller than that reported for reactions of saccharin,67 (KIE 15.1, C.3.e.ii.), is greater than the KIE value of 2.7 observed in the FeCl3·6H2O/t-BuOOH procedure (C.3.b.iii.).62 This led to the proposal that C–H bond cleavage is involved in the rate-limiting step. Mechanistically, this reaction is likely to proceed via a process similar to that shown in Scheme 55, where hypoiodite or iodite can be formed through the action of the peroxide on n-Bu4N+I−. These species then result in a radical intermediate from the ether, further oxidation of which by the hypoiodite/iodite can lead to an oxocarbenium ion that is finally trapped by the tetrazole, possibly with the participation of a released hydroxide ion.
The reactions of ethers, and predominantly of anisole derivatives, were also investigated using the catalytic n-Bu4N+I−/peroxide system.70 Optimization reactions were conducted using anisole and 5-phenyl-2H-tetrazole at 80–100 °C, with t-BuOOH in decane or in water, aqueous H2O2, K2S2O8, (t-BuO)2, (PhCOO)2, t-BuOOCOPh or PhI(OAc)2. t-BuOOH in decane gave a low product yield and the others gave no detectable product. n-Bu4N+Br−, n-Bu4N+Cl−, n-Bu4N+F−, KI and I2 were evaluated in place of n-Bu4N+I−, but no to low conversions were observed. A good product yield was observed with 10 mol% of n-Bu4N+I− and 3 equiv. of t-BuOOH in decane at 90 °C, and the reaction did not proceed in the absence of either the iodide salt or the peroxide. These reactions were conducted neat in the ethers, which were either liquids or low-melting solids. Anisole and its derivatives gave good product yields (Scheme 56). Also, p-methoxyphenetole and p-propoxyanisole underwent good conversions to their respective products.
 | ||
| Scheme 56 Reactions of anisole and its derivatives with 5-phenyl-2H-tetrazole. | ||
The reaction also proceeded well with 2,3-dihydro-1,4-benzodioxene and dibenz[b,e]oxepin-11(6H)-one (Fig. 47). When challenged with a 1° versus 2° carbon center, the reaction occurred predominantly at the latter (Fig. 47). As may be anticipated, (2-ethoxyethoxy)benzene gave a lower selectivity as compared with (2-methoxyethoxy)benzene because of both greater radical and oxocarbenium ion stabilization at the 2° center. A notable exception was the reactivity of 4-methylanisole, where the reaction occurred at the benzylic methyl group rather than at the methoxy carbon atom.
 | ||
| Fig. 47 Reactions of other ethers and an unusual reaction of 4-methylanisole. | ||
At the next stage, 11 other reactions were conducted: 8 involving anisole and different 5-aryl-2H-tetrazoles, 1 with p-bromoanisole and 2 with 1H-benzotriazole and anisole as well as p-bromoanisole. These results are shown in Fig. 48. Notably, the use of 5-(4-methylphenyl)-2H-tetrazole led to not just the CDC product but also to the concomitant oxidation of the benzylic methylene to the carbonyl (in the grey oval). With benzotriazole, 4 equiv. of aqueous t-BuOOH and a higher temperature of 100 °C were necessary to obtain the indicated yields.
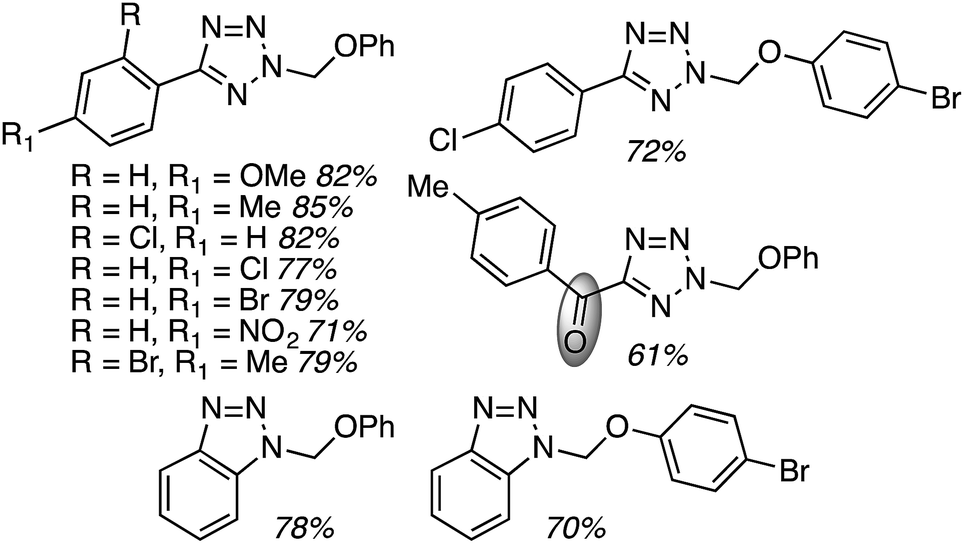 | ||
| Fig. 48 Reactions of other 5-aryl-2H-tetrazoles and 1H-benzotriazole. | ||
A detailed mechanistic analysis is not described here because this reaction also parallels that of phthalimide66 shown in Scheme 55, and that in the immediately preceding section.68 In this study, the use of TEMPO in the reaction of anisole with 5-phenyl-2H-tetrazole suppressed the reaction greatly and a TEMPO–anisole adduct was detected using HRMS. Although the products here and those in the immediately preceding section67 have the same substitution pattern around the tetrazole, in the present work one compound was analyzed using X-ray crystallography to confirm the structure.70
The use of n-Bu4N+I− was studied for oxidative-amination reactions of benzimidazoles, benzotriazole, triazoles and pyrazoles.71 In initial tests on the reactivity of benzimidazole and THF, combinations of 70% aqueous solution of t-BuOOH and several iodides (catalytic n-Bu4N+I−, stoichiometric NaI and stoichiometric KI) as well as stoichiometric iodine were evaluated. DCE, EtOAc and THF itself were tested as solvents, at temperatures ranging from room temperature to 80 °C. In every case, product formation was observed in yields ranging from 50 to 72%. Only when peroxide was absent was no product formed.
