
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
“On demand” redox buffering by H2S contributes to antibiotic resistance revealed by a bacteria-specific H2S donor†
Prashant
Shukla‡
ae,
Vinayak S.
Khodade‡
b,
Mallojjala
SharathChandra
b,
Preeti
Chauhan
b,
Saurabh
Mishra
a,
Shivakumara
Siddaramappa
c,
Bulagonda Eswarappa
Pradeep
 d,
Amit
Singh
*a and
Harinath
Chakrapani
*b
d,
Amit
Singh
*a and
Harinath
Chakrapani
*b
aDepartment of Microbiology and Cell Biology, Centre for Infectious Disease and Research, Indian Institute of Science, Bangalore 5600012, Karnataka, India. E-mail: asingh@mcbl.iisc.ernet.in
bDepartment of Chemistry, Indian Institute of Science Education and Research Pune, Dr Homi Bhabha Road, Pashan, Pune 411 008, Maharashtra, India. E-mail: harinath@iiserpune.ac.in
cInstitute of Bioinformatics and Applied Biotechnology, Bengaluru 5600100, Karnataka, India
dSri Sathya Sai Institute of Higher Learning, Vidyagiri, Prasanthi Nilayam, Andhra Pradesh, India
eInternational Centre for Genetic Engineering and Biotechnology, New Delhi, India
First published on 27th April 2017
Abstract
Understanding the mechanisms of antimicrobial resistance (AMR) will help launch a counter-offensive against human pathogens that threaten our ability to effectively treat common infections. Herein, we report bis(4-nitrobenzyl)sulfanes, which are activated by a bacterial enzyme to produce hydrogen sulfide (H2S) gas. We found that H2S helps maintain redox homeostasis and protects bacteria against antibiotic-triggered oxidative stress “on demand”, through activation of alternate respiratory oxidases and cellular antioxidants. We discovered, a hitherto unknown role for this gas, that chemical inhibition of H2S biosynthesis reversed antibiotic resistance in multidrug-resistant (MDR) uropathogenic Escherichia coli strains of clinical origin, whereas exposure to the H2S donor restored drug tolerance. Together, our study provides a greater insight into the dynamic defence mechanisms of this gas, modes of antibiotic action as well as resistance while progressing towards new pharmacological targets to address AMR.
Introduction
Maintenance of redox homeostasis is fundamental to cellular growth and survival. Induction of dysfunctional redox environment is a common mechanism used against pathogens by immune cells.1 In the past several decades, it has been well established that gases such as hydrogen sulfide (H2S) and nitric oxide (NO) affect cellular redox balance.2 Bacteria-derived-H2S through microbiota contribute significantly to repair mechanisms and are vital for the health of the gastrointestinal tract.3 Bacterial H2S is also implicated as a cytoprotective agent against antibiotic-induced stress, thereby enhancing antibiotic tolerance.4 Oxidant remediation by bacterial H2S is operational, but precise mechanisms of protection remain to be completely elucidated.5 Mapping out these cytoprotective mechanisms will help progress towards new strategies to combat the growing threat of antimicrobial resistance (AMR).6 Due to the dwindling arsenal of antibiotics, AMR is possibly the biggest problem that this current generation will face. In order to address this complex socioeconomic public health problem, multiple methodologies are necessary including a better understanding of the mechanisms of antibiotic action and factors contributing to antibiotic resistance.7 Herein, we systematically investigated the dynamic effects of H2S in protecting bacteria from antibiotic-induced stress and the role of H2S in modulating AMR.Being a gaseous species, reliable detection8 as well as controlled and site-specific generation of H2S within cells is fundamental to understanding its biology.9 Numerous donors of H2S (ref. 3) are in development but none, to our knowledge, distinguish one type of cells over others.10 Enzymes, as metabolic triggers for activation of donors, offer distinct advantages as they facilitate localization of H2S. A H2S generating functional group is tethered to a substrate for an enzyme that is normally expressed in cells of interest (Fig. 1a). Upon entry into cells, metabolism by the target enzyme frees up the active H2S generator inside cells thus achieving localized delivery. Recently, two esterase-activated H2S donors were reported with wide potential applications in cellular studies (Fig. 1b).11 However, generating H2S specifically in certain cells over others might be problematic when using esterase as a trigger. We chose E. coli nitroreductase (NTR), an oxygen-insensitive bacterial enzyme that is frequently expressed in bacteria but not in mammalian cells.12 Geminal dithiols are reported to undergo hydrolysis in buffer to produce H2S;13 4-nitroaryl groups are known substrates for NTR. We hence designed 1, an NTR-activated H2S donor (Fig. 1b).
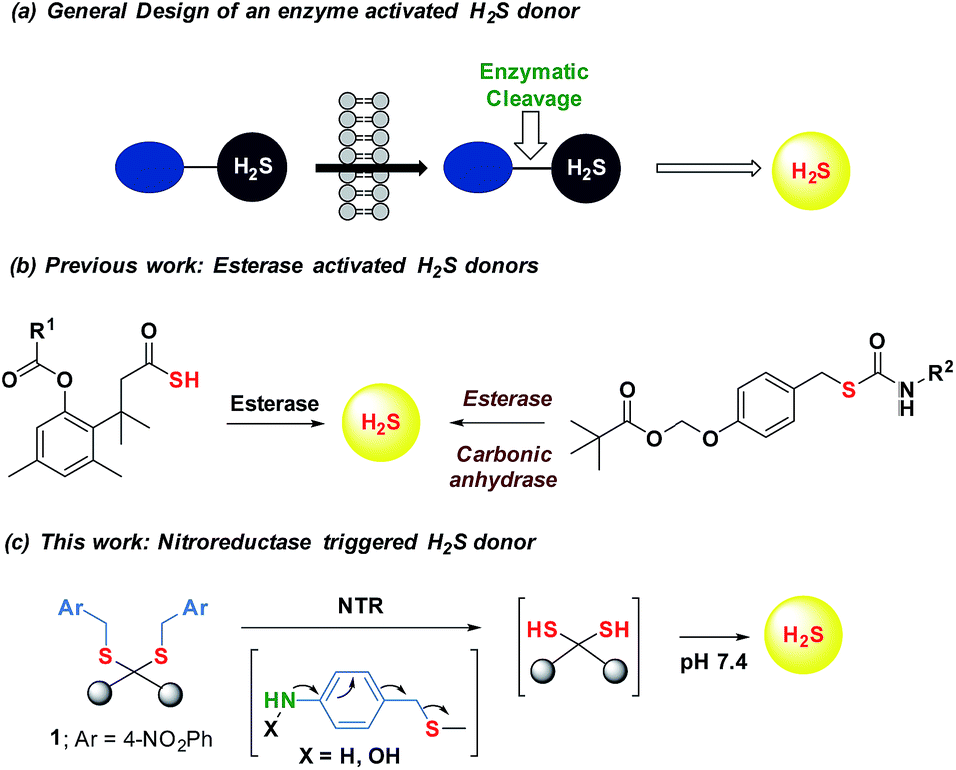 | ||
| Fig. 1 (a) General design of an enzyme activated H2S donor. (b) Esterase sensitive H2S donor; (b) esterase activated H2S and COS/H2S donor; R1 can be a non-steroidal anti-inflammatory drug (NSAID) while R2 is an aryl group or benzyl (c) bacterial enzyme nitroreductase (NTR) activated H2S donor and inset contains compounds synthesized in this study. | ||
Results and discussion
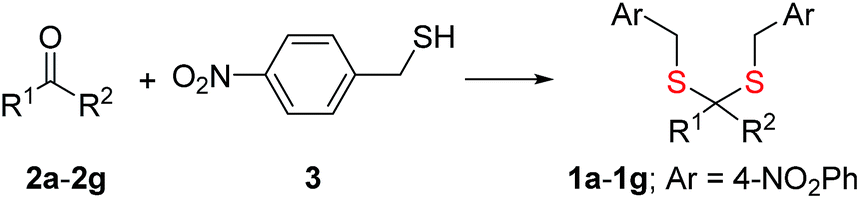
Synthesis of 1 is achieved by the reaction of a variety of ketones (2) with 4-nitrobenzyl thiol (3) (Table 1). A H2S-sensitive dye BODIPY-azide 4a was employed to detect H2S. BODIPY-azide is known to be reduced by H2S to produce a fluorescent amine 4b.14 Compounds 1a–1g were independently exposed to NTR and all compounds were found to generate H2S under these conditions (Fig. 2a). The cyclopentyl derivative 1c was found to be slightly better than the cohort of donors tested, and this compound was used for further studies.|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Ketone | Prod | Yield % |
| 1 | CH3 | CH3 | 2a | 1a | 80 |
| 2 | Et | Et | 2b | 1b | 78 |
| 3 | Cyclopentyl | 2c | 1c | 70 | |
| 4 | Cyclohexyl | 2d | 1d | 71 | |
| 5 | Ph | CH3 | 2e | 1e | 60 |
| 6 | 4-FPh | CH3 | 2f | 1f | 42 |
| 7 | Thiophene | CH3 | 2g | 1g | 38 |
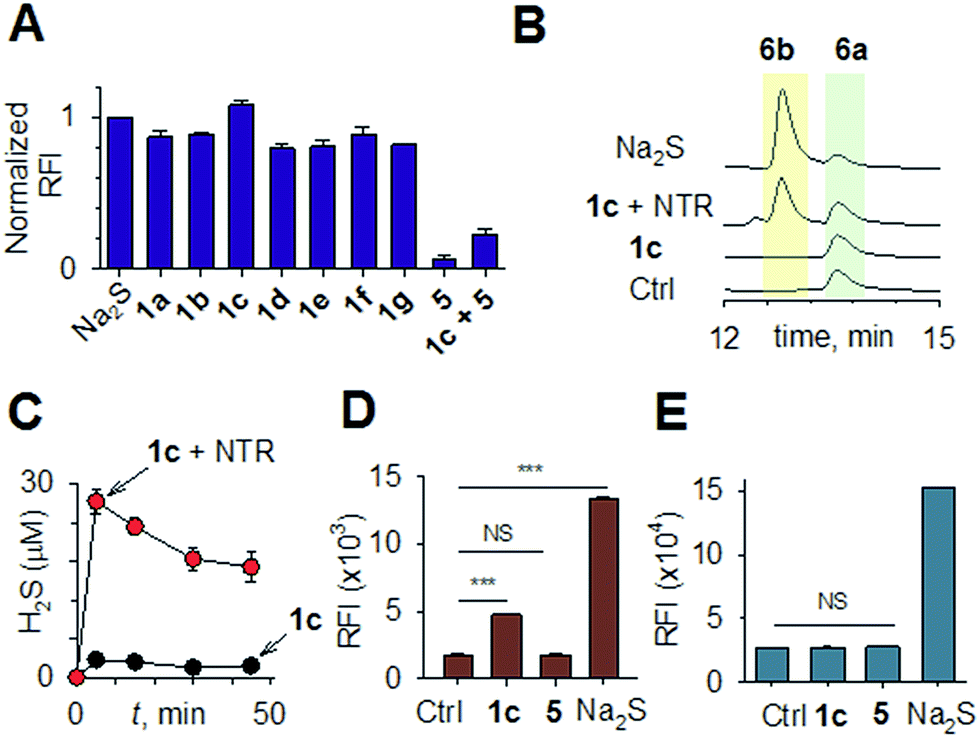 | ||
| Fig. 2 (a) Hydrogen sulfide generated during incubation of 1a–1g (50 μM) with NTR in HEPES buffer pH 7.4 was estimated using a BODIPY-based sensor. (b) A monobromobimane (6a) method for the estimation of hydrogen sulfide. The formation of thioether 6b supports the intermediacy of H2S. (c) Time course of H2S generation during incubation of 1c in pH 7.4 buffer alone and in the presence of NTR was assessed by BODIPY-based sensor 4a. (d) Flow cytometry analysis of intrabacterial H2S generation in E. coli using the probe 4a. 1c was incubated for 20 minutes ***p-value = 0.0001; NS = not significant. (e) Flow cytometry analysis of intracellular H2S generation in THP-1 cells. 1c was incubated for 20 minutes (100 μM) and 4a was used for H2S detection NS = not significant. | ||
A monobromobimane (mBBr, 6a) assay (with some modifications) was next used to confirm the production of H2S.15 The electrophile mBBr reacts with sulfide anion to produce a thioether, which contains two bimane units (6b). When 6a was treated with Na2S in a pH 7.4 buffer, as expected, 6b was formed (Fig. 2b). Under similar conditions, when 1c was co-incubated with 6a and NTR, we found evidence for the formation of 6b again supporting 1c as a source of H2S when incubated with NTR (Fig. 2b). Next, NBD-fluorescein, a H2S sensitive dye, was synthesized and incubated in the presence of 1c and NTR.16 Again, we found a distinct increase in fluorescence attributable to H2S generation (Fig. S1, ESI†). Thus, the formation of H2S was validated by several independent assays suggesting that this compound is a reliable donor of H2S.
The H2S donor 1c was able to maintain elevated levels of H2S over 45 min (Fig. 2c). The formation of a hydroxylamino- or amino-aryl derivative (Fig. 1c, Scheme S2, ESI†), which self-immolates to generate a geminal dithiol, was likely. This geminal dithiol should hydrolyze to produce H2S and a ketone.13 Accordingly, when 1e was incubated in buffer in the presence of Zn and ammonium formate, acetophenone was formed, supporting the proposed mechanism (Fig. S2, ESI†).
Having established that 1c generated H2S in cell-free conditions in the presence of a bacterial enzyme, the ability of this compound to permeate cells to be metabolized by NTR to generate H2S was evaluated. First, an HPLC-based method was used: E. coli cells were incubated with the H2S-sensitive dye 4a and 1c. Cells were lysed and HPLC analysis of the lysate revealed the formation of 4b supporting H2S generation (Fig. S3, ESI†); a similar result was recorded for a variety of bacteria supporting the broad utility of this donor. Next, flow cytometry analysis revealed the generation of H2S inside intact bacterial cells when treated with 1c, supporting the ability of this donor to enhance H2S levels in live bacterial cells (Fig. 2d). Next, 4-nitrobenzylbenzoate 5 (a likely competitive inhibitor) was synthesized using a previously reported method.17 This compound was by itself incapable of generating H2S in the presence of NTR and also inhibited H2S generation from 1c (Fig. 2a). The negative control 5 did not generate H2S within the bacteria, suggesting that the metabolism of the nitro group does not contribute to H2S production (Fig. 2d). The H2S donor 1c was ineffective in generating H2S in E. coli strains lacking NTR (Fig. S4, ESI†), confirming NTR-specificity in vivo. As NTR is predominantly produced in bacteria but not in mammalian cells, the H2S donor 1c must selectively enhance H2S in bacteria. Human monocytic cells (THP-1) were treated with 1c, and H2S levels were assessed by flow cytometry. Herein, we find that while Na2S was capable of enhancing H2S levels within THP-1 cells, 1c remained completely ineffective (Fig. 2e). Thus, 1c was selective in its ability to enhance H2S in bacteria over mammalian cells (Fig. S5, ESI†). To our knowledge, this is the first example of a H2S donor with species selectivity. Thus, this study lays the foundation for novel methodologies for site-specific enhancement of H2S using this class of H2S donors, for example, selectively enhancing H2S in microbiota to study the effects of this gas on colorectal cancer and other similar pathophysiologies is possible.2
To begin understanding the mechanisms of H2S-mediated oxidation remediation, we used a non-invasive redox biosensor (roGFP2) and assessed dynamic changes in the cytoplasmic redox potential of E. coli in response to oxidative stress.18 An increase in 405/488 nm excitation ratio of roGFP2 indicates oxidative stress, while the reverse suggests reductive changes.18b We first exposed E. coli expressing roGFP2 to 1c and measured the roGFP2 biosensor response, and no significant changes in 405/488 ratio were observed (Fig. 3a). Hence, H2S alone did not affect ambient redox-potential of E. coli. In contrast, oxidative challenge with H2O2, a reactive oxygen species (ROS), rapidly increased the 405/488 ratio, and pre-treatment with 1c significantly reversed this response (Fig. 3a). However, pre-treatment with either Na2S or 5 had no influence on H2O2-induced oxidative changes in the biosensor response (Fig. S6, ESI†).
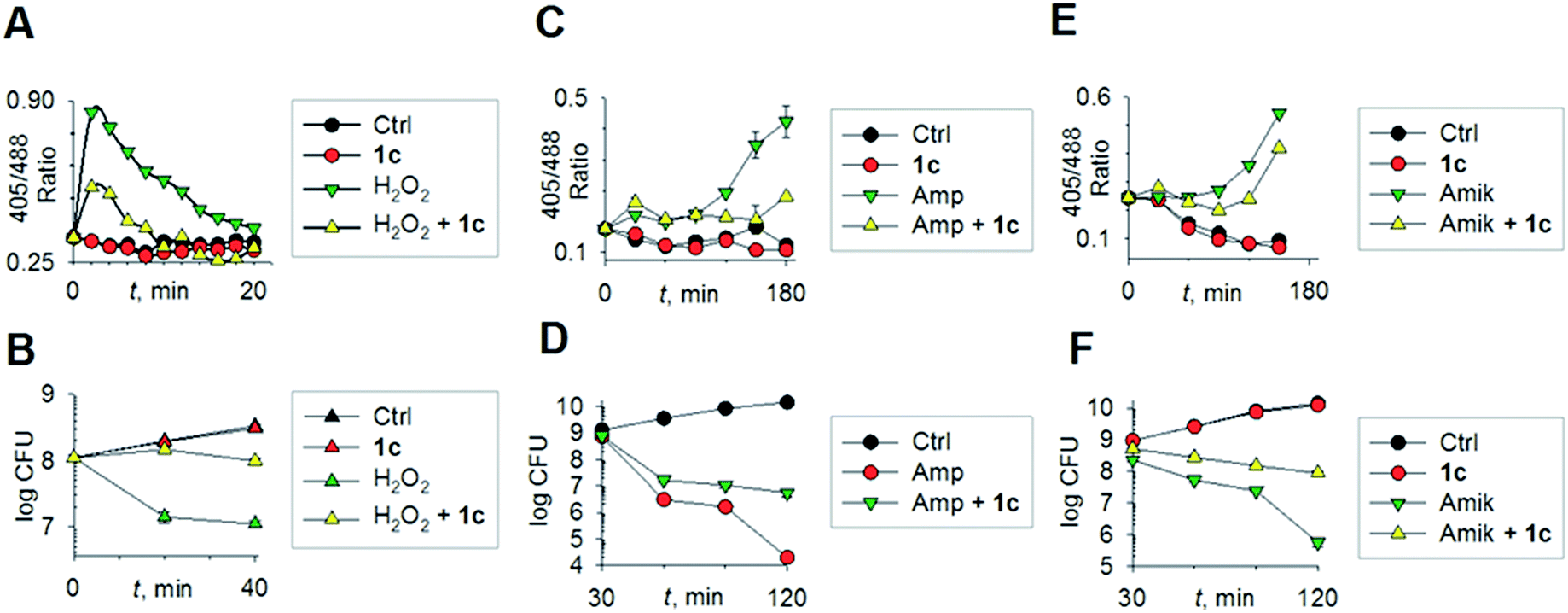 | ||
| Fig. 3 (a) Reduction–oxidation sensitive GFP (roGFP2) was used to measure dynamic changes in cytoplasmic redox potential of E. coli upon exposure to: H2O2, 1 mM; 1c, 100 μM; (b) time-kill analysis of E. coli treated with hydrogen peroxide (1 mM) and 1c (100 μM) during 40 min. (c) Dynamic changes in cytoplasmic redox potential of E. coli upon exposure to: Amp, 5 μg mL−1; 1c, 100 μM; (d) time-kill analysis of E. coli treated with Amp (5 μg mL−1) and 1c (100 μM) during 120 min. (e) Dynamic changes in cytoplasmic redox potential of E. coli upon exposure to: Amik, 20 μg mL−1; 1c, 100 μM; (f) time-kill analysis of E. coli treated with Amik (20 μg mL−1) and 1c (100 μM) during 120 min. | ||
Importantly, protective influence of 1c on intrabacterial redox potential translated into significantly higher resistance displayed by E. coli against bactericidal concentrations of H2O2 (Fig. 3b). Interestingly, H2S itself did not have any significant effect on the growth of E. coli. Thus, intervention by H2S occurs when other endogenous oxidant–remediation systems are overwhelmed. This property is consistent with the lower reduction potential of H2S, when compared with major cellular thiols such as glutathione,19 and thus affords H2S a unique role in cellular redox chemistry. Furthermore, in contrast with other routinely used antioxidants in redox biology, such as thiourea and bipyridyl, 1c does not significantly affect bacterial growth (Fig. 3b), suggesting that this tool would be appropriate for studying H2S-mediated response to dynamic redox alterations during antibiotic-induced stress and lethality.
The emerging model for antibiotic lethality involves the induction of complex redox and metabolic alterations as a consequence of drugs' interaction with their specific targets.5,20 Thus, it is important to understand the dynamic effects of H2S in mitigating antibiotic-induced redox stress.21 To accomplish this, we exposed E. coli to clinically relevant concentrations of the bactericidal antibiotic ampicillin (Amp; cell wall targeting), and an oxidative shift was recorded (Fig. 3c).21a,22 More importantly, pre-treatment with 1c reduced the degree of oxidative shift induced by Amp, resulting in significant tolerance to antibiotics (Fig. 3d). Amp-mediated increase in roGFP2 ratios emerged earlier than the time points at which significant killing was observed, indicating that oxidative stress precedes death, and 1c-derived H2S protects bacteria by maintaining cytoplasmic redox potential.
Similar results were recorded for amikacin (Fig. 3e–f), a translation inhibitor, and ciprofloxacin, a replication inhibitor (Fig. S7, ESI†). Altogether, these results demonstrate that elevating endogenous H2S levels can arrest antibiotics-triggered redox stress and killing.
Mechanisms of H2S-mediated protection from antibiotic-induced lethality are poorly understood. It has been shown that H2S elevates cellular antioxidant capacity and suppresses iron load in order to mitigate antibiotic-linked ROS production.23 Since sulfide is a potent ligand of copper and heme moieties, H2S efficiently inhibits aerobic respiration by targeting copper-heme containing cytochrome bo oxidase (CyoA).24 Under these conditions, respiration becomes primarily dependent upon less energy-efficient cytochrome bd oxidase (CydB).24 Interestingly, modulation of cytochrome oxidases expression is known to influence antibiotic toxicity.21b Therefore, we assessed whether terminal oxidases are important contributory factors in H2S-mediated antibiotic tolerance. First, quantitative reverse transcription-PCR (qRT-PCR) analysis of E. coli cells in the absence or presence of 1c was conducted (see ESI†). A significant down-regulation of the genes encoding CyoA was observed with 1c-treated bacteria (Fig. 4a). The expression of alternate oxidases was however either maintained (cytochrome bd oxidase I [cydB]) or highly induced (cytochrome bd oxidase II [appY]) by H2S (Fig. 4a). During growth under low-O2 tension, E. coli down-regulated cyo operon and upregulated cyd and app operons, indicating that H2S triggered genetic and physiological changes comparable to O2-limitation.25 Amp treatment reversed the influence of H2S on the expression of cytochrome oxidases as cyoA and cydB transcripts were significantly induced and repressed, respectively, compared to untreated cells (Fig. 4a). The appY transcript remained down-regulated in response to Amp. Data suggest that Amp treatment promotes respiration via energetically efficient CyoA, which is consistent with a recent study demonstrating acceleration in aerobic respiration in response to bactericidal antibiotics.21b
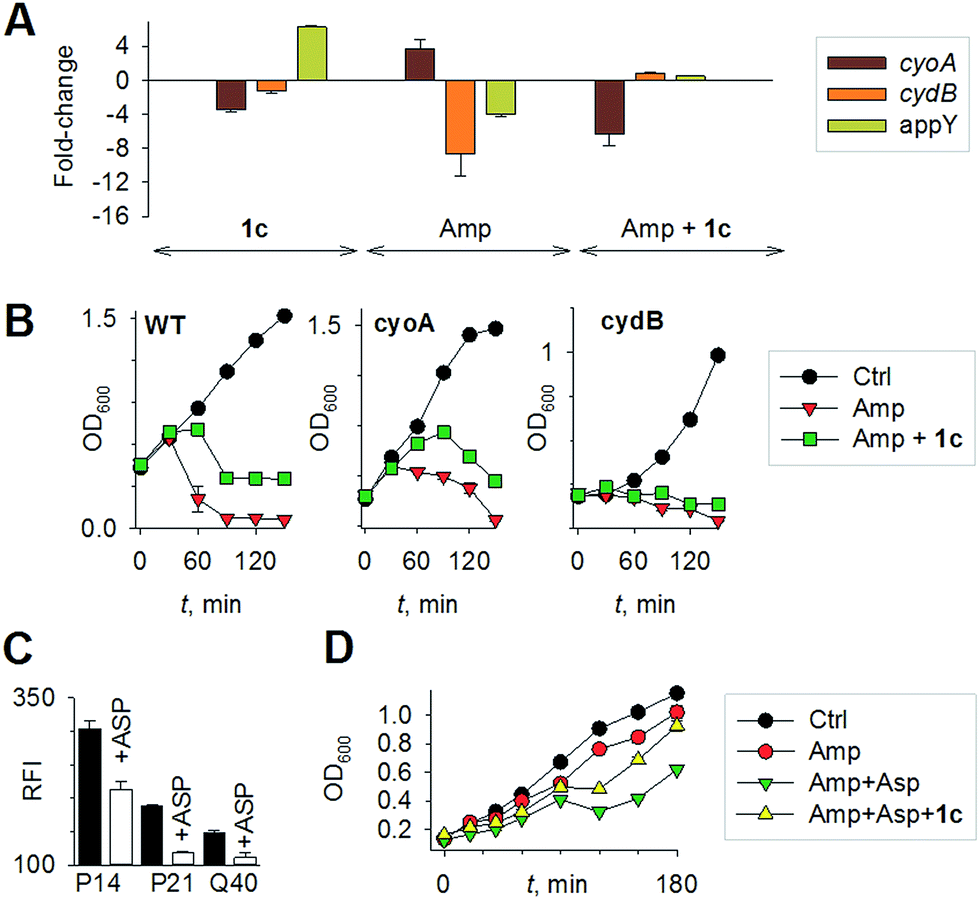 | ||
| Fig. 4 (a) qRT-PCR analysis of E. coli cells treated with 1c alone, Amp alone. The fold-change is with respect to untreated control; 1c + Amp where fold-change is with respect to Amp-treated cells. Fold change for each transcript was calculated by normalizing expression with the housekeeping 16SrRNA transcript. A 2-fold (p < 0.05) increase or decrease in expression was considered significant in all cases. (b) Growth curves of E. coli treated with Amp (5 μg mL−1) with and without 1c (100 μM); Ctrl is untreated bacteria: wild-type (WT); cyoA mutant; cydB mutant. (c) H2S levels measured using the H2S-sensitive dye 4a in the presence of ASP, a biosynthesis inhibitor of H2S generation (d) bacterial growth measurements: Amp (100 μg mL−1); Asp (4 mM) and 1c (100 μM) were used in this study. | ||
Having observed divergent effects of H2S and Amp on cytochrome oxidases gene expression, we next examined the outcome of H2S and Amp combination on transcription. qRT-PCR analysis of E. coli pre-treated with 1c, followed by exposure to Amp showed severely down-regulated expression cyoA, whereas expression of cydB and appY was robustly maintained (Fig. 4a) compared to that of Amp alone. Thus, maintenance of a respiratory flux through cytochrome bd oxidase I/II in response to H2S treatment may be a key trait that permits adaptation upon subsequent exposure to antibiotics. To examine this possibility, we assessed cell-killing in respiratory mutants lacking either cyoA or cydB. While 1c-pretreatment resulted in a significant attenuation of Amp lethality in the case of CyoA mutant (like WT strain), it was completely ineffective in protecting cydB mutant (Fig. 4b). In LB medium, WT, cyoA, and cydB strains showed comparable growth profiles in the absence or presence of 1c, indicating that the differences in Amp susceptibility are not a consequence of reduced growth rates. Sustenance of cydB expression in response to H2S-Amp combination, coupled with maintenance of H2S-mediated antibiotic tolerance in cyo mutant (where aerobic respiration is mainly carried out by CydB) but not in cydB mutant, suggests that the H2S effect is likely to be dependent upon cydB.24 Along with its role in respiration, CydB from E. coli has been shown to reduce H2O2 by displaying catalase and quinol peroxidase activities.26 Therefore, maintenance of cydB expression by H2S can potentiate antibiotic tolerance by bolstering the bacterial antioxidant capacity. In concurrence with this result, displayed heightened sensitivity to H2O2 compared to cyoA and WT strains (Fig. S8, ESI†).
In addition, consistent with previous results,4d we observed that H2S is ineffective in protecting cells that lack cytoplasmic catalase and peroxidase (KatA, KatE, ahpCF; HPX-) from Amp-induced lethality (Fig. S9, ESI†). Altogether, data implicate a central role for cytochrome bd oxidase and oxidant-remediation mechanisms in diminishing the effectiveness of the antibiotic by H2S (Fig. 5).27
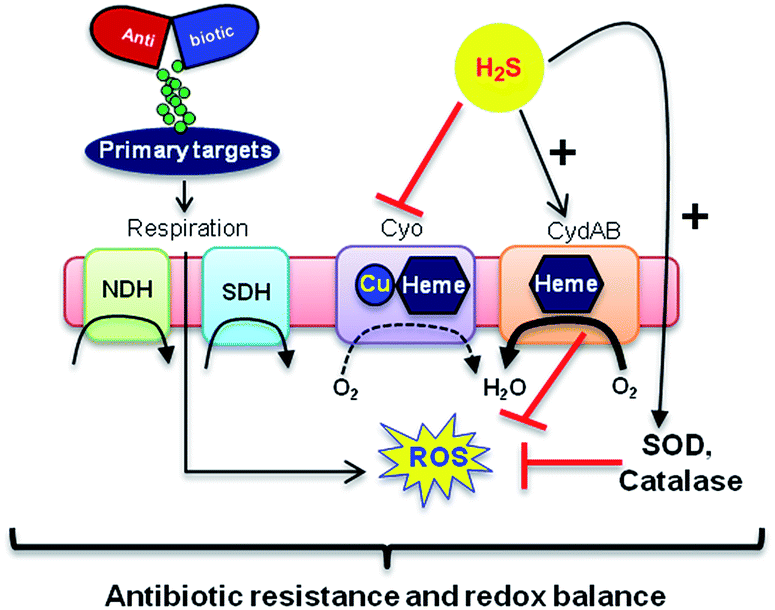 | ||
| Fig. 5 Evolving model for H2S mediated antibiotic tolerance. Bactericidal antibiotics alters respiration and metabolism to elevate the production of endogenous ROS. Based on our findings, we propose that bacterial H2S provides tolerance to antibiotics by two mechanisms; (i) down-regulation of energy-efficient cytochrome bo oxidase (Cyo) and induction of less-energy efficient cytochrome bd oxidase I/II (CydAB/AppBC) to maintain respiratory flux and redox balance, and (ii) augmentation of antioxidant capacity by elevating catalase and superoxide dismutase (SOD) activities. NDH: NADH dehydrogenase and SDH: succinate dehydrogenase. | ||
Finally, in order to examine if elevated endogenous H2S levels are associated with drug resistance in the physiological context of human infections, we measured the intracellular H2S levels of several multidrug-resistant (MDR) E. coli strains isolated from patients (Table S2, ESI† for resistance profile) suffering from urinary tract infections (UTI). The endogenous H2S levels were considerably higher than WT indicating a possible functional role for H2S in antibiotic resistance (Fig. S10, ESI†).28 In the presence of a well-established 3-mercaptopyruvates sulfurtransferase (3-MST) inhibitor (aspartate, Asp), we found that H2S levels significantly diminished (Fig. 4c).4d To understand the functional relevance of endogenous H2S levels in drug resistance, we monitored resistance of P14 strain to Amp. We found that pre-treatment with Asp efficiently inhibited the growth in response to Amp (Fig. 4d). More-importantly, co-treatment with Asp and 1c, significantly restored resistance to Amp in the strain P14 (Fig. 4d). Altogether, these findings revealed a previously unknown contribution of H2S in cooperating with the genetic mechanisms of antibiotic resistance (Fig. 5). Further study is needed to examine H2S-mediated mechanisms contributing to the emergence of drug-resistance in clinical strains.5,28 Amongst the major infectious diseases, UTI affects millions and is further complicated by conditions such as diabetes. E. coli has now become resistant to most major classes of antibiotics and therefore there is an urgent need to develop new therapeutics. Recently, Berkowitz and co-workers developed a CBS inhibitor that helps to prevent the deleterious effects of enhanced H2S such as neuronal cell death during episodes of stroke.29 The inhibitors that were developed in their study showed a marked diminution in neuronal cell death compared with an untreated control. It is likely that inhibitors of 3-MST may find similar application in sensitizing resistant pathogens.30 Our results provide a sound pharmacological basis for the design of inhibitors of biosynthesis of H2S as a possible adjuvant.29–31 Furthermore, we identified critical aspects of bacterial physiology that could be exploited as part of new potentiation strategies. For examples, targeting antioxidant enzymes and alternate respiratory complexes (Cyd/App) is likely to enhance the killing potential of antibiotics. A combination of molecules/drugs targeting H2S, antioxidants, and respiration could have a remarkable impact on drug-resistance and clinical outcomes.
Conclusions
In summary, we report a new H2S donor that reliably and selectively enhances H2S within bacteria. An application of our new tool clearly revealed that H2S is a key player in the maintenance of intracellular redox balance of bacteria to counteract a lethal degree of oxidative stress induced by antibiotics. The critical role that H2S played in modulating drug resistance is also shown. Antibiotic resistance is emerging as possibly the biggest global health challenge for this generation. Therefore understanding pathogen defence mechanisms and their consequences in drug resistance is critical. From the evolutionary perspective, H2S generating enzymes are prevalent in most sequenced bacterial genomes including environmental bacteria, indicating a naturally conserved role of H2S in ensuring survival. It is likely that H2S producing capability is under the selective pressure in diverse environmental bacteria that arises due to antimicrobials secreted by other bacteria and fungi inhabiting the same niche. Our study presents significant advances towards a complete understanding of antibiotic-induced stress and cytoprotective mechanisms of H2S.Acknowledgements
The authors thank the Department of Science and Technology (DST, Grant number EMR/2015/000668), DBT-IISc program (AS), and the Council for Scientific and Industrial Research (CSIR) for financial support. The authors thank James Imlay, University of Illinois at Urbana-Champaign and Antonio Valle Gallardo, Universidad de Cádiz for E. coli mutants. This work was supported by the Wellcome-DBT India Alliance grant, WT-DBT/500034-Z-09-Z and IA/S/16/2/502700 (A. S.), and DBT-IISc program. A. S. is a Wellcome DBT India Alliance Senior Fellow. The authors thank Vasista Adiga, IISc for help with FACS analysis.Notes and references
- F. C. Fang, Nat. Rev. Microbiol., 2004, 2, 820 CrossRef CAS PubMed.
- C. Szabo, Nat. Rev. Drug Discovery, 2007, 6, 917 CrossRef CAS PubMed.
- J. L. Wallace and R. Wang, Nat. Rev. Drug Discovery, 2015, 14, 329 CrossRef CAS PubMed.
- (a) L. Luhachack and E. Nudler, Curr. Opin. Microbiol., 2014, 21, 13 CrossRef CAS PubMed; (b) I. Gusarov and E. Nudler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13855 CrossRef CAS PubMed; (c) I. Gusarov, K. Shatalin, M. Starodubtseva and E. Nudler, Science, 2009, 325, 1380 CrossRef CAS PubMed; (d) K. Shatalin, E. Shatalina, A. Mironov and E. Nudler, Science, 2011, 334, 986 CrossRef CAS PubMed.
- D. J. Dwyer, J. J. Collins and G. C. Walker, Annu. Rev. Pharmacol. Toxicol., 2015, 55, 313 CrossRef CAS PubMed.
- D. J. Dwyer, M. A. Kohanski and J. J. Collins, Curr. Opin. Microbiol., 2009, 12, 482 CrossRef CAS PubMed.
- A. R. M. Coates, G. Halls and Y. Hu, Br. J. Pharmacol., 2011, 163, 184 CrossRef CAS PubMed.
- (a) Z. Liang, T.-H. Tsoi, C.-F. Chan, L. Dai, Y. Wu, G. Du, L. Zhu, C.-S. Lee, W.-T. Wong, G.-L. Law and K.-L. Wong, Chem. Sci., 2016, 7, 2151 RSC; (b) X. Wang, J. Sun, W. Zhang, X. Ma, J. Lv and B. Tang, Chem. Sci., 2013, 4, 2551 RSC; (c) Y. Qian, L. Zhang, S. Ding, X. Deng, C. He, X. E. Zheng, H.-L. Zhu and J. Zhao, Chem. Sci., 2012, 3, 2920 RSC.
- Y. Zhao, T. D. Biggs and M. Xian, Chem. Commun., 2014, 50, 11788 RSC.
- (a) Y. Zhao and M. D. Pluth, Angew. Chem., Int. Ed., 2016, 55, 14638 CrossRef CAS PubMed; (b) J. Kang, Z. Li, C. L. Organ, C.-M. Park, C.-t. Yang, A. Pacheco, D. Wang, D. J. Lefer and M. Xian, J. Am. Chem. Soc., 2016, 138, 6336 CrossRef CAS PubMed; (c) S. Le Trionnaire, A. Perry, B. Szczesny, C. Szabo, P. G. Winyard, J. L. Whatmore, M. E. Wood and M. Whiteman, MedChemComm, 2014, 5, 728 RSC.
- (a) Y. Zheng, B. Yu, K. Ji, Z. Pan, V. Chittavong and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 4514 CrossRef CAS PubMed; (b) P. Chauhan, P. Bora, G. Ravikumar, S. Jos and H. Chakrapani, Org. Lett., 2017, 19, 62 CrossRef CAS PubMed.
- (a) P. R. Race, A. L. Lovering, R. M. Green, A. Ossor, S. A. White, P. F. Searle, C. J. Wrighton and E. I. Hyde, J. Biol. Chem., 2005, 280, 13256 CrossRef CAS PubMed; (b) S. Zenno, H. Koike, M. Tanokura and K. Saigo, J. Biochem., 1996, 120, 736 CrossRef CAS PubMed; (c) A. B. Mauger, P. J. Burke, H. H. Somani, F. Friedlos and R. J. Knox, J. Med. Chem., 1994, 37, 3452 CrossRef CAS PubMed.
- (a) N. O. Devarie-Baez, P. E. Bagdon, B. Peng, Y. Zhao, C.-M. Park and M. Xian, Org. Lett., 2013, 15, 2786 CrossRef CAS PubMed; (b) Y. Zhao, J. Kang, C.-M. Park, P. E. Bagdon, B. Peng and M. Xian, Org. Lett., 2014, 16, 4536 CrossRef CAS PubMed.
- T. Saha, D. Kand and P. Talukdar, Org. Biomol. Chem., 2013, 11, 8166 CAS.
- X. Shen, C. B. Pattillo, S. Pardue, S. C. Bir, R. Wang and C. G. Kevil, Free Radical Biol. Med., 2011, 50, 1021 CrossRef CAS PubMed.
- C. Wei, Q. Zhu, W. Liu, W. Chen, Z. Xi and L. Yi, Org. Biomol. Chem., 2014, 12, 479 CAS.
- A. T. Dharmaraja and H. Chakrapani, Org. Lett., 2014, 16, 398 CrossRef CAS PubMed.
- (a) P. Tyagi, A. T. Dharmaraja, A. Bhaskar, H. Chakrapani and A. Singh, Free Radical Biol. Med., 2015, 84, 344 CrossRef CAS PubMed; (b) A. Bhaskar, M. Munshi, S. Z. Khan, S. Fatima, R. Arya, S. Jameel and A. Singh, J. Biol. Chem., 2014 Search PubMed; (c) A. Bhaskar, M. Chawla, M. Mehta, P. Parikh, P. Chandra, D. Bhave, D. Kumar, K. S. Carroll and A. Singh, PLoS Pathog., 2014, 10, e1003902 Search PubMed.
- Q. Li and J. R. Lancaster Jr, Nitric Oxide, 2013, 35, 21 CrossRef CAS PubMed.
- (a) M. A. Kohanski, D. J. Dwyer, J. Wierzbowski, G. Cottarel and J. J. Collins, Cell, 2008, 135, 679 CrossRef CAS PubMed; (b) P. Belenky, J. D. Ye, C. B. M. Porter, N. R. Cohen, M. A. Lobritz, T. Ferrante, S. Jain, B. J. Korry, E. G. Schwarz, G. C. Walker and J. J. Collins, Cell Rep., 2015, 13, 968 CrossRef CAS PubMed.
- (a) D. J. Dwyer, P. A. Belenky, J. H. Yang, I. C. MacDonald, J. D. Martell, N. Takahashi, C. T. Y. Chan, M. A. Lobritz, D. Braff, E. G. Schwarz, J. D. Ye, M. Pati, M. Vercruysse, P. S. Ralifo, K. R. Allison, A. S. Khalil, A. Y. Ting, G. C. Walker and J. J. Collins, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E2100 CrossRef CAS PubMed; (b) M. A. Lobritz, P. Belenky, C. B. M. Porter, A. Gutierrez, J. H. Yang, E. G. Schwarz, D. J. Dwyer, A. S. Khalil and J. J. Collins, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8173 CrossRef CAS PubMed.
- M. A. Kohanski, D. J. Dwyer, B. Hayete, C. A. Lawrence and J. J. Collins, Cell, 2007, 130, 797 CrossRef CAS PubMed.
- G. Wu, N. Li, Y. Mao, G. Zhou and H. Gao, Front. Microbiol., 2015, 6, 314 Search PubMed.
- S. Korshunov, K. R. C. Imlay and J. A. Imlay, Mol. Microbiol., 2016, 101, 62 CrossRef CAS PubMed.
- K. A. Salmon, S.-p. Hung, N. R. Steffen, R. Krupp, P. Baldi, G. W. Hatfield and R. P. Gunsalus, J. Biol. Chem., 2005, 280, 15084 CrossRef CAS PubMed.
- S. Al-Attar, Y. Yu, M. Pinkse, J. Hoeser, T. Friedrich, D. Bald and S. de Vries, Sci. Rep., 2016, 6, 27631 CrossRef CAS PubMed.
- Y. Liu and J. A. Imlay, Science, 2013, 339, 1210 CrossRef CAS PubMed.
- R. T. Jones, L. P. Thai and R. P. Silver, Antimicrob. Agents Chemother., 1978, 14, 765 CrossRef CAS PubMed.
- C. D. McCune, S. J. Chan, M. L. Beio, W. Shen, W. J. Chung, L. M. Szczesniak, C. Chai, S. Q. Koh, P. T. H. Wong and D. B. Berkowitz, ACS Cent. Sci., 2016, 2, 242 CrossRef CAS PubMed.
- K. Hanaoka, K. Sasakura, Y. Suwanai, S. Toma-Fukai, K. Shimamoto, Y. Takano, N. Shibuya, T. Terai, T. Komatsu, T. Ueno, Y. Ogasawara, Y. Tsuchiya, Y. Watanabe, H. Kimura, C. Wang, M. Uchiyama, H. Kojima, T. Okabe, Y. Urano, T. Shimizu and T. Nagano, Sci. Rep., 2017, 7, 40227 CrossRef CAS PubMed.
- (a) J. K. Holden, H. Li, Q. Jing, S. Kang, J. Richo, R. B. Silverman and T. L. Poulos, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18127 CrossRef CAS PubMed; (b) J. RubenMorones-Ramirez, J. A. Winkler, C. S. Spina and J. J. Collins, Sci. Transl. Med., 2013, 5, 190 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc00873b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |