
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective propargylic [1,3]-rearrangements: copper-catalyzed O-to-N migrations toward C–N bond formation†
Li-Jie
Cheng
,
Alexander P. N.
Brown
and
Christopher J.
Cordier
 *
*
Department of Chemistry, Imperial College London, South Kensington, London, SW7 2AZ, UK. E-mail: ccordier@imperial.ac.uk
First published on 31st March 2017
Abstract
We have identified an enantioselective copper-catalyzed O-to-N formal [1,3]-rearrangement to form N-propargylic-2-pyridones. This enantioconvergent O-to-N propargylic rearrangement occurs rapidly at ambient temperature and high enantioselectivity is observed for a range of 3-alkyl-substituted substrates. Stereochemical features include a mild kinetic enantioenrichment of the substrate and a non-linear relationship between product and ligand enantiopurity. Based on kinetic analyses and cross-over experiments, we put forward a mechanistic proposal involving Cu-acetylides as well as bimetallic intermediates in which coordination to the pyridyl nitrogen is likely a crucial binding interaction.
Introduction
Thermal suprafacial O-to-N [3,3]-sigmatropic rearrangements1 are well documented for allylic acetimidates,2 phosphorimidates3 and cyanates.4 Transition metal catalyzed formal [3,3]-sigmatropic rearrangements of this type can be performed at ambient temperatures,5 and chiral metal–ligand complexes have allowed enantioselective processes to be developed.6 Thermal O-to-N [3,3]-rearrangements of 2-allyloxypyridines are also documented, providing N-allyl-2-pyridones,7 and Pd catalysts with chiral ligands have enabled enantioselective processes from achiral substrates (eqn (1)).8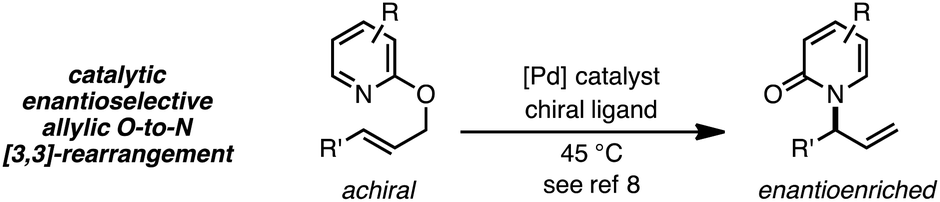 | (1) |
 | (2) |
 | (3) |
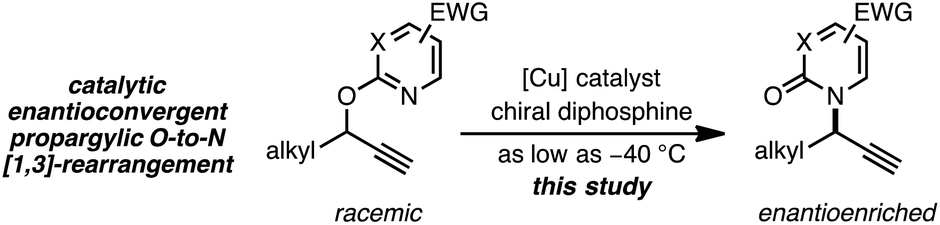
As a juxtaposition, suprafacial [1,3]-sigmatropic rearrangements are thermally disallowed9 but Ru10 and Ir11 catalysts can promote formal sigmatropic [1,3]-rearrangements of 2-benzyloxypyridine derivatives at elevated temperatures (eqn (2)).12 To our knowledge, enantioselective methods for O-to-N rearrangements yielding propargylic products have not been reported13 and, moreover, enantioselective O-to-N [1,3]-rearrangements are extremely rare.14 Here we report that a chiral Cu–diphosphine complex can promote the formal [1,3]-rearrangement of 2-propargyloxypyridines to enantioenriched N-propargylic-2-pyridones at temperatures as low as −40 °C (eqn (3)).
 | (4) |
 | (5) |
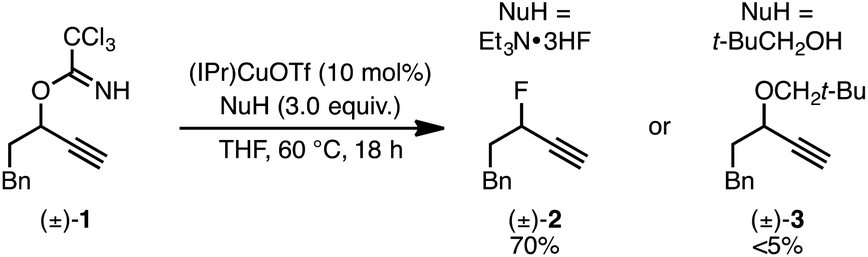
We recently reported a Cu-catalyzed fluorination protocol for the preparation of secondary and tertiary propargylic fluorides from the corresponding sulfonate esters and trichloroacetimidates.15 An N-heterocyclic carbene ligand was crucial to expanding the breadth of propargylic substitutions16 from N-,17C-,18 and O-nucleophiles19 to include fluoride. We decided to explore NHC–Cu complexes as catalysts for propargylic etherification of trichloroacetimidate (±)-1 (eqn (4)). Under the same conditions used for propargylic fluorination, direct replacement of Et3N·3HF with neopentyl alcohol resulted in trace (±)-3 (eqn (4)). A broader survey of imidate-like leaving groups led us to 2-propargyloxypyridine derivatives. We found that (IPr)CuOTf catalyzed etherification of (±)-4a at room temperature (eqn (5a)). A control experiment designed to contrast the influence of this NHC ligand with phosphine ligands led to a different result. Using catalytic (PPh3)3CuOTf, pyridone (±)-5a was formed in 51% yield, constituting a formal [1,3]-sigmatropic rearrangement (eqn (5b)). Furthermore, etherification of enantioenriched (S)-4a (98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 er) led to racemic product under the same conditions. This observation is indicative of achiral intermediates20 and presented the possibility to develop an enantioconvergent O-to-N [1,3]-rearrangement by employing chiral phosphine ligands.
:2 er) led to racemic product under the same conditions. This observation is indicative of achiral intermediates20 and presented the possibility to develop an enantioconvergent O-to-N [1,3]-rearrangement by employing chiral phosphine ligands.
Results and discussion
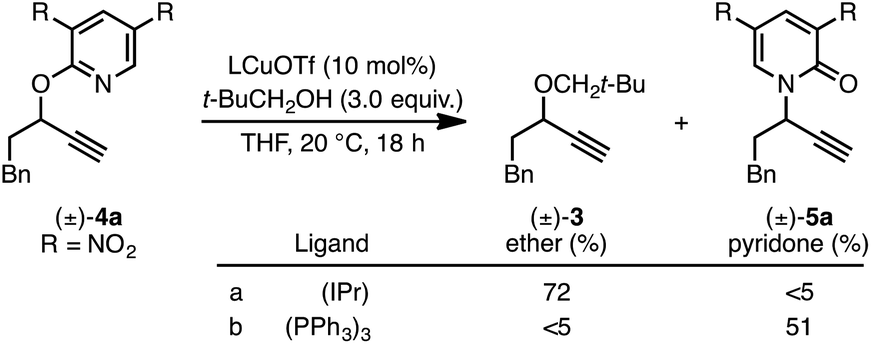
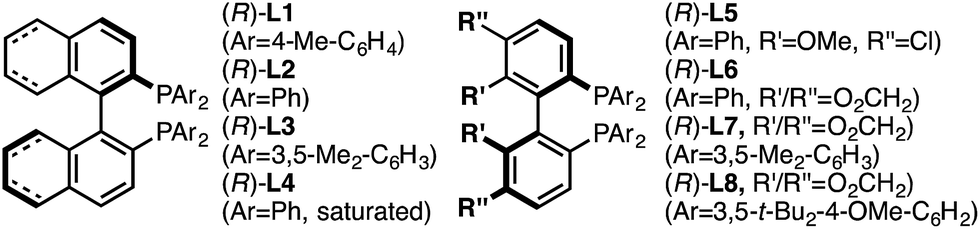
Investigation of a range of parameters21 showed that a complex derived from CuTC and (R)-L1 can catalyze the formation of (R)-5a at −40 °C in just 3 hours (Table 1, entry 2).|
|
||||
|---|---|---|---|---|
| Entry | Variation from ‘standard’ conditions | Conv.b (%) | Yieldb (%) | erc |
| a All data are the average of two experiments performed using 0.1 mmol substrate. For entries in which incomplete conversion was observed, reaction times were 24 h. b Determined by analysis of the crude reaction mixtures by 1H NMR using CH2Br2 as an internal standard. c Determined by chiral stationary phase HPLC. CuTC = copper(I)-thiophenecarboxylate. Bn = benzyl. | ||||
| 1 | R = H | 0 | — | — |
| 2 | None | >98 | 90 |
97.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2.5 :2.5
|
| 3 | No CuTC | 0 | — | — |
| 4 | No (R)-L1 | 0 | — | — |
| 5 | CuOTf·0.5PhH, instead of CuTC | 66 | 50 | 77:23 |
| 6 | CuI, instead of CuTC | 88 | 50 | 47:53 |
| 7 | Cu(MeCN)4BF4, instead CuTC | 72 | 60 | 46:54 |
| 8 | (R)-L2, instead of (R)-L1 | >98 | 94 | 96.5:3.5 |
| 9 | (R)-L4, instead of (R)-L1 | >98 | 97 | 95.5:4.5 |
| 10 | (R)-L5, instead of (R)-L1 | >98 | 95 | 96.5:3.5 |
| 11 | (R)-L6, instead of (R)-L1 | >98 | 92 | 96.5:3.5 |
| 12 | (R)-L3, instead of (R)-L1 | 39 | 24 | 19:81 |
| 13 | (R)-L7, instead of (R)-L1 | 80 | 40 | 50:50 |
| 14 | (R)-L8, instead of (R)-L1 | <2 | — | — |
| 15 | THF, instead of PhMe | >98 | 80 | 96:4 |
| 16 | Addition of i-Pr2NEt (2.0 equiv.) | >98 | 82 | 96.5:3.5 |
| 17 | 5 mol% CuTC, 6 mol% (R)-L1 (7 h) | >98 | 85 | 97.5:2.5 |
| 18 | −20 °C (1 h), instead of −40 °C | >98 | 96 | 96:4 |
| 19 | 20 °C (10 min), instead of −40 °C | >98 | 87 |
94:6
|
|
||||
Under our standard protocol, nitro-substituents on the pyridine were found to be required (entry 1).21b However, substrate (±)-4a underwent an efficient enantioconvergent O-to-N rearrangement, providing pyridone (R)-5a in 90% yield and 97.5:2.5 er (entry 2). In the absence of CuTC or (R)-L1, no conversion of (±)-4a was observed (entries 3 and 4). The copper source had a strong influence on both conversion and enantioselectivity (entries 5–7). Other ligand architectures faired similarly well in terms of yield and enantioselectivity compared with L1 (entries 8–11). Effects of the phosphine aryl-substituents were more pronounced; diphenyl- and di-4-methylphenyl-phosphines (L1–L2, L4–L6) resulted in high conversion and stereoinduction. However, under the same conditions bis-3,5-dimethylphenyl analogues led to low product enantiomeric ratios (entries 12 and 13) and the highly bulky ligand L8 resulted in no conversion (entry 14). Performing the transformation in THF (entry 15) offered similar results to toluene. Since related propargylic substitutions normally require base, we note that base is not required for this process (entry 16). Employing less catalyst did not impact enantioselectivity but did influence yield and reaction time (entry 17). If the transformation is performed at higher temperature, enantioselectivity is affected minimally (entry 18). Significantly, the rearrangement is complete in less than 10 minutes at ambient temperature (entry 19).
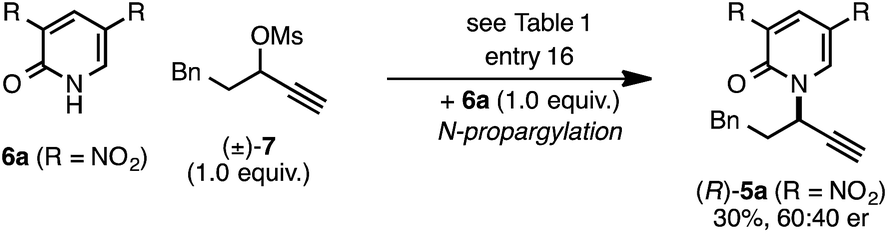
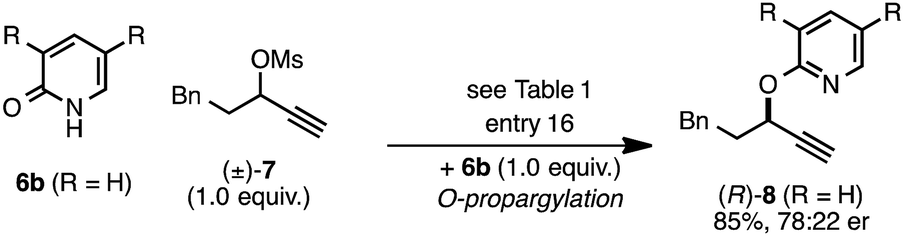
The rearrangement approach toward enantioselective propargylic C–N bond formation complements Cu-catalyzed substitution methods.17 In particular 3-alkyl substrates typically require longer reaction times during substitutions with N-nucleophiles than 3-aryl substrates require when using Cu–diphosphine or Cu–pybox catalyst systems; the current method occurs far faster and occurs at remarkably lower temperatures.22 To examine the efficacy of our conditions toward propargylic substitutions, we decided to use 2-(1H)-pyridones 6a and 6b in reaction with mesylate (±)-7 (eqn (6) and (7)).23
 | (6) |
 | (7) |
N-propargylated product (R)-5a was formed but with substantially reduced enantioselectivity (eqn (6)).24 Reaction of unsubstituted pyridone 6a led exclusively to O-propargylation, forming (R)-8 in good yield and with modest enantioselectivity (eqn (7)).25 The observed O- vs. N-alkylation selectivity may arise from initial O-alkylation of 7 with 6a or 6b but with only intermediate 4a undergoing the rearrangement process. The sense of stereoinduction during these substitutions matches that for the rearrangement process, indicating a similar pathway for enantiocontrol but with markedly reduced efficacy under these conditions.
Following established protocols for modifying the pyridone core26 we demonstrated that (S)-5a serves as a surrogate for α-nitroacetanilide (S)-9, and can be readily converted into amine derivative (S)-10 (Scheme 1). While related substitution methods are not suitable for the preparation of propargylic amides or primary amines, the rearrangement approach to (S)-5a represents a precursor to (S)-10, an intermediate in synthesis of the potent cysteine protease inhibitor K777 (11).27
 | ||
| Scheme 1 Synthetic utility of pyridone (S)-5a. (a) NH3 in MeOH (7 M), cyclohexane (2.0 equiv.), 65 °C, 3 h; (b) aq. HCL, 90 °C, 24 h; (c) Boc2O (1.5 equiv.), NaHCO3 (2.0 equiv.), DCM, RT, 18 h. | ||
Having established a useful protocol for an enantioselective C–N bond formation, we elected to move forward to explore the influence of 3-substituents on enantioselectivity in this transformation (Table 2). Preparative scale reaction of substrate (±)-4a proceeded smoothly (entry 1) and a reaction performed on 5 mmol scale provided gram quantities of (R)-5a (81%). Rearrangements were successful using substrates containing an unfunctionalized alkyl chain, an alkene, and a primary alkyl chloride (entries 2–4). A benzyl ether and silyl ether were both tolerated (entries 5 and 6) but an unprotected alcohol negatively affected both conversion and stereoinduction (entry 7). The presence of an acetal, an unprotected aldehyde and a methyl ester had minimal effects on enantioselectivity (entries 8–10). Assessing the influence of bulkier 3-substituents, a 3-benzyl group was tolerated very poorly while a 3-cyclohexyl appendage was accommodated smoothly (entries 11 and 12). Reaction of a highly sterically hindered 3-(1-adamantyl) substrate suffered from low conversion and poor enantioselectivity (entry 13). A benzylic substrate was compatible with our standard conditions but led to product with mediocre er (entry 14). Furthermore, attempts to conduct this rearrangement using a substrate bearing an internal alkyne failed, and recovered starting material was observed even after prolonged heating.
|
|
|||
|---|---|---|---|
| Entry | R′ | Yieldb (%) | erc |
| a All data are the average of two experiments performed using 0.4 mmol substrate, unless otherwise stated. b Isolated yields. c Determined by chiral stationary phase HPLC. d Performed using 5.0 mmol of substrate. e The Cahn–Ingold–Prelog terminology for product 5n denotes the (S)-configuration. Bn = benzyl; TBS = tert-butyldimethylsilyl; 1-Adam = 1-adamantyl. | |||
| 1 (4a) |
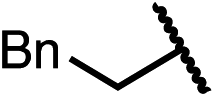
|
90 | 97.5:2.5 |
| 81d | 97.5:2.5d |
||
| 2 (4b) |

|
76 | 97:3 |
| 3 (4c) |
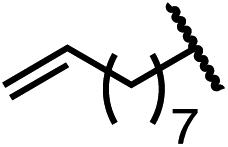
|
88 | 95.5:4.5 |
| 4 (4d) |
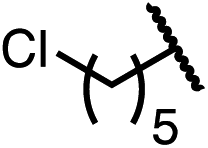
|
92 | 97:3 |
| 5 (4e) |
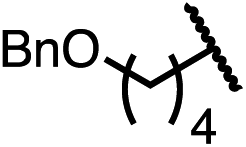
|
90 | 96.5:3.5 |
| 6 (4f) |
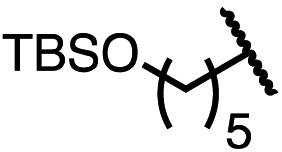
|
75 | 96.5:3.5 |
| 7 (4g) |
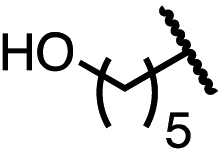
|
52 | 63:37 |
| 8 (4h) |

|
88 | 97:3 |
| 9 (4i) |
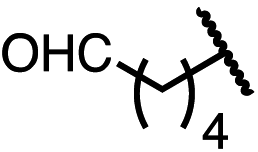
|
89 | 96.5:3.5 |
| 10 (4j) |
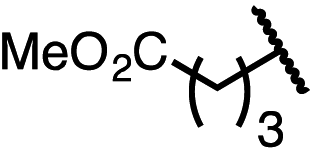
|
90 | 96:4 |
| 11 (4k) |
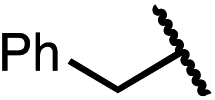
|
49 | 62:38 |
| 12 (4l) |
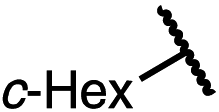
|
80 | 95.5:4.5 |
| 13 (4m) |

|
30 | 55:45 |
| 14e (4n) |
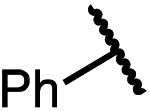
|
70 | 85:15 |
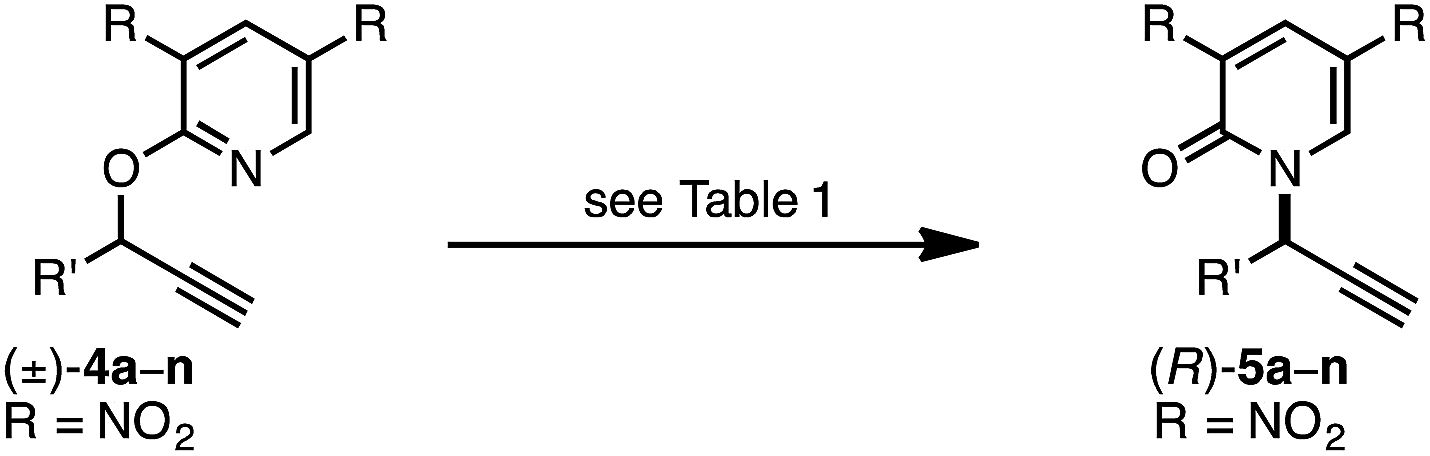
Next, we conducted a range of experiments designed to reveal stereochemical features about this transformation. Quenching our standard reaction of (±)-4a after 1 h (70% conversion) led to recovered starting material (S)-4a with 61:39 er, indicating a mild kinetic resolution28 (s factor = 2.7) associated with C–O bond cleavage (eqn (8)).29 We note that the enantiomeric ratio of (R)-5a is constant throughout the course of the reaction. To examine the extent of enantioconvergence during our rearrangement, we subjected enantioenriched substrate (S)-4a to our standard rearrangement conditions using (R)-L1 (eqn (9)) and (S)-L1 (eqn (10)). The stereochemistry of the pyridone product was dependent primarily on the stereochemistry of the ligand rather than of the starting propargyloxypyridine. However, when employing (S)-L1 net stereoretention was observed to occur with a small but measurable enhancement in product er (99:1 cf. 97:3), indicating a non-zero degree of stereochemical transfer during this transformation.28 A positive non-linear effect was observed when employing non-enantiomerically pure L1 (Fig. 1); this observation may indicate dinuclear, or higher order, species involved in the stereochemistry determining step30 or may be a kinetic consequence off-cycle polynuclear species.31
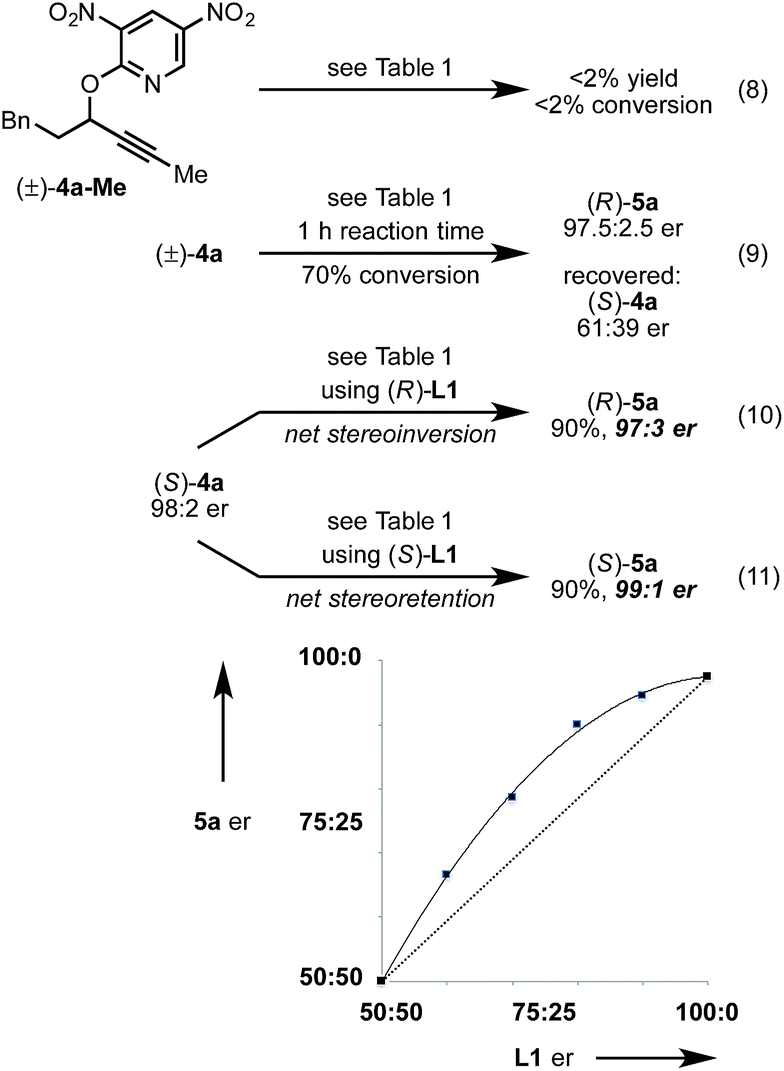 | ||
| Fig. 1 Non-linear er relationship between L1 and 5a under standard conditions using (±)-4a. | ||
Next, we conducted reaction progress kinetic analysis to elucidate some kinetic features of this transformation,32 using the rearrangement of 4a as a representative substrate. Concentration profiles of 4a were generated by following the rearrangement using 1H NMR in d8-PhMe at [CuTC–Tol-BINAP] = 10, 15, and 20 mM (Fig. 2A). Applying the Burés method33 for variable time normalization analysis, using t[cat]Tn, shows that the transformation is far from half- or first-order (Fig. 2B and C, respectively) but more closely approximate to second order (Fig. 2D)34 in catalyst for [CuTC–Tol-BINAP] between the 10–20 mM range examined. These data are consistent with the results obtained following nonlinear experiments and imply that bimetallic species are involved in the turnover-limiting and stereochemistry-determining steps.
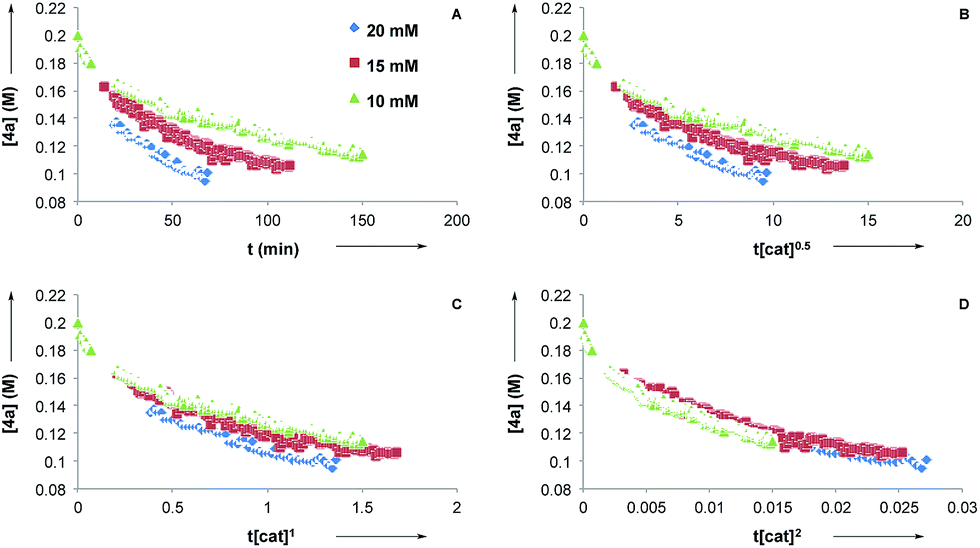 | ||
| Fig. 2 (a) Reaction profiles for initial conditions: [CuTC–Tol-BINAP] = 20, 15, 10 mM. Determinations of catalyst order using time-normalization profiles: (b) half order in catalyst; (c) first order in catalyst; (d) second order in catalyst. | ||
We used 31P NMR to identify catalyst species present in the reaction mixture: non-complexed ligand, Tol-BINAP, showed a signal at −16.47 ppm; the CuTC–Tol-BINAP adduct showed a signal at −3.93 ppm, and addition of the product led to a signal at −4.30 ppm. At ca. 20% conversion, 31P NMR of the reaction mixture showed signals corresponding to the non-complexed ligand, the CuTC–Tol-BINAP adduct, and the presence of two additional signals at −3.56 and −0.94 ppm. We assign these signals to potential bimetallic species of the catalyst resting state.
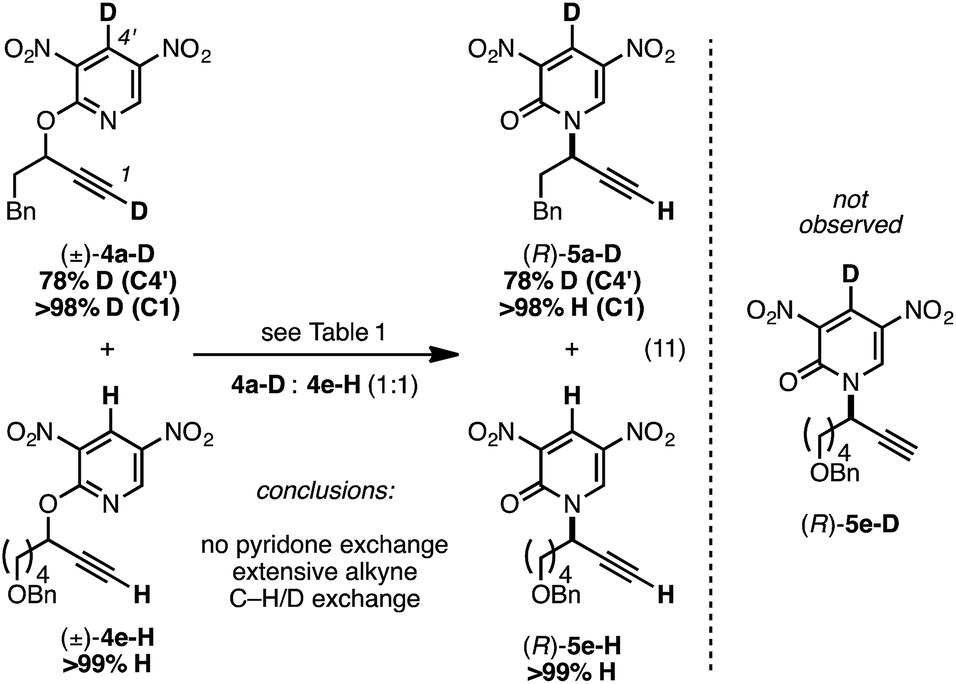
We prepared the deuterium-labeled substrate 4a-D in order to examine the extent of pyridone–alkyne dissociation during the rearrangement process (eqn (11)). Treatment of a 1:1 mixture of 4a-D and 4e-H to our standard conditions led to rearranged products 5a-D and 5e-H; the ‘cross-over’ adduct 5e-D was not observed. These results are consistent with a mechanism that involves no dissociation between the alkyne component of 4a-D and the deuterium-labeled pyridone moiety. Alternatively solvent-cage effects may kinetically favor C–N bond formation over diffusion-based separation of the pyridone and alkyne species. During our synthetic preparation of 4a-D, the terminal alkyne was also labeled with a deuteron. Performing the rearrangement of 4a-D, either in the presence or absence 4e-H, led to 5a-D bearing a terminal proton; performing this rearrangement in d8-toluene and monitoring progress by 1H NMR demonstrated that terminal deuteron/proton exchange was rapid and the source of the proton in eqn (11) was likely adventitious moisture.35
We conclude from the data presented that bimetallic intermediates are likely involved and speculate that Cu-coordination to the pyridyl nitrogen of (±)-4a may be an important binding interaction during C–O bond cleavage (Scheme 2). Thus, complexation of a second catalyst to Cu-acetylide 12 leads to bimetallic intermediate 13 (the putative resting state). We assign the heterolytic C–O bond cleavage as the turnover-limiting state, leading to bimetallic copper-pyridone intermediate 14/14′, similar to that proposed by Nishibayashi during propargylic etherifications;36 collapse of this intermediate permits C–N bond formation leading to Cu-acetylide 15. The cycle may close by means of protodecupration or, alternatively, alkyne exchange with another substrate molecule.
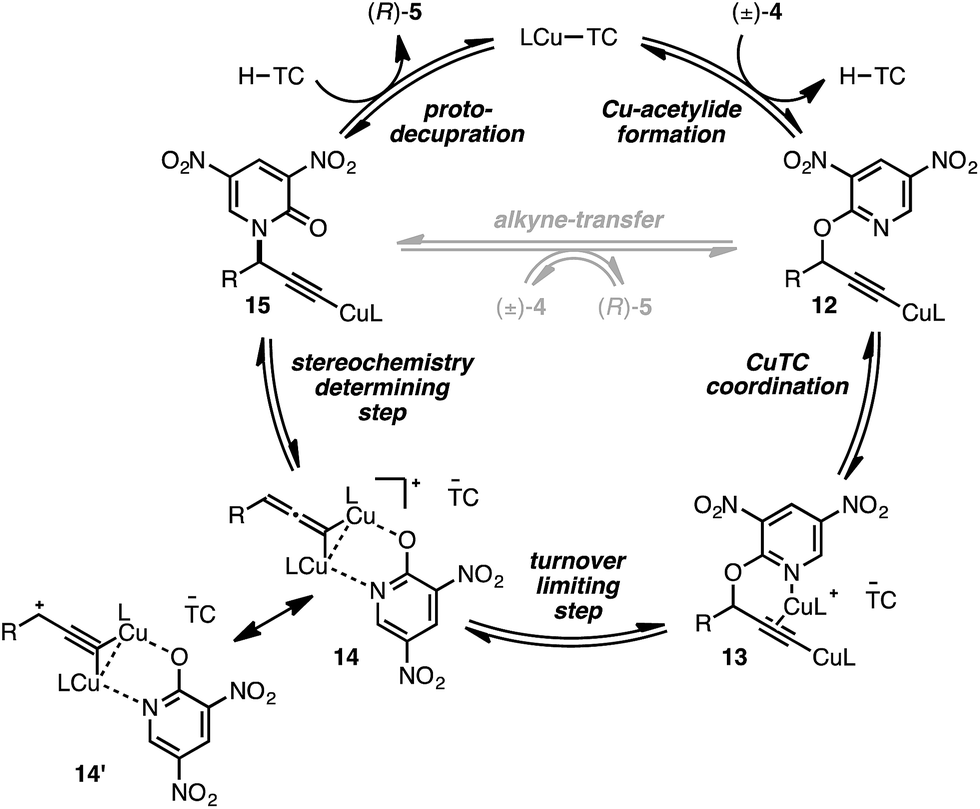 | ||
| Scheme 2 Proposed mechanistic pathway. | ||
Conclusions
In summary, we have developed the first enantioconvergent O-to-N [1,3]-rearrangement of a propargylic substrate, specifically, the enantioselective Cu-catalyzed rearrangement of electron-deficient 2-propargyloxy-pyridines. High enantioselectivity is observed with a range of 3-alkyl substituents, and short reaction times were noted in all cases. The pathway for stereoconvergence in the present method likely involves heterolytic C–O bond cleavage promoted by bimetallic Cu-species. Additional investigations to elucidate the mechanism of this transformation are underway, and methods that expand upon this [1,3]-rearrangement concept to include other propargylic bond formations will be reported in due course.Acknowledgements
We knowledge financial support from Imperial College London, the EPSRC (EP/L00352X/1), and the Royal Society for a University Research Fellowship (C. J. C.). We thank Prof. Alan Spivey and his research group for useful discussion following a double-blind repetition of the reaction in Table 2, entry 2. We would also like to thank Peter Haycock for assistance during our use of 1H NMR for kinetic analyses. L.-J. Cheng would like to dedicate this manuscript to Prof. Qi-Lin Zhou on the occasion of his 60th Birthday.References
- For a review concerning thermal O-to-N [3,3]-rearrangements of trichloroacetimidates, see: L. E. Overman, Acc. Chem. Res., 1980, 13, 218–224 CrossRef CAS.
- For seminal work, see: L. E. Overman, J. Am. Chem. Soc., 1974, 96, 597–599 CrossRef CAS.
- For seminal work, see: (a) E. E. Lee and R. A. Batey, Angew. Chem. Int. Ed., 2004, 43, 1865–1868 ( Angew. Chem. , 2004 , 116 , 1901–1904 ) CrossRef CAS PubMed; (b) B. Chen and A. K. Mapp, J. Am. Chem. Soc., 2004, 126, 5364–5365 CrossRef CAS PubMed.
- For seminal work, see: (a) Y. Ichikawa, Synlett, 1991, 4, 238–240 CrossRef; (b) Y. Ichikawa, M. Yamazaki and M. Isober, J. Chem. Soc., Perkin Trans. 1, 1993, 2429–2432 RSCFor recent applications, see: (c) M. Chwastek, M. Pleczykolan and S. Stecko, J. Org. Chem., 2016, 81, 9046–9074 CrossRef CAS PubMed; (d) S. Henrion, B. Carboni, F. P. Cossio, T. Roisnei, J. M. Villalgordo and F. Carreaux, J. Org. Chem., 2016, 81, 4633–4644 CrossRef CAS PubMed.
- (a) For a review concerning Hg and Pd-catalyzed [3,3]-rearrangements, see: L. E. Overman, Angew. Chem. Int. Ed., 1984, 23, 579–586 ( Angew. Chem. , 1984 , 96 , 565–573 ) CrossRef; (b) B. Chen and A. K. Mapp, J. Am. Chem. Soc., 2005, 127, 6712–6718 CrossRef CAS PubMed.
- For enantioselective variants, see: (a) M. Calter, T. K. Hollis, L. E. Overman, J. Ziller and G. G. Zipp, J. Org. Chem., 1997, 62, 1449–1456 CrossRef CAS; (b) Y. Donde and L. E. Overman, J. Am. Chem. Soc., 1999, 121, 2933–2934 CrossRef CAS; (c) C. E. Anderson and L. E. Overman, J. Am. Chem. Soc., 2003, 125, 12412–12413 CrossRef CAS PubMed; (d) E. E. Lee and R. A. Batey, J. Am. Chem. Soc., 2005, 127, 14887–14893 CrossRef CAS PubMed; (e) M. P. Watson, L. E. Overman and R. G. Bergman, J. Am. Chem. Soc., 2007, 129, 5031–5044 CrossRef CAS PubMed.
- F. J. Dinan and H. Tiechelmann, J. Org. Chem., 1964, 29, 892–895 CrossRef CAS.
- A. Rodrigues, E. E. Lee and R. A. Batey, Org. Lett., 2010, 12, 260–263 CrossRef CAS PubMed.
- For a discussion of molecular rearrangements through thermal [1,3] carbon shifts, including the stereochemical consequences of such processes, see: J. E. Baldwin and P. A. Leber, Org. Biomol. Chem., 2008, 6, 36–47 CAS.
- For Ru-catalyzed O-to-N [1,3]-rearrangements of primary benzylic substrates, see: C. S. Yeung, T. H. H. Hsieh and V. M. Dong, Chem. Sci., 2011, 2, 544–551 RSC.
- For Ir-catalyzed O-to-N [1,3]-rearrangements of secondary benzylic substrates, including two unactivated secondary substrates, see: S. Pan, N. Ryu and T. Shibata, Org. Lett., 2013, 15, 1902–1905 CrossRef CAS PubMed.
- For LiI-promoted O-to-N [1,3]-rearrangements of primary benzylic substrates, see: (a) E. L. Lanni, M. A. Bosscher, B. D. Ooms, C. A. Shandro, B. A. Ellsworth and C. E. Anderson, J. Org. Chem., 2008, 73, 6425–6428 CrossRef CAS PubMedLiI-promoted O-to-N [1,3]-rearrangements of alkyl substrates, see: (b) S. Z. Tasker, M. A. Bosscher, C. A. Shandro, E. L. Lanni, K. A. Ryu, G. S. Snapper, J. M. Utter, B. A. Ellsworth and C. E. Anderson, J. Org. Chem., 2012, 77, 8220–8230 CrossRef CAS PubMed.
- (a) For studies relating to thermal [3,3]-sigmatropic rearrangements of secondary propargylic trichloroacetimidates, leading to racemic allenamide derivatives that subsequently form 1,3-diene amides, see: L. E. Overman and L. A. Clizbe, J. Am. Chem. Soc., 1976, 98, 2352–2354 CrossRef CAS For non-enantioselective examples, see: (b) S. Z. Tasker, B. M. Brandsen, K. A. Ryu, G. S. Snapper, R. J. Staples, R. L. DeKock and C. E. Anderson, Org. Lett., 2011, 13, 6224–6227 CrossRef CAS PubMed; (c) S. Z. Tasker, M. A. Bosscher, C. A. Shandro, E. L. Lanni, K. A. Ryu, G. S. Snapper, J. M. Utter, B. A. Ellsworth and C. E. Anderson, J. Org. Chem., 2012, 77, 8220–8230 CrossRef CAS PubMed.
- (a) For an organocatalytic enantioselective O-to-N [1,3]-rearrangements of allylic trichloroacetimidates bearing electron-withdrawing groups in the 2-position, see: S. Kobbelgaard, S. Brandes and K. A. Jorgensen, Chem.–Eur. J., 2008, 14, 1464–1471 CrossRef CAS PubMed; (b) For one example of a Rh-catalyzed O-to-N [1,3]-rearrangements of allyloxy-2-pyridines, see: C. Li, M. Kähny and B. Breit, Angew. Chem. Int. Ed., 2014, 53, 13780–13784 ( Angew. Chem. , 2014 , 126 , 14000–14004 ) Search PubMed; (c) For two examples of O-to-N [1,3]-rearrangements of allyloxy-2-pyridines, see: X. Zhang, Z.-P. Yang, L. Huang and S.-L. You, Angew. Chem. Int. Ed., 2015, 54, 1873–1876 ( Angew. Chem. , 2015 , 127 , 1893–1896 ) CrossRef CAS PubMed.
- L.-J. Cheng and C. J. Cordier, Angew. Chem. Int. Ed, 2015, 54, 13734–13738 ( Angew. Chem. , 2015 , 127 , 13938–13942 ) CrossRef CAS PubMed.
- For reviews of catalytic propargylic substitution reactions, see: (a) Y. Miyake, S. Uemura and Y. Nishibayashi, ChemCatChem, 2009, 1, 342–356 CrossRef CAS; (b) Y. Nishibayashi, Synthesis, 2012, 44, 489–503 CrossRef CAS; (c) D.-Y. Zhang and X.-P. Hu, Tetrahedron Lett., 2015, 56, 283–295 CrossRef CAS; (d) A. F. Adeleke, A. P. N. Brown, L.-J. Cheng, K. A. M. Mosleh and C. J. Cordier, Synthesis, 2017, 49, 790–801 CrossRef CAS.
- For selected examples of N-nucleophiles, see: (a) Y. Imada, M. Yuasa, I. Nakamura and S.-I. Murahashi, J. Org. Chem., 1994, 59, 2282–2284 CrossRef CAS; (b) R. J. Detz, M. M. E. Delville, H. Hiemstra and J. H. van Maarseveen, Angew. Chem. Int. Ed., 2008, 47, 3777–3780 ( Angew. Chem. , 2008 , 120 , 3837–3840 ) CrossRef CAS PubMed; (c) G. Hattori, H. Matsuzawa, Y. Miyake and Y. Nishibayashi, Angew. Chem. Int. Ed, 2008, 47, 3781–3783 ( Angew. Chem. , 2008 , 120 , 3841–3843 ) CrossRef CAS PubMed; (d) G. Hattori, K. Sakata, H. Matsuzawa, Y. Tanabe, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2010, 132, 10592–10608 CrossRef CAS PubMed; (e) A. Yoshida, G. Hattori, Y. Miyake and Y. Nishibayashi, Org. Lett., 2011, 13, 2460–2463 CrossRef CAS PubMed; (f) C. Chang, Y.-H. Wang, X.-H. Hu, Z. Zheng, J. Xu and X.-P. Hu, Adv. Synth. Catal., 2012, 354, 2854–2858 CrossRef; (g) Z. Zheng, G. Deng and Y. Liang, RSC Adv., 2016, 6, 103478–103481 RSC; (h) Z.-T. Liu, Y.-H. Wang, F.-L. Zhu and X.-P. Hu, Org. Lett., 2016, 18, 1190–1193 CrossRef CAS PubMed; (i) Y. Zhou, F.-L. Zhu, Z.-T. Liu, X.-M. Zhou and X.-P. Hu, Org. Lett., 2016, 18, 2734–2737 CrossRef CAS PubMed; (j) C. Zhang, Y.-Z. Hui, D.-Y. Zhang and X.-P. Hu, RSC Adv., 2016, 6, 14763–14767 RSC.
- For selected examples of C-nucleophiles, see: (a) C. Zhang, X.-H. Hu, Y.-H. Wang, Z. Zheng, J. Xu and X.-P. Hu, J. Am. Chem. Soc., 2012, 134, 9585–9588 CrossRef CAS PubMed; (b) F.-L. Zhu, Y. Zou, D.-Y. Zhang, Y.-H. Wang, X.-H. Hu, S. Chen, J. Xu and X.-P. Hu, Angew. Chem. Int. Ed., 2014, 53, 1410–1414 ( Angew. Chem. , 2014 , 126 , 1434–1438 ) CrossRef CAS PubMed; (c) F.-L. Zhu, Y.-H. Wang, D.-Y. Zhang, J. Xu and X.-P. Hu, Angew. Chem. Int. Ed., 2014, 53, 10223–10227 ( Angew. Chem. , 2014 , 126 , 10387–10391 ) CrossRef CAS PubMed; (d) W. Shao, H. Li, C. Liu, C.-J. Liu and S.-L. You, Angew. Chem. Int. Ed, 2015, 54, 7684–7687 ( Angew. Chem. , 2015 , 127 , 7794–7797 ) CrossRef CAS PubMed; (e) Q. Wang, T.-R. Li, L.-Q. Lu, M.-M. Li, K. Zhang and W.-J. Xiao, J. Am. Chem. Soc., 2016, 138, 8360–8363 CrossRef CAS PubMed.
- For selected examples of O-nucleophiles, see: (a) K. Nakajima, M. Shibata and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 2472–2475 CrossRef CAS PubMed; (b) L. Shao, D.-Y. Zhang, Y.-H. Wang and X.-P. Hu, Adv. Synth. Catal., 2016, 358, 2558–2563 CrossRef CAS.
- Previous investigations have suggested copper–allenylidene intermediates may be involved in similar transformations. (a) For a review on the reactivity of metal–allenylidene complexes, see: V. Cadierno, M. P. Gamasa and J. Gimeno, Eur. J. Inorg. Chem., 2001, 571–591 CrossRef CAS; (b) For computational and experimental evidence relating to copper–allenylidenes as well as bimetallic intermediates, see ref. 16d and 18.
- (a) See ESI† for further details of control experiments, optimization data, and the use of other pyridine derivatives as substrates; ; (b) At elevated temperatures, the use of other pyridine derivatives as substrates led to virtually racemic rearrangement products: The 3-nitro-5-H analogue of (±)-4a (30% yield at 50 °C); the 3-H-5-nitro analogue of (±)-4a (19% yield at 100 °C); the unsubstituted 2-alkoxypyridine analogue of (±)-4a (0% yield at 100 °C); the 2-alkoxypyrimidine analogue of (±)-4a (33% yield at 100 °C).
- An explanation for this enantioselectivity trend has been speculated to involve key aryl–aryl interactions between catalyst ligand and substrate, which are absent in the case of 3-alkyl substrates. see ref. 16 for more details.
- Under our rearrangement conditions, the corresponding propargylic acetate, methylcarbonate, 4-nitrobenzoate, and chloride of (±)-7 were unreactive toward substitution using 6a or 6b. No background reaction between 6a or 6b with (±)-7a was observed at rt. Base is required to sequester the acid by-product.
- Low yield is likely due to poor solubility of 6a in toluene; similar results were observed when the transformation was conducted in THF, in which 6a is more soluble.
- For discussions relating to O-alkylation vs. N-alkylation of pyridones, see: (a) D. L. Comins and G. Jianhua, Tetrahedron Lett., 1994, 35, 2819–2822 CrossRef CAS; (b) W. R. Bowman and C. F. Bridge, Synth. Commun., 1999, 29, 4051–4059 CrossRef CAS; (c) C. Li, M. Kähny and B. Breit, Angew. Chem. Int. Ed., 2014, 53, 3780–13784 ( Angew. Chem. , 2014 , 126 , 14000–14004 ) CrossRef PubMed; (d) X. Hao, Z. Xu, H. Lu, X. Dai, T. Yang, X. Lin and F. Ren, Org. Lett., 2015, 17, 3382–3385 CrossRef CAS; (e) S. Gibson, R. Fernando, H. K. Jacobs and A. S. Gopalan, Tetrahedron, 2015, 71, 9271–9281 CrossRef CAS PubMed.
- Y. Tohda, M. Eiraku, T. Nakagawa, Y. Usami, M. Ariga, T. Kawashima, K. Tani, H. Watanabe and Y. Mori, Bull. Chem. Soc. Jpn., 1990, 63, 2820–2827 CrossRef CAS.
- E. R. Kiemele, M. Wathier, P. Bichler and J. A. Love, Org. Lett., 2016, 18, 492–495 CrossRef CAS PubMed.
- For examples of substrate enantioenrichment during related Cu-catalyzed propargylic substitutions, see ref. 16c (s = 2.7) and17d (s = 3.0).
-
(a) An alternative explanation could involve a dynamic kinetic resolution; ;
(b) Quenching the reaction in (eqn (9)) after 1 hour led to 80% (R)-5a with 97:3 er and 16% recovered (S)-4a with 90:10 er. Quenching the reaction in (eqn (10)) after 1 hour led to 90% (S)-5a in 99:1 er and 5% recovered (S)-4a in 56:44 er.
- D. Guillaneaux, S.-H. Zhou, O. Samuel, D. Rainford and H. B. Kagan, J. Am. Chem. Soc., 1994, 116, 9430–9439 CrossRef.
- For a review concerning the kinetic aspects of nonlinear effects, see: D. G. Blackmond, Acc. Chem. Res., 2000, 33, 402–411 CrossRef CAS PubMed.
- For a review, see: D. G. Blackmond, Angew. Chem. Int. Ed., 2005, 44, 4302–4320 ( Angew. Chem. , 2005 , 117 , 4374–4393 ) CrossRef CAS PubMed.
- (a) J. Burés, Angew. Chem. Int. Ed., 2016, 55, 2028–2031 ( Angew. Chem. , 2016 , 128 , 2068–2071 ) CrossRef PubMed; (b) J. Burés, Angew. Chem. Int. Ed., 2016, 55, 16084–16087 ( Angew. Chem. , 2016 , 128 , 16318–16321 ) CrossRef PubMed.
- t[cat]1.9 also provides a close overlay of the variable time normalized data.
- See ESI.†.
- (a) See ref. 19a; ; (b) evidence for alkenyl dicopper species related to 14 have been reported for copper-catalyzed azide–alkyne couplings: B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc01042g |
| This journal is © The Royal Society of Chemistry 2017 |