The optimized conditions (10 mol% of n-Bu4N+I− and 3.5 equiv. of 70% aqueous t-BuOOH in DCE at 80 °C) were then applied to the reactions of benzimidazole, 5,6-dimethylbenzimidazole and purine (Fig. 49). Modest to high yields were obtained in the reaction of THF, 1,4-dioxane, Et2O, thiolane and 1,3-dithiolane, in reaction times of 5–15 h. 5,6-Dimethylbenzimidazole gave higher product yields than benzimidazole. 2-Methyl THF is stated to give a “mixture”71 for which details have not been described. Methyl neopentyl ether and diisopropyl ether did not yield products derived from benzimidazole. Similarly, indole and 7-azaindole did not yield products with THF.
 | ||
| Fig. 49 Products obtained from the reactions of benzimidazoles and purine. | ||
Other azoles were tested and these data are summarized in Table 6. In these analyses, unlike a successful outcome with benzimidazole, the reaction of BtH with 1,3-dithiolane did not return any product. The reactions of BtH and 1,2,3-triazoles gave regioisomeric mixtures of products.
| Ether | Products |
|---|---|
| a Reactions were conducted with 10 mol% of n-Bu4N+I− and 3.5 equiv. of 70% t-BuOOH, in DCE at 80 °C. Reaction times were 5–16 h. | |

|

|

|
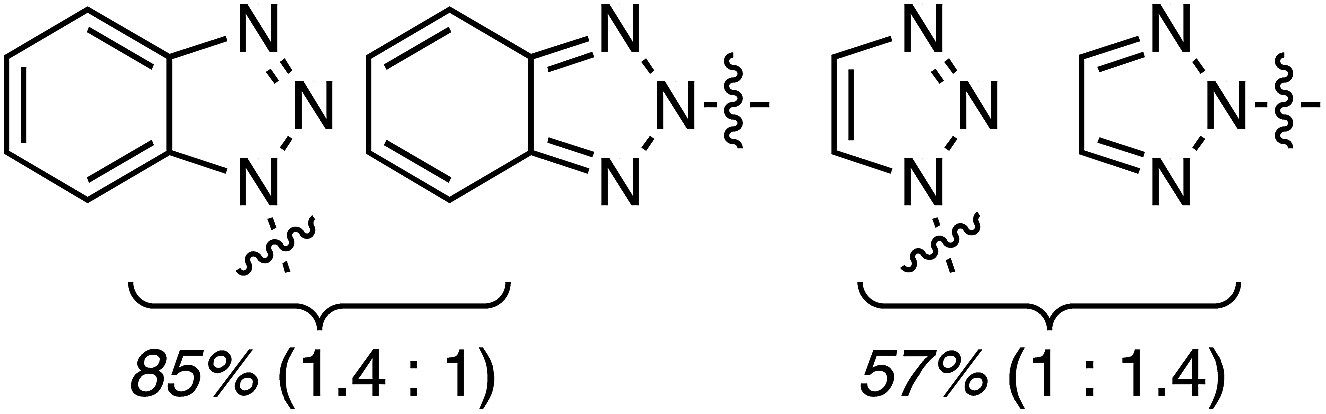
|
|

|

|

|

|

|
The reaction was postulated to proceed via a free radical mechanism because the yield of the product formed from benzimidazole and THF was reduced in the presence of TEMPO, and a TEMPO–THF adduct was formed and characterized. However, even 10 molar equiv. did not completely suppress product formation, leading the authors to propose rapid conversion of an initially formed radical to an oxocarbenium ion. Mechanistically, this transformation is related to the other examples in this section.
Because several systems have been utilized for reactions of heteroaryls with ethers, a side-by-side comparison of the outcomes is possible. These data are summarized in Table 7. While differences are to be expected, a notable point is in the reactions of benzotriazole. In the RuCl3·3H2O catalyzed reactions,65 the N1 and N2 isomers were always isolated in every case. However, with FeCl3·6H2O only the N1 isomer was isolated.61 In contrast, with n-Bu4N+I− the N1 and N2 isomers were obtained in all cases except with THP.71 In this context we note that our work65 has demonstrated the possible conversion of N2 alkyl benzotriazoles to the N1 isomers, potentially via oxocarbenium ions. We wonder, therefore, if the absence of the N2 isomer in the other literature cases is because they were not formed, were lost as minor components, or they simply isomerized at some point.
| Reaction partners | FeCl3·6H2O + t-BuOOH (ref. 59) | FeCl3·6H2O + t-BuOOH (ref. 61) | RuCl3·3H2O + t-BuOOH (ref. 65) | n-Bu4N+I− + t-BuOOH (ref. 71) |
|---|---|---|---|---|
| Benzimidazole + THF | 86–89% | 78% | 83% | 72% |
| Benzimidazole + 1,4-dioxane | — | 77% | — | 30% |
| Benzimidazole + Et2O | 93% | — | — | 55% |
| Benzimidazole + 2-Me THF | 65% 1:1 mixture of diastereomers |
— | — | Mixture, no data provided |
| Imidazole + THF | 88–90% | — | 56% | — |
| Benzotriazole + THF | — | 74% N1 isomer only | 84% N1/N2: 3.2:1 |
98% N1/N2: 2:1 |
| Benzotriazole + 1,4-dioxane | — | 55% N1 isomer only | 46% N1/N2: 1.7:1 |
85% N1/N2: 1.4:1 |
| Benzotriazole + THP | — | — | 65% N1/N2: 7.1:1 |
60% N1 isomer only |
| Benzotriazole + Et2O | — | — | 73% N1/N2: 1.7:1 |
65% N1/N2: 5.5:1 |
| 1,2,3-Triazole + THF | — | — | 57% single isomer isolated | — |
| 1,2,4-Triazole + THF | 72% single isomer | — | 61% single isomer | — |
C.3.f.i. Reactions of purines. Just as with the uncatalyzed C–C reactions with heteroaryls, comparable reactions have been studied for C–N bond formation.72,73 The purine skeleton is a highly privileged structure in medicinal chemistry and, thus, methods for its modification are always important. In the course of developing N-alkylated purine derivatives,72 the reaction of 2,6-dichloropurine with THF was investigated (Scheme 57). The use of 2 equiv. each of PhI(OAc)2 and THF in DMF, DMSO, CH2Cl2 or EtOAc at elevated temperatures gave no product. In cyclohexane and MeCN, low 10 and 20% yields, respectively, were obtained. This improved to 41% in DCE and to 45% in THF itself as a solvent. Interestingly, the use of a trace of I2 produced a substantial yield improvement (94%), which remained unaltered when the solvent was DCE (90%). A reaction in THF at room temperature did not progress and a lower 50 °C, rather than 70 °C, gave a lowered yield.
 | ||
| Scheme 57 Reaction of 2,6-dichloro-9H-purine with THF. | ||
The reaction with a trace amount of I2 but without PhI(OAc)2 did not proceed, while lowered amounts of PhI(OAc)2 gave lowered yields (25% with 1 equiv. and 68% with 1.5 equiv.). Using 2 equiv. of PhI(OAc)2 and a trace of I2, a series of purines were reacted with THF (used as the solvent), at 70 °C. In the case of 2-chloro-6-N,N-dimethylamino-9H-purine, DCE was used as the solvent for solubility reasons. The products as well as yields are shown in Fig. 50. Notably a purinyl iodide atom is unaffected under what are presumably radical conditions.
 | ||
| Fig. 50 Products from the reactions of THF with various purine derivatives. | ||
On the basis of these data, reactions of 2,6-dichloro-9H-purine and other ethers were investigated. However, one more addition was made to the reaction conditions to maximize the product yields: irradiation with a 200 W lamp. Under these conditions, increasing the ring size to six, as with THP, caused a reduction in yield by about 9%. Aliphatic ethers reacted well, with a yield lowering observed with increasing chain length. For a reasonable reaction of methyl t-butyl ether, 1 equiv. of I2 was necessary, but diisopropyl ether did not react. These results are shown in Fig. 51.
 | ||
| Fig. 51 Products from the reactions of 2,6-dichloro-9H-purine with other ethers. | ||
A generic mechanism has been proposed wherein a purinyl radical centered at the N9 atom reacts with the ether through hydrogen atom transfer. The ether radical so formed is proposed to undergo oxidation to an oxocarbenium ion that is captured by the purine, with loss of a proton. What is interesting about this chemistry is the apparent formation of single regioisomeric products, represented as the N9-alkylated products. Alkylation reactions of purines can produce N9/N7 regioisomers,74 with predominance of the N9 isomer, that need separation in many instances. Also, from the mechanism shown, the N9-centered radical is one resonance form. Thus, one must wonder about the reasons leading to such exquisite regioselectivity, although it may be argued that the C6 substituent discourages reactivity at the N7 atom. This could be have been probed with the reaction of just 9H-purine itself.
C.3.f.ii. Reactions of heteroaryls, phthalimide and pyrimidinones. The oxidative-amination of ethers with alkyl and aryl sulfonamides, and amides was investigated.73 Using N-benzylmethanesulfonamide and THF, various conditions were evaluated. As a first step, deprotonation of the sulfonamide was performed with NaH over 1 h and then PhI(OAc)2 was added, and the reaction was continued at room temperature for 10 h. Of note, this is the same oxidant used for the reaction of purines in C.3.f.i.,72 and it led to a reasonable 43% yield. However, in the absence of the oxidant no reaction was observed. With PhI(OAc)2/I2, on the other hand, a lower 36% product yield was obtained. Just I2, K2S2O8 and O2 as oxidants gave no product. Next, Ph2I+TfO− and Ph2I+PF6− were evaluated, resulting in excellent and comparable 85% and 87% product yields, respectively. Bases other than NaH, such as NaOH, Cs2CO3 and t-BuOK, gave inferior results (18–38% yields). The yields were negatively affected by solvents other than THF, the reactant-solvent (<1–66% with DCE, PhMe, CH2Cl2, EtOAc and MeCN). A reaction with Ph2I+TfO− in the absence of NaH led to no product.
Thus, NaH (1 equiv.) and Ph2I+PF6− were used for the reactions of various sulfonamides and amides with THF, with yields ranging from 39–95% (data not shown). For the reactions of sulfonamides, 1.2 equiv. of the hypervalent iodine reagent was used, whereas with amides 1.5 equiv. was needed. Of particular relevance to this perspective were reactions of phthalimide, imidazole, benzimidazole, indole, carbazole, 5-methyluracil and 5-fluorouracil. These reactions were conducted in two steps: (a) deprotonation with NaH (1 equiv., 1–3 h at room temperature) and (b) reaction with Ph2I+PF6− (1.5–1.7 equiv., 6 h at room temperature). The reaction of phthalimide with THF required 40 °C (12 h), whereas that with 1,4-dioxane required 60 °C (10 h). The products obtained from these reactions are shown in Fig. 52.
 | ||
| Fig. 52 Products of the uncatalyzed oxidative-amination with phthalimide, imidazole, benzimidazole, indole, carbazole, 5-methyluracil and 5-fluorouracil. | ||
Reactions of phthalimide can be compared to the n-Bu4N+I−/t-BuOOH reactions described in section C.3.e.i., where the yields obtained were 92% with THF and 54% with THP.66 The yields in the uncatalyzed process were lower. The yield from the reaction of imidazole and THF was comparable to that from the RuCl3·3H2O/t-BuOOH reaction (56%),65 but lower than that obtained with FeCl3·6H2O/t-BuOOH (88–90%).59 The yield from the reaction of benzimidazole with THF was comparable to those obtained with FeCl3·6H2O/t-BuOOH (86–89%),59 and RuCl3·3H2O/t-BuOOH (83%).65 Please refer to Table 7 for additional comparisons. Although CuCl2/bipy/(t-BuO)2 has been used for the reaction of carbazole with THF (90% yield in comparison to the 65% with the uncatalyzed process), a comparable reaction of indole is not available.63
Among the pyrimidine reactions, the p-methoxybenzyl group was used to protect thymine (the authors state this as p-methoxybenzoyl but the alkyl, rather than acyl protection, was discerned from the supporting information to that work).73 However, this group proved difficult to remove and the Boc protection was used for the other substrates. The product from the reaction of 5-fluorouracil and THF is in fact a Boc-protected version of tegafur, a prodrug that produces 5-fluorouracil upon metabolism.
This reaction is inhibited by 10 mol% of TEMPO and the measured KIE in a reaction of acetanilide with THF/THF-d8 was 4.6. Thus, the process is a radical reaction where C–H bond cleavage is likely involved in the rate-limiting step. As shown in Scheme 58, deprotonation of the amide and reaction with Ph2I+PF6− can be followed by salt metathesis. Oxidation of the amidyl anion by the hypervalent iodine reagent can then produce an aminyl radical and Ph2I˙. The latter can then decompose via formation of PhI and Ph˙ that can react with THF to produce a radical. Reaction of the THF radical with the deprotonated amide can lead to a radical anion that can be oxidized by Ph2I+ to give the final product (route A). Alternatively, the THF radical can be oxidized to an oxocarbenium ion with Ph2I+. The oxocarbenium ion can finally be trapped by the amidyl anion, resulting in the product (route B). In both routes, the formed Ph2I˙ can propagate the radical process.
 | ||
| Scheme 58 Possible reaction mechanisms for the uncatalyzed oxidative amination. | ||
C.3.f.iii. Reactions of heteroaryls with alcohols. In contrast to reactions of ethers, which are more abundant, the use of alcohols is more limited. Several preceding sections describe C–C bond formation with alcohols. Recently, C–N bond formation between heteroaryls and alcohols has been investigated.75 In this context, the reaction of BtH and EtOH with t-BuOOH at 120 °C gave a product that would otherwise arise through a CDC reaction between BtH and Et2O (Scheme 59).65,71
 | ||
| Scheme 59 Uncatalyzed reaction of BtH with EtOH. | ||
The use of (t-BuO)2, (PhCOO)2, t-BuOOCOPh, K2S2O8, NIS and H2O2 gave no product. Traces of product were observed with 1,4-benzoquinone and dicumylperoxide. The use of metal catalysts such as Mn(OAc)3, Fe(acac)3 and AgNO3 returned trace to 15% product. Pd(OAc)2 gave a better 56% yield of product, but no condition was better than the use of only t-BuOOH, where a 76% yield of product was obtained. Using 2 equiv. of t-BuOOH at 120 °C, four BtHs were reacted with alcohols and the results are summarized in Fig. 53. Generally, good reactions were observed but bromomethanol (!) and trifluoroethanol did not react.
 | ||
| Fig. 53 Products from the reactions of benzotriazoles with alcohols. | ||
The conditions were then applied to the reactions of 5-aryl-1H-tetrazoles. The resulting 1,5-disubstituted tetrazoles (Fig. 54) are complementary to the tetrazoles described in section C.3.e.iii.68 In these reactions, while a nitro group on the aryl ring gave a good product yield, the monochloro and dichloro aryl precursors reacted poorly.
 | ||
| Fig. 54 Products obtained from the reactions of 5-aryl-1H-tetrazoles with ethers. | ||
The reactions of EtOH with carbazole, 3-chloro-1H-indazole, 3-methyl-6-nitro-1H-indazole, benzimidazole and indole were evaluated as well. Among these, carbazole gave the highest product yield of 48%. Between the indazoles, 3-chloro-1H-indazole gave a 16% yield of product and 3-methyl-6-nitro-1H-indazole gave a 20% yield. By contrast to these, both benzimidazole and indole did not give any product.
In mechanistic studies, the reactions of BtH with EtOH proceeded well under nitrogen or air (78 and 76% product yield, respectively), but the reaction did not proceed under oxygen. The lack of reactivity under oxygen hints at a possible radical reaction. A trapping reaction with TEMPO only showed 2,2,6,6-tetramethyl-1-piperidinol by GC/MS. A reaction of BtH with acetaldehyde gave a good 60% yield of the product. The authors, therefore, propose two possible routes to the product. As in the reactions of alcohols described in C.1.f.vii. and C.1.f.viii.,50,51t-BuOOH likely produces a radical from EtOH. This initial radical can form acetaldehyde that can directly react with BtH to yield a hemiaminal, or it can react with EtOH. The latter can form an oxocarbenium ion that can be captured by BtH. These pathways are summarized in Scheme 60.
 | ||
| Scheme 60 A possible mechanism for the reaction of BtH with EtOH. | ||
C.3.f.iv. Metal-free cascade cyclizations involving alcohols leading to oxindoles. Related to the Fe-catalyzed cascade CDC reactions described in section C.2.a.ii.,53 similar reactions of alcohols have been studied, but in the presence of only an oxidant.76 Reactions of N-methyl-N-phenylmethylacrylamide and EtOH (see Scheme 61) were evaluated with 2 equiv. of t-BuOOCOPh, (t-BuO)2, dicumylperoxide and t-BuOOH. At 100 °C, no product formation was observed with (t-BuO)2 and dicumylperoxide. With BuOOCOPh, a 65% yield of the product was obtained (Scheme 61, R = CH(OH)Me). In comparison to 5–6 M t-BuOOH in decane, a 70% aqueous solution gave a better yield (76 versus 86%). At 80 °C a substantially lower yield was observed (28%), whereas at 120 °C the yield was slightly lower (73%). No product formation occurred in the absence of the oxidant.
 | ||
| Scheme 61 Cascade cyclization reactions of N-methyl-N-phenylmethylacrylamide with alcohols. | ||
Using the optimized conditions, several alcohols were reacted with N-methyl-N-phenylmethylacrylamide (Scheme 61). Generally good to high yields were obtained. However, with MeOH a low 24% yield of product was observed, and with propargyl alcohol only a trace amount of product resulted. Several oxindoles were synthesized through reactions with iPrOH and the product structures are shown in Fig. 55. While most substrates reacted well (55–91% product yields), only a trace amount of product was formed when a phenyl substituent was present on the olefin. An unsubstituted nitrogen atom and a hydroxymethyl substituent on the olefin were not tolerated.
 | ||
| Fig. 55 Various oxindoles synthesized through the cascade cyclization with iPrOH. | ||
In an experiment with an ortho-deuterated precursor and iPrOH, kH/kD = 1.0 was observed (Scheme 62). Similarly the reaction of a 1:1 mixture of the protio and pentadeutereo precursors with iPrOH gave kH/kD = 1.0. However, kH/kD = 6.7 was observed when a reaction was conducted with a 1:1 mixture of MeOH and MeOH-d4. Thus, cleavage of the C–H bond in the alcohol likely contributes to the rate-determining step.
 | ||
| Scheme 62 Experiments to determine kinetic isotope effects. | ||
On the basis of these results a proposed mechanism (Scheme 63) involves the abstraction of a hydrogen radical α to the oxygen atom in the alcohol. Addition of this radical to the acrylamide olefin, cascade cyclization followed by loss of hydrogen radical and rearomatization, leads to the final product. This mechanism is similar to that in the Fe-catalyzed addition of radicals derived from ethers to N-arylacrylamides discussed in C.2.a.ii.53
 | ||
| Scheme 63 Proposed mechanism for the cascade reaction with alcohols leading to oxindoles. | ||
C.4. CDC reactions for the introduction of CF3 groups into heteroaromatic systems
In a very recent development,77 TFE (CF3CH2OH) has been used in oxidative-amination type reactions with indoles and pyrroles. This represents the first attempt to introduce a CF3 group through such reactions with fluorinated alcohols. In order to identify optimal conditions, initial reactions were conducted with indole and TFE in the presence of 5 mol% of CuBr and 2 equiv. of an oxidant, at 130–140 °C. Dicumylperoxide, t-BuOOH and t-BuOOCOPh were tested under various conditions, and all led to reaction at the C3 position of indole (Fig. 56, R = R1 = R2 = R3 = H). The use of t-BuOH as a cosolvent was found to be important as was the ratio of t-BuOH/TFE. The best conditions were 2 equiv. of dicumyl peroxide in a 3:2 ratio of t-BuOH/TFE at 140 °C.
Using these, a series of products were synthesized. Some variations in reaction conditions were necessary for optimal results with some substrates. The product structures are summarized in Fig. 56.
 | ||
| Fig. 56 Products from the CDC reactions with TFE. | ||
Because reactions occur at the C3 and not the C2 position, initially, a Friedel–Crafts type process was speculated. To probe the mechanism, independent reactions of indole were conducted with CF3CHO and CF3CH(OH)OEt. Both reactions led to efficient product formation (Fig. 56, R = R1 = R2 = R3 = H). Radical trapping experiments did not lead to the identification of a radical from TFE and GC/MS indicated the presence of 1-(t-butoxy)-2,2,2-trifluoroethan-1-ol. Thus, a carbon-centered radical arising from TFE may be very unstable. However, ESR experiments indicated the presence of a β-hydroxy carbon-centered radical as well as CuII.
On the basis of these collective data, an interesting mechanism has been proposed. CuI could be oxidized to CuII through electron transfer to dicumyl peroxide. The ensuing PhMe2C˙ radical can collapse to acetophenone and a methyl radical, which in turn can abstract a hydrogen radical from TFE. The ensuing TFE radical can undergo oxidation to CF3CHO and then react with t-BuOH, leading to 1-(t-butoxy)-2,2,2-trifluoroethan-1-ol, which was identified using GC/MS. Such a hemiacetal can react with indole through an electrophilic mechanism. This is summarized in Scheme 64. It should be noted though that the mechanism does not show how CuII is reduced to the catalytic CuI. We, therefore, suggest that the oxidation of the TFE radical to CF3CHO could also occur with CuII (shown as an alternative in Scheme 64).
 | ||
| Scheme 64 A possible mechanism for the reaction of indole with TFE. | ||
Two of the products were elaborated to other CF3-substituted compounds. These conversions (Scheme 65) go towards demonstrating the potential versatility of the products in deriving other high-value products.
 | ||
| Scheme 65 Use of the CDC products for the synthesis of other CF3-substituted compounds. | ||
D. Conclusions
This perspective is a comprehensive survey of CDC processes using both metal and organic catalysts, and reactions without the involvement of metals. Two complementary types of reactions are described: (a) C–C bond forming reactions between aromatic and heteroaromatic compounds with ethers and (b) oxidative amination reactions of heteroaryls containing NH groups with ethers leading to C–N bond formation. Among these reactions, comparable chemistry involving alcohols is also discussed. What we have found in the course of this survey is that the reactivity of any ether is not assured and ethers do not all react in comparable manners. For example, cyclic ethers are not all similar in their reactivities. Part of the reason for this may be due to the bond dissociation energies of the C–H bonds of ethers. Also, stereoelectronic effects have been shown to influence hydrogen atom abstraction from ethers.78 In this context, there are examples where the unreactivity of certain ethers is either noted by the reporting authors, or in other cases obvious ether substrates are missing from the generality analyses. The latter leads one to wonder about negative results with such ethers. What should be kept in mind is that a lack of reactivity of various aryls or heteroaryls with particular ethers should not necessarily be construed as a limitation of the approach, because these types of reactions offer some of the most expeditious routes to relatively complex, chiral molecules. It is our view that there are a large number of heteroaryls whose reactivity under CDC and oxidative-amination conditions are yet to be explored. These as yet unstudied reactions will lead to a greater understanding and appreciation of reactivity manifolds, and at the same time expand the palette of functionalized heterocyclic systems. While this manuscript was being prepared, two other reviews on Csp3 bond activation α to the oxygen atoms in ethers and alcohols were published.79,80 While there is some unavoidable overlap between those reviews and this perspective, there are substantial differences. Further, this perspective also covers up to the most recently published data and we have attempted to make this a one-stop reference for this chemistry.Looking ahead, the development of enantioselective versions of the reactions described herein will substantially advance the utility of this chemistry, in that enantioenriched molecules can be accessed via relatively simple reactions. Whilst this is a challenging undertaking, approaches to chiral CDC reactions are already being realized.81–89 Thus, it is likely only a matter of time before enantioselective versions of many transformations described herein become chemically feasible.
Acknowledgements
Our efforts in this area have been supported by NSF Grant CHE-1265687 and infrastructural support at CCNY has been provided by NIH Grant G12MD007603 from the National Institute on Minority Health and Health Disparities. We thank Mr Kirill Korvinson for drawing our attention to ref. 78.References
- J. Miao and H. Ge, Eur. J. Org. Chem., 2015, 7859–7868 CrossRef CAS.
- C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed.
- S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed.
- X. Shang and Z.-Q. Liu, Chem. Soc. Rev., 2013, 42, 3253–3260 RSC.
- C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed.
- X. Bugaut and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 7479–7481 CrossRef CAS PubMed.
- L. Yang and H. Huang, Catal. Sci. Technol., 2012, 2, 1099–1112 CAS.
- R. Rohlmann and O. G. Mancheño, Synlett, 2013, 24, 6–10 CrossRef CAS.
- S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC.
- C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS PubMed.
- M. Klussmann and D. Sureshkumar, Synthesis, 2011, 353–369 CrossRef CAS.
- J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540–548 RSC.
- C. J. Scheuermann, Chem.–Asian J., 2010, 5, 436–451 CrossRef CAS PubMed.
- S.-Y. Zhang, F.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2011, 40, 1937–1949 RSC.
- D. Liu and A. Lei, Chem.–Asian J., 2015, 10, 806–823 CrossRef CAS PubMed.
- C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- A. A. O. Sarhan and C. Bolm, Chem. Soc. Rev., 2009, 38, 2730–2744 RSC.
- C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed.
- W. Han and A. R. Ofial, Synlett, 2011, 1951–1955 CAS.
- J. Le Bra and J. Muzart, Chem. Rev., 2011, 111, 1170–1214 CrossRef PubMed.
- K. Hirano and M. Miura, Chem. Commun., 2012, 48, 10704–10714 RSC.
- F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31–41 CAS.
- S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886–896 RSC.
- Z. Li and C.-J. Li, J. Am. Chem. Soc., 2005, 127, 6968–6969 CrossRef CAS PubMed.
- M. Ghobrial, K. Harhammer, M. D. Mihovilovic and M. Schnürch, Chem. Commun., 2010, 46, 8836–8838 RSC.
- M. Ghobrial, M. Schnürch and M. D. Mihovilovic, J. Org. Chem., 2011, 76, 8781–8793 CrossRef CAS PubMed.
- S. J. Park, J. R. Price and M. H. Todd, J. Org. Chem., 2012, 77, 949–955 CrossRef CAS PubMed.
- E. Shirakawa, N. Uchiyama and T. Hayashi, J. Org. Chem., 2011, 76, 25–34 CrossRef CAS PubMed.
- R. P. Pandit and Y. R. Lee, Adv. Synth. Catal., 2014, 356, 3171–3179 CrossRef CAS.
- D. Liu, C. Liu, H. Li and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 4453–4456 CrossRef CAS PubMed.
- W. Muramatsu, K. Nakano and C.-J. Li, Org. Lett., 2013, 15, 3650–3653 CrossRef CAS PubMed.
- W. Muramatsu and K. Nakano, Org. Lett., 2014, 16, 2042–2045 CrossRef CAS PubMed.
- W. Muramatsu and K. Nakano, Org. Lett., 2015, 17, 1549–1552 CrossRef CAS PubMed.
- M. Shibuya, Y. Sasano, M. Tomizawa, T. Hamada, K. Kozawa, N. Nagahama and Y. Iwabuchi, Synthesis, 2011, 3418–3425 CAS.
- K. Qvortrup, D. A. Rankic and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 626–629 CrossRef CAS PubMed.
- A. Tyagi, T. Matsumoto, T. Kato and H. Yoshida, Catal. Sci. Technol., 2016, 6, 4577–4583 CAS.
- D. Liu, C. Liu, H. Li and A. Lei, Chem. Commun., 2014, 50, 3623–3626 RSC.
- Z. Xie, Y. Cai, H. Hu, C. Lin, J. Jiang, Z. Chen, L. Wang and Y. Pan, Org. Lett., 2013, 15, 4600–4603 CrossRef CAS PubMed.
- A. Correa, B. Fiser and E. Gómez-Bengoa, Chem. Commun., 2015, 51, 13365–13368 RSC.
- Y. Li, M. Wang, W. Fan, F. Qian, G. Li and H. Lu, J. Org. Chem., 2016, 81, 11743–11750 CrossRef CAS PubMed.
- T. He, L. Yu, L. Zhang, L. Wang and M. Wang, Org. Lett., 2011, 13, 5016–5019 CrossRef CAS PubMed.
- Y. Zhang, K. B. Teuscher and H. Ji, Chem. Sci., 2016, 7, 2111–2118 RSC.
- Z. Wu, C. Pi, X. Cui, J. Bai and Y. Wu, Adv. Synth. Catal., 2013, 355, 1971–1976 CrossRef CAS.
- E. Kianmehr, N. Faghih, S. Karaji, Y. A. Lomedasht and K. M. Khan, J. Organomet. Chem., 2016, 801, 10–13 CrossRef CAS.
- W. Sun, Z. Xie, J. Liu and L. Wang, Org. Biomol. Chem., 2015, 13, 4596–4604 CAS.
- G. Deng, K. Ueda, S. Yanagisawa, K. Itami and C.-J. Li, Chem.–Eur. J., 2009, 15, 333–337 CrossRef CAS PubMed.
- J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565–1569 CrossRef CAS PubMed.
- A. P. Antonchick and L. Burgmann, Angew. Chem., Int. Ed., 2013, 52, 3267–3271 CrossRef CAS PubMed.
- S. Liu, A. Liu, Y. Zhang and W. Wang, Chem. Sci., 2017, 8, 4044–4050 RSC.
- C. A. Correia, L. Yang and C.-J. Li, Org. Lett., 2011, 13, 4581–4583 CrossRef CAS PubMed.
- H. Jiang, J. Xie, A. Lin, Y. Cheng and C. Zhu, RSC Adv., 2012, 2, 10496–10498 RSC.
- X. Guo, S. Pan, J. Liu and Z. Li, J. Org. Chem., 2009, 74, 8848–8851 CrossRef CAS PubMed.
- W.-T. Wei, M.-B. Zhou, J.-H. Fan, W. Liu, R.-J. Song, Y. Lu, M. Hu, P. Xie and J.-H. Li, Angew. Chem., Int. Ed., 2013, 125, 3726–3729 CrossRef.
- L.-K. Jin, L. Wan, J. Feng and C. Cai, Org. Lett., 2015, 17, 4726–4729 CrossRef CAS PubMed.
- L. Jin, J. Feng, G. Lu and C. Cai, Adv. Synth. Catal., 2015, 357, 2105–2110 CrossRef CAS.
- Q. Yang, P. Y. Choy, Y. Wu, B. Fan and F. Y. Kwong, Org. Biomol. Chem., 2016, 14, 2608–2612 CAS.
- Z. Gu, Y. Tang and G.-F. Jiang, J. Org. Chem., 2017, 82, 5441–5448 CrossRef CAS PubMed.
- D. Lee and R. D. Otte, J. Org. Chem., 2004, 69, 3569–3571 CrossRef CAS PubMed.
- S. Pan, J. Liu, H. Li, Z. Wang, X. Guo and Z. Li, Org. Lett., 2010, 12, 1932–1935 CrossRef CAS PubMed.
- (a) D. H. R. Barton and V. N. Le Gloahec, Tetrahedron, 1998, 54, 15457–15648 CrossRef CAS; (b) P. A. MacFaul, D. D. M. Wayner and K. U. Ingold, Acc. Chem. Res., 1998, 31, 159–162 CrossRef CAS; (c) D. H. R. Barton, Chem. Soc. Rev., 1996, 237–239 RSC; (d) D. H. R. Barton and D. Doller, Acc. Chem. Res., 1992, 25, 504–512 CrossRef CAS.
- X. Liu, Y. Chen, K. Li, D. Wang and B. Chen, Chin. J. Chem., 2012, 30, 2285–2291 CrossRef CAS.
- K.-q. Zhu, L. Wang, Q. Chen and M.-y. He, Tetrahedron Lett., 2015, 56, 4943–4946 CrossRef CAS.
- Q. Yang, P. Y. Choy, W. C. Fu, B. Fan and F. Y. Kwong, J. Org. Chem., 2015, 80, 11193–11199 CrossRef CAS PubMed.
- D. Wang, R. Zhang, R. Deng, S. Lin, S. Guo and Z. Yan, J. Org. Chem., 2016, 81, 11162–11167 CrossRef CAS PubMed.
- M. K. Singh, H. K. Akula, S. Satishkumar, L. Stahl and M. K. Lakshman, ACS Catal., 2016, 6, 1921–1928 CrossRef CAS PubMed.
- L. Dian, S. Wang, S. Z. Negrerie, Y. Du and K. Zhao, Chem. Commun., 2014, 50, 11738–11741 RSC.
- K. Sun, X. Wang, G. Li, Z. Zhu, Y. Jiang and B. Xiao, Chem. Commun., 2014, 50, 12880–12883 RSC.
- L. Wang, K.-q. Zhu, W.-t. Wu, Q. Chen and M.-y. He, Catal. Sci. Technol., 2015, 5, 2891–2896 CAS.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (c) B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- S. Rajamanickam, G. Majji, S. K. Santra and B. K. Patel, Org. Lett., 2015, 17, 5586–5589 CrossRef CAS PubMed.
- H. Aruri, U. Singh, S. Sharma, S. Gudup, M. Bhogal, S. Kumar, D. Singh, V. K. Gupta, R. Kant, R. A. Vishwakarma and P. P. Singh, J. Org. Chem., 2015, 80, 1929–1936 CrossRef CAS PubMed.
- H. M. Guo, C. Xia, H.-Y. Niu, X.-T. Zhang, S.-N. Kong, D.-C. Wang and G.-R. Qu, Adv. Synth. Catal., 2011, 353, 53–56 CrossRef CAS.
- I. Buslov and X. Hu, Adv. Synth. Catal., 2014, 356, 3325–3330 CrossRef CAS.
- See for example: (a) Z. Kazimierczuk, H. B. Cottam, G. R. Revankar and R. K. Robins, J. Am. Chem. Soc., 1984, 106, 6379–6382 CrossRef CAS; (b) C. Hildebrand and G. E. Wright, J. Org. Chem., 1992, 57, 1808–1813 CrossRef CAS.
- J. Sun, Y. Zhang, S. Mathan, Y. Wang and Y. Pan, J. Org. Chem., 2016, 81, 3380–3385 CrossRef CAS PubMed.
- Y. Meng, L.-N. Guo, H. Wang and X.-H. Duan, Chem. Commun., 2013, 49, 7540–7542 RSC.
- Z. Xu, Z. Hang, L. Chai and Z.-Q. Liu, Org. Lett., 2016, 18, 4662–4665 CrossRef CAS PubMed.
- V. Malatesta and K. U. Ingold, J. Am. Chem. Soc., 1981, 103, 609–614 CrossRef CAS.
- S.-r. Guo, P. S. Kumar and M. Yang, Adv. Synth. Catal., 2017, 359, 2–25 CrossRef CAS.
- A. Batra, P. Singh and K. N. Singh, Eur. J. Org. Chem. DOI:10.1002/ejoc.201700341.
- Z. Li and C.-J. Li, Org. Lett., 2004, 6, 4997–4999 CrossRef CAS PubMed.
- Z. Li, P. D. MacLeod and C.-J. Li, Tetrahedron: Asymmetry, 2006, 17, 590–597 CrossRef CAS.
- G. Zhang, Y. Zhang and R. Wang, Angew. Chem., Int. Ed., 2011, 50, 10429–10432 CrossRef CAS PubMed.
- J. Zhang, B. Tiwari, C. Xing, X. Chen and Y. R. Chi, Angew. Chem., Int. Ed., 2012, 51, 3649–3652 CrossRef CAS PubMed.
- A. J. Neel, J. P. Hehn, P. F. Tripet and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 14044–14047 CrossRef CAS PubMed.
- S. Sun, C. Li, P. E. Floreancig, H. Lou and L. Liu, Org. Lett., 2015, 17, 1684–1687 CrossRef CAS PubMed.
- Z. Xie, X. Liu and L. Liu, Org. Lett., 2016, 18, 2982–2985 CrossRef CAS PubMed.
- H. Yamamoto and H. Tsuji, Synfacts, 2016, 12, 0948 CrossRef.
- Q. Yang, L. Zhang, C. Ye, S. Luo, L.-Z. Wu and C.-H. Tung, Angew. Chem., Int. Ed., 2017, 56, 3694–3698 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |