
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formylation or methylation: what determines the chemoselectivity of the reaction of amine, CO2, and hydrosilane catalyzed by 1,3,2-diazaphospholene?†
Yu
Lu‡
a,
Zhong-Hua
Gao‡
b,
Xiang-Yu
Chen
b,
Jiandong
Guo
a,
Zheyuan
Liu
a,
Yanfeng
Dang
a,
Song
Ye
 *b and
Zhi-Xiang
Wang
*a
*b and
Zhi-Xiang
Wang
*a
aSchool of Chemistry and Chemical Engineering, University of the Chinese Academy of Sciences, Beijing 100049, China. E-mail: zxwang@ucas.ac.cn
bInstitute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: songye@iccas.ac.cn
First published on 11th September 2017
Abstract
DFT computations have been performed to gain insight into the mechanisms of formylation/methylation of amines (e.g. methylaniline (1a)/2,2,4,4-tetramethylpiperidine (2a)) with CO2 and hydrosilane ([Si]H2, [Si] = Ph2Si), catalyzed by 1,3,2-diazaphospholene ([NHP]H). Different from the generally proposed sequential mechanism for the methylation of amine with CO2, i.e. methylation proceeds via formylation, followed by further reduction of formamide to give an N-methylated amine, the study characterized a competition mechanism between formylation and methylation. The chemoselectivity originates from the competition between the amine and [NHP]H hydride to attack the formyloxy carbon of [Si](OCHO)2 (the insertion product of CO2 into [Si]H2). When the attack of an amine (e.g.1a) wins, the transformation affords formamide (1b) but would otherwise (e.g.2a) result in an N-methylated amine (2c). The reduction of formamide by [Si]H2 or [NHP]H is highly unfavorable kinetically, thus we call attention to the sequential mechanism for understanding the methylation of amine with CO2. In addition, the study has the following key mechanistic findings. The activation of CO2 by [NHP]H establishes an equilibrium: [NHP]H + CO2 ⇄ [NHP]OCHO ⇄ [NHP]+ + HCO2−. The ions play catalytic roles to promote formylation via HCO2− or methylation via[NHP]+. In 1a formylation, HCO2− initiates the reaction, giving 1b and silanol byproducts. However, after the initiation, the silanol byproducts acting as hydrogen transfer shuttles are more effective than HCO2− to promote formylation. In 2a methylation, [NHP]+ promotes the generation of the key species, formaldehyde and a carbocation species (IM17+). Our experimental study corroborates our computed mechanisms.
1. Introduction
The rising concentration of carbon dioxide (CO2) in the atmosphere is one of the key factors for global warming, leading to great efforts to develop effective catalytic routes that convert CO2 to value-added chemicals.1–3 Formylation and methylation of amines with CO2 are promising synthetic strategies to use CO2 as a C1 carbon source.4 In 1998, Vaska and coworkers developed the first Pt-catalyzed formylation of amine with CO2 and H2.5 This study has encouraged further developments using other transition metal catalysts6 or metal-free catalysts.7 In 2012, Cantat and coworkers achieved the first organocatalytic formylation of amines with CO2 and hydrosilane, catalyzed by triazabicyclodecene (TBD).8 Since then, more similar transformations were reported.9 In 2013, Beller and coworkers reported the first methylation of amine with CO2 and hydrosilane, catalyzed by a ruthenium complex.10 More similar transformations were later developed.11 It is worth mentioning that Cantat et al. also developed metal-free methylation of CO2 with amines.12 Furthermore, transition metal catalyzed methylation of amines with CO2 and H2 has also been accomplished by several groups.13Previously, we studied the catalytic mechanisms of CO2 reduction to methanol14 and methane.15 In this context, we were intrigued by the catalytic reactions developed by Kinjo and coworkers.16 They used 1,3,2-diazaphospholene ([NHP]H) to catalyze the formylation of amines ([N]H) with CO2 and hydrosilane (Ph2SiH2 = [Si]H2) (e.g. eqn (1) in Scheme 1). Interestingly, two amines (2a and 3a) were found to be exceptional, affording N-methylated amines (2c and 3c). They attributed 2c and 3c to the further reductions of 2b and 3b, respectively, complying with the general consideration that methylation takes place sequentially through formylation, giving formamide, followed by the reduction of formamide.10,17 Nevertheless, we conceived that this mechanism may not be true in the present system. First, due to the smaller steric effect of 1b compared to 2b, 1b should be reduced more easily than 2b, but eqn (1) affords 1b rather than 1c. Second, if the methylation mechanism is true, N-methylated amines could be at least detected in eqn (1). In addition, Cantat et al.18 showed that in the TBD-catalyzed aminal synthesis from amine, CO2, and hydrosilane, which is somewhat similar to methylation, the formation of an aminal product takes place after forming [Si]OCH2O[Si] via two sequential 2-electron reductions of CO2 with hydrosilane and the HC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O[Si] intermediate resulting from the first 2-electron reduction of CO2 with hydrosilane does not react with amine to give formamide. Thus, the formation of aminal does not pass formamide as an intermediate. Given these analyses, we carried out a DFT mechanistic study to deeply understand the catalytic system, in combination with experimental verifications. To our knowledge, there has been no systematic study on the mechanisms of formylation and methylation of amines with CO2, although Cantat and coworkers reported some computational results in their experimental study.19
O)O[Si] intermediate resulting from the first 2-electron reduction of CO2 with hydrosilane does not react with amine to give formamide. Thus, the formation of aminal does not pass formamide as an intermediate. Given these analyses, we carried out a DFT mechanistic study to deeply understand the catalytic system, in combination with experimental verifications. To our knowledge, there has been no systematic study on the mechanisms of formylation and methylation of amines with CO2, although Cantat and coworkers reported some computational results in their experimental study.19
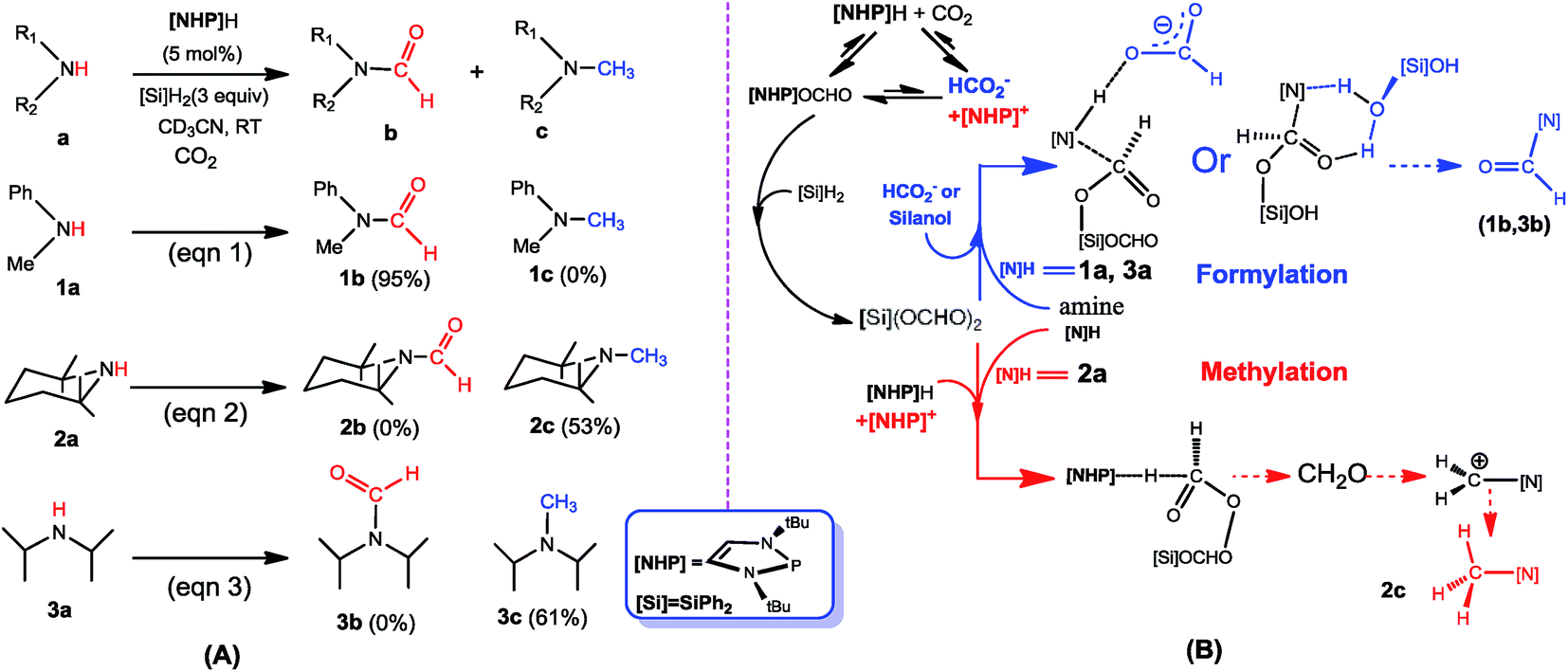 | ||
| Scheme 1 (A) Formylation (eqn (1)) and methylation (eqn (2) and (3)) of amines with CO2 and hydrosilane ([Si]H2 = Ph2SiH2), reported by Kinjo et al. (B) Schematic illustration of our proposed mechanism. | ||
Scheme 1B sketches our computed mechanisms. CO2 first inserts into the P–H bond of [NHP]H, giving [NHP]OCHO. The insertion is only slightly exergonic and the insertion product can easily dissociate into HCO2− and [NHP]+ ions, thus resulting in a microscopic equilibrium: [NHP]H + CO2 ⇄ [NHP]OCHO ⇄ [NHP]+ + HCO2−. Subsequently, [NHP]OCHO reacts with [Si]H2, giving [Si](OCHO)2. Finally, [Si](OCHO)2 reacts with amine, giving either a formamide or an N-methylated amine, with the chemoselectivity controlled by the competition between the amine nucleophilic attack (blue pathway) and [NHP]H hydride transfer (red pathway). For small amines such as 1a, the blue pathway is preferred, giving formamide (e.g.1b) under the catalytic effect of HCO2− or silanol (e.g. [Si](OH)2). For bulky amines (e.g.2a), the red pathway is favored, giving the N-methylated amine (e.g.2c) with the involvement of [NHP]H and [NHP]+. Instead of formamide being the intermediate of methylation, formaldehyde and a carbocation species were found to be the key intermediates of the methylation. Note that our results show that 3a prefers formylation, giving 3b rather than 3c, as reported previously (eqn (3)).
2. Computational details
Experimentally, the reactions were carried out in a polar solvent (acetonitrile, ε = 35.7). Considering the possible significant effects of the strong polar solvent, all geometries were optimized and characterized as minima (no imaginary frequency) or transition states (TSs, having one unique imaginary frequency) at the M06-2X20/6-31G(d,p) level with the solvation effect of acetonitrile simulated by the SMD21 solvent model. At the M06-2X/6-31G(d,p) geometries, the energies were further refined by M06-2X/6-311++G(d,p) single-point energy calculations with the solvent effect accounted for by the SMD solvent model. All DFT calculations adopted ultrafine integration grids (Int = ultrafine) to ensure stable numerical integrations. The M06-2X/6-31G(d,p) frequencies were used for thermal and entropic corrections at 298.15 K and 1 atm. It should be emphasized that such a correction approach is based on the ideal gas phase model, which inevitably overestimates entropy contributions to free energies for reactions in solvent, in particular for reactions involving a multicomponent change, because they ignore the suppressing effect of solvent on the rotational and transitional freedoms of substrates. The entropy overestimation of the approach was also demonstrated experimentally.22,23 While no standard quantum mechanics-based method is available to accurately calculate entropy in solution, approximate methods were proposed. According to the proposal of Martin et al.24 we previously applied a correction of (n − m) × 4.3 kcal mol−1 for a process from m- to n-components and found that such corrected free energies were more reasonable than enthalpies and uncorrected free energies,15,25 although the protocol is by no means accurate. Other correction factors (e.g. 1.9,26 2.6,3a,27 and 5.4 kcal mol−1 (ref. 28)) were adopted in the literature depending on the approximate approaches. As will be seen, our studied reactions involve multicomponent changes. As a conservative consideration, we applied a correction factor of 1.9 kcal mol−1 in this study. The corrected free energies are discussed and the uncorrected ones are given in the parentheses for references, unless otherwise specified. Note that using a correction factor of 4.3 kcal mol−1 does not alter our conclusions except for the numerical values. Natural bond orbital (NBO) analyses were performed at the M06-2X/6-311++G(d,p) level to assign partial atomic charges (Q).29 All calculations were carried out using Gaussian 09.303. Results and discussion
In this study, we use eqn (1) as a representative to compute the formylation mechanism of amine 1a (Section 3.1). In Section 3.2, using eqn (2), we investigate the methylation mechanism of amine (2a). After characterizing the mechanisms of formylation and methylation, we discuss the origins of chemoselectivity and experimentally verify our proposed mechanism in Section 3.3. Our computed mechanisms involve ionic species, thus we explicitly label the charges of all species when applicable for simplicity of the descriptions.3.1 Mechanism for 1a formylation (eqn (1))
The catalytic cycle for 1a formylation (eqn (1)) consists of three stages, namely, hydrophosphination of CO2 (stage I), formation of diformyloxysilane (stage II), and C–N bond formation (stage III). We below characterize how these stages proceed in order.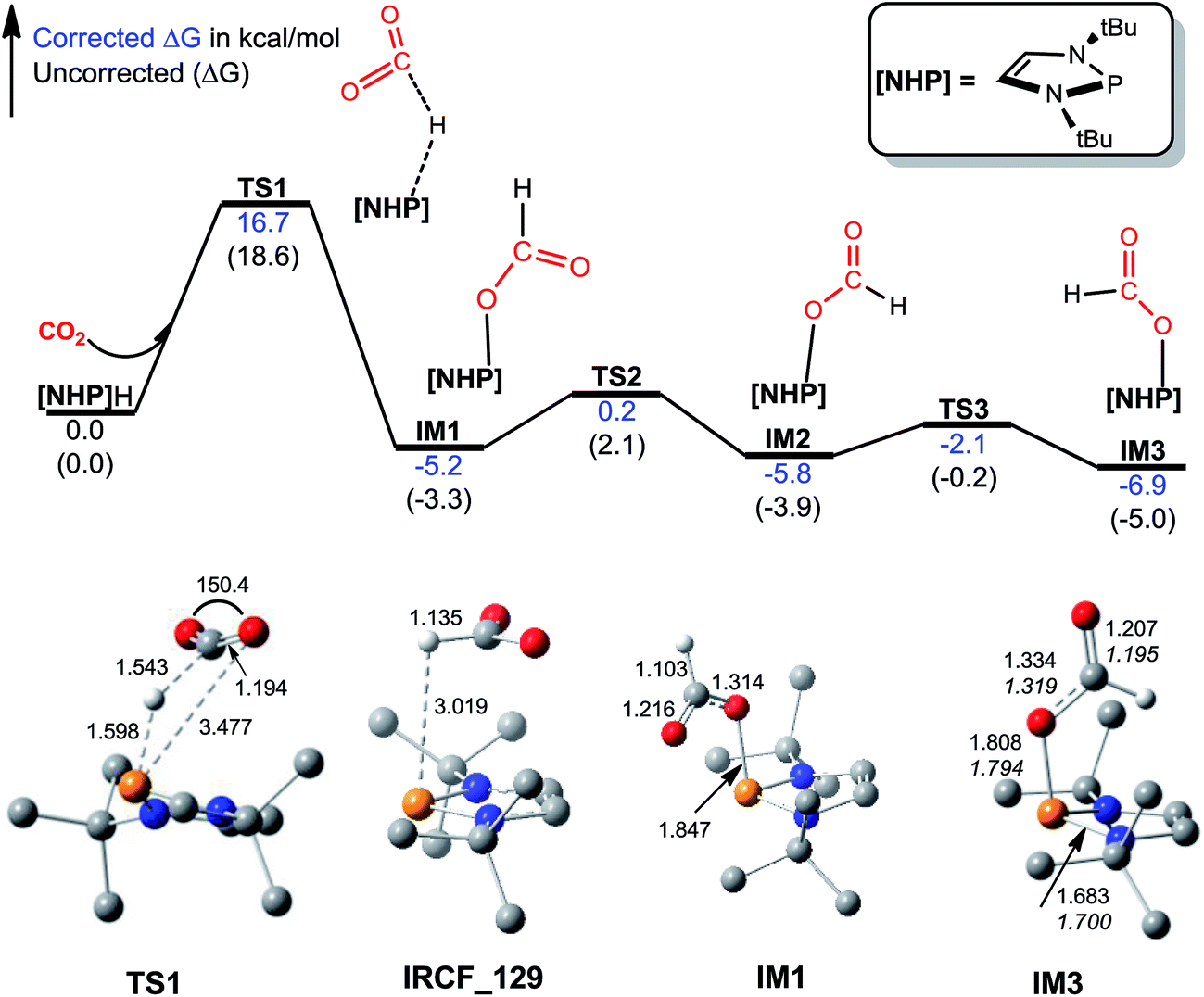 | ||
| Fig. 1 Free energy profile for hydrophosphination of CO2, together with key optimized structures with key bond lengths in angstroms and bond angles in degrees. All optimized structures are displayed in Fig. S2.† The italic values in IM3 are X-ray geometric parameters. | ||
Kinjo et al. observed zwitterionic character of IM3. Consistently, the [NHP] and HCO2 moieties in IM3 bear charges of 0.658 and −0.658e, respectively. Because of the zwitterionic nature, we conceived that IM3 can dissociate easily in the strong polar acetonitrile solvent, as demonstrated by the small dissociation energy (4.6 kcal mol−1, see Scheme 2). Thus a microscopic equilibrium is expected in this catalytic system. As will be shown, the free [NHP]+ and HCO2− ions play catalytic roles to mediate subsequent steps of the transformation.
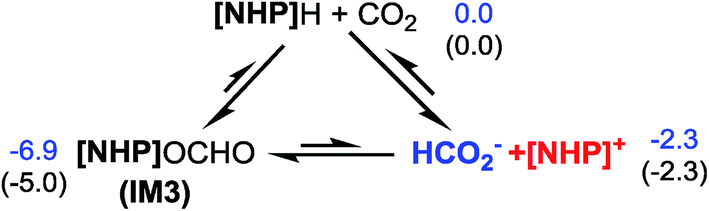 | ||
| Scheme 2 Microscopic equilibrium in the system. Values are relative free energies. | ||
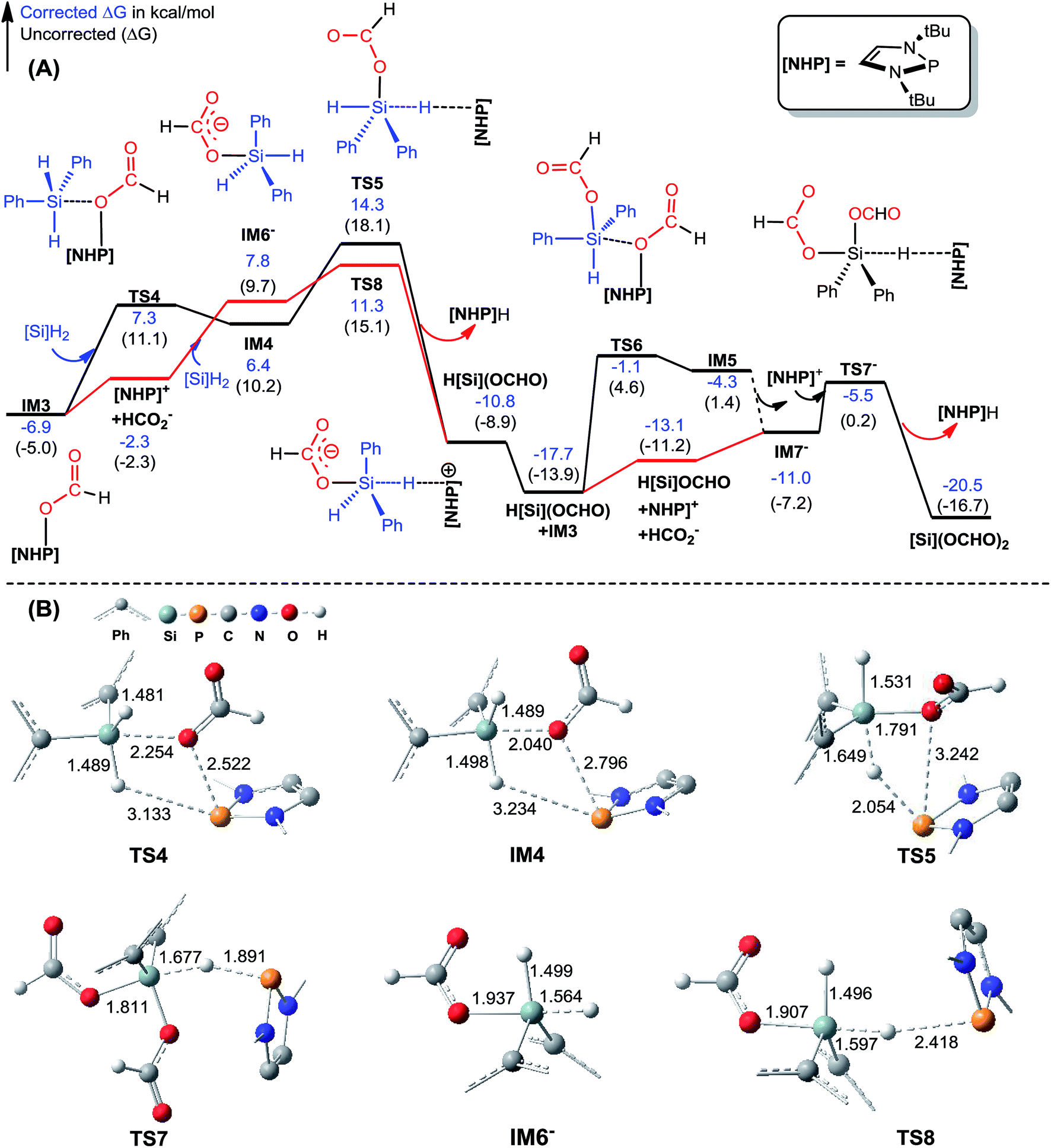 | ||
| Fig. 2 (A) Free energy profiles for the formation of [Si](OCHO)2. Energies are relative to [NHP]H, CO2, and [Si]H2 and are mass balanced. (B) Key optimized structures with key bond lengths given in angstroms. Other optimized structures are given in Fig. S4.† The details for the isomerization of IM5 to IM7− are given in Fig. S3.† | ||
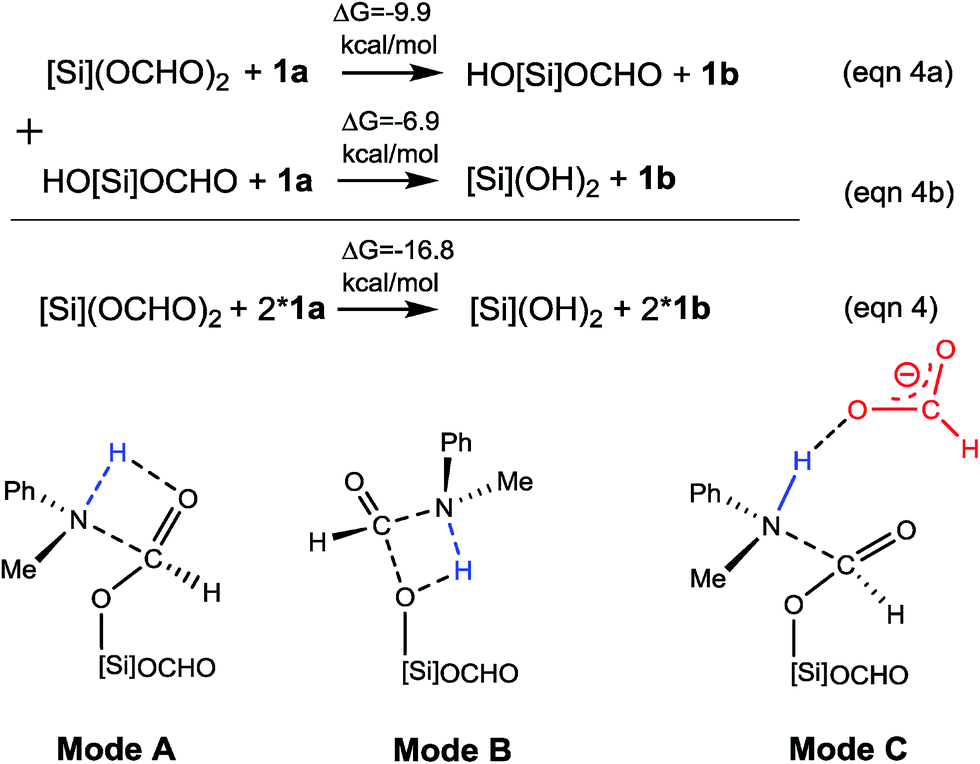 | ||
| Scheme 3 C–N bond formation stage (eqn (4)) and possible modes to form the bond. | ||
C–N bond formation catalyzed by HCO2−. As discussed above, HCO2− is available via microscopic equilibrium (Scheme 2). Thus, we considered whether a HCO2− ion can facilitate the C–N bond formation via H-bonding to the N–H bond of 1a (i.e. mode C in Scheme 3), because the bonding of the anionic species can enhance the nucleophilicity of amine 1a. Fig. 3 depicts the mechanism for eqn (4a) under the catalytic effect of HCO2−, along with key optimized structures. First, HCO2− and 1a form a H-bond complex IM8−, then the complex attacks [Si](OCHO)2viaTS9−, giving IM9− with a C–N bond formed. Interestingly, the C–N bond formation shifts the N–H1⋯O3 H-bond pattern (R(N–H1)/R(H1⋯O3) = 1.033/1.791 Å) in IM8− to the N⋯H1–O3 pattern (R(N⋯H1)/R(H–O3) = 1.617/1.031 Å) in IM9−. Meanwhile, the formal negative charge of HCO2− is shifted to the O1C1O2 moiety, as reflected by the bond equalization of the two C–O bonds from 1.348/1.198 Å in [Si](OCHO)2 to 1.379/1.396 Å in IM9−. The charge transfer shortens the O2⋯Si distance to 1.741 Å due to the attraction of Siδ+ and (O2)δ− and elongates the Si–O1 bond (from 1.683 to 1.816 Å) because of the disruption of the original Si–O1 single bond, resulting in the four-membered ring (SiO1C1O2) in IM9−. Subsequently, the HCO2H moiety in IM9− swings to the O2 site by crossing a lower barrier (TS10−, 2.7 kcal mol−1 relative to IM9−), giving IM10−, in which the four-membered SiO1C1O2 ring and the O2⋯H1–O3 H-bond pattern (R(O2⋯H1)/R(H1–O3) = 1.569/1.011 Å) are maintained. TS11− leads IM10− to the formamide product (1b) and IM11−. In addition to breaking the C–O2 and Si–O1 bonds to give 1b, TS11− alters the O2⋯H1–O3 H-bond pattern in IM10− to the O2–H1⋯O3 H-bond pattern (R(O2–H1)/R(H1⋯O3) = 1.045/1.455 Å) in IM11−. The dissociation of HCO2− from IM11− to regenerate the active HCO2− species costs only 5.0 kcal mol−1. The mechanism discussed above indicates that HCO2− is not just a H-bond partner to enhance the nucleophilicity of amine 1a. By altering the H-bond pattern between X⋯H–O and X–H⋯O (X = N or O) and shifting the charge between the HCO2− and O1C1O2 unit, HCO2− catalyzes bond formations (i.e. C–N and Si–O2 bonds in IM9−) and cleavages (i.e. C–O2 and Si–O1 bonds in IM10−). It is interesting that CO2 can be activated to an active species to facilitate its transformation. Following the same mechanism in Fig. 3, eqn (4b) takes place, producing another formamide (1b) and silanol [Si](OH)2. Without going into detail (see Fig. S5† for the energy profile of eqn (4b)), we mention that the RDS barrier of eqn (4b) is 27.3 kcal mol−1, 5.5 kcal mol−1 higher than that of eqn (4a).
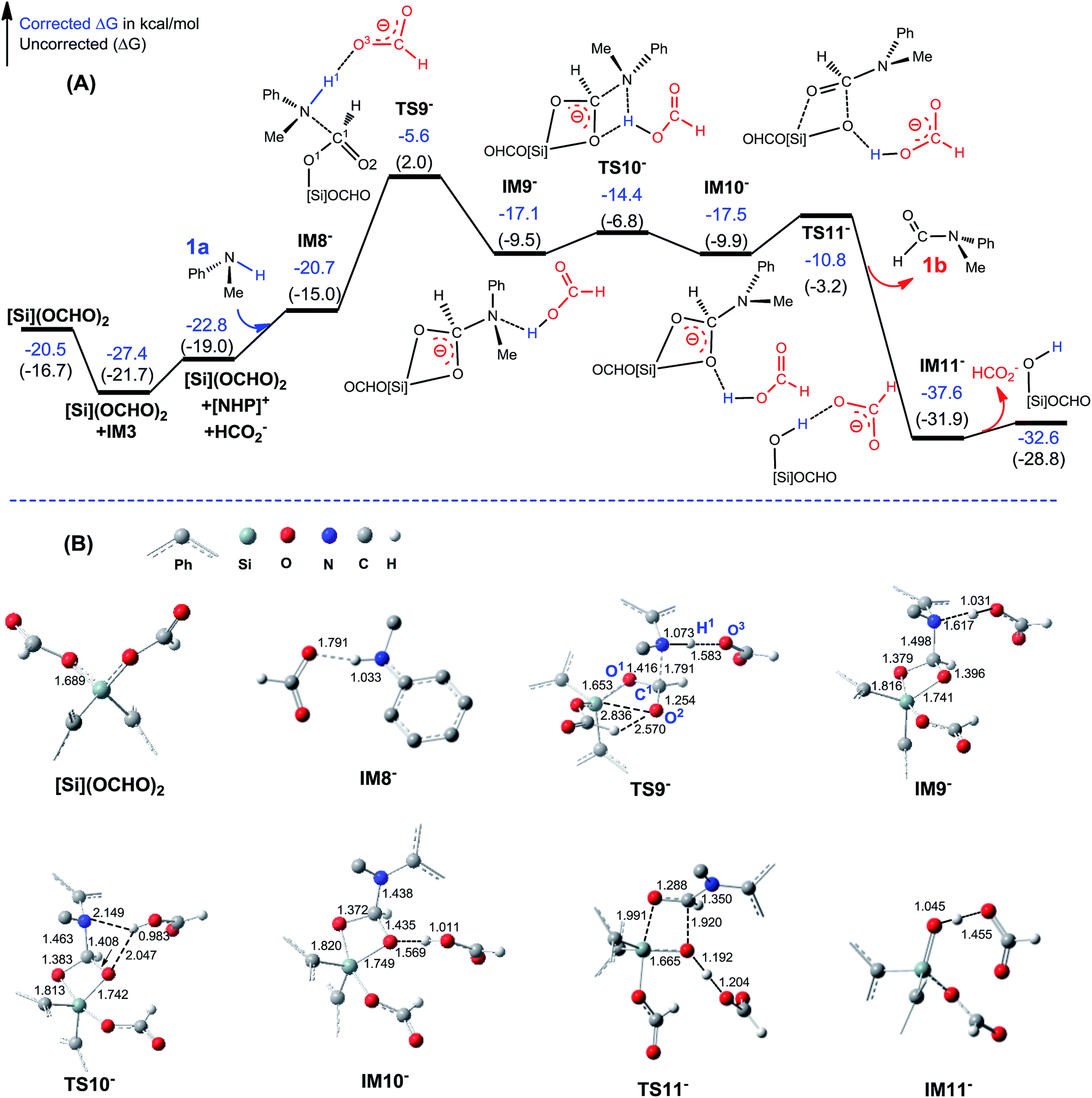 | ||
| Fig. 3 (A) Free energy profile for eqn (4a). Energies are relative to [NHP]H, CO2, 1a, and [Si]H2 and are mass balanced. (B) Key optimized structures with key bond lengths in angstroms. Other optimized structures are given in Fig. S4.† | ||
C–N bond formation facilitated by hydrogen transfer shuttle. The C–N bond formation through mode A and B involves a four-membered TS featuring hydrogen transfer (see Scheme 3). Thus a protic molecule such as water may act as a hydrogen transfer shuttle (H-shuttle)32,33 to facilitate the stage. In the present system, the possible H-shuttles could be water (trace water could not be excluded absolutely), N-methylaniline 1a, and silanol (HO[Si]OCHO and [Si](OH)2), which are available when the reaction is initiated. Using water as a representative, we characterize the H-shuttle-aided pathway (eqn (4)) through mode A, as illustrated in Fig. 4. Without going into detail, we mention that the water-aided C–N bond formation involves two hydrogen transfer steps, sequentially forming C–N and breaking C–O (CO2 deoxygenation) bonds, as described by TS12 and TS13 for eqn (4a), respectively.
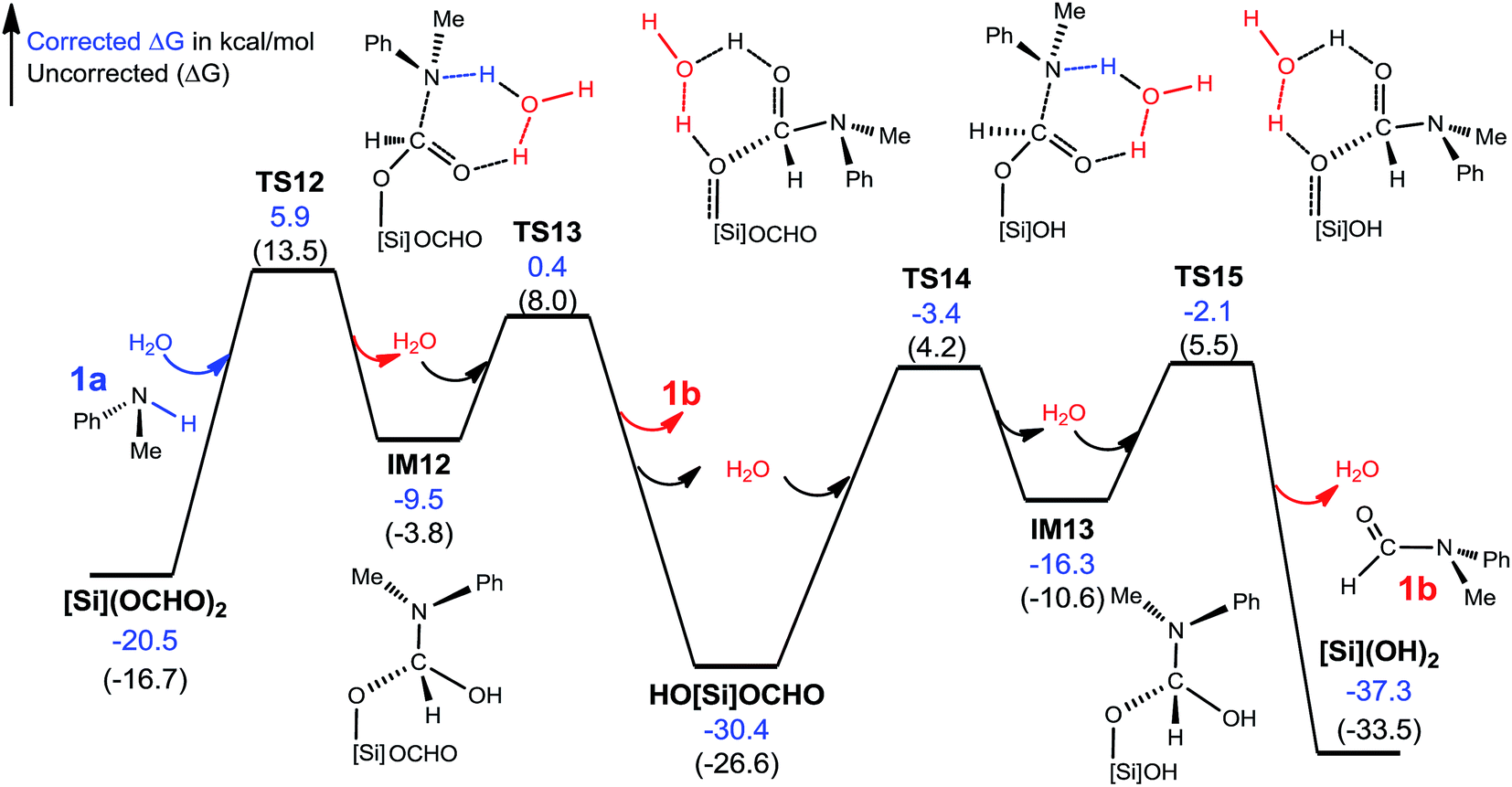 | ||
| Fig. 4 Free energy profile for the conversion of [Si](OCHO)2 + 2 × 1a→ 2 × 1b + [Si](OH)2. Optimized structures of key stationary points are displayed in Fig. S7.† Energies are relative to [NHP]H, CO2, 1a, H2O, and [Si]H2 and are mass balanced. | ||
Table 1 compares the RDS barriers for eqn (4a) and (4b), mediated by various H-shuttles and HCO2−. Note that, because the hydrogen transfers do not involve IM3 or [NHP]+/HCO2− ions, their RDS barriers were measured relative to [Si](OCHO)2 for eqn (4a) or HO[Si](OCHO) for eqn (4b). As compared, water is a more effective H-shuttle than amine 1a, which is consistent with our previous study of C–N bond formation in the dehydrogenative coupling of alcohol and amine.25d Both HO[Si]OCHO and [Si](OH)2 are better than water with HO[Si]OCHO being even better, which is due to the more polar O–H bond in silanol compared to that in water (see Fig. S6†). HCO2− is more effective than water but less effective than silanol.
| Mediator | Eqn (4a) | Eqn (4b) |
|---|---|---|
| a Complete pathway is given in Fig. S8. b Complete pathway is given in Fig. S9. c Complete pathway is given in Fig. S10. ND: not determined. | ||
| HCO2− | 21.8(23.7) | 27.3(29.2) |
| No (mode A) | 41.1(43.0) | ND |
| Water (mode A) | 26.4(30.2) | 28.3(32.1) |
| Amine 1a (mode A)a | 28.7(32.5) | 34.1(37.9) |
| HO[Si]OCHO (mode A)b | 18.8(22.6) | 19.9(23.7) |
| [Si](OH)2 (mode A)c | 20.4(24.2) | 24.8(28.6) |
| No (mode B) | 31.6(33.5) | ND |
| Water (mode B) | 30.5(34.3) | ND |
| HO[Si]OCHO (mode B) | 27.3(31.1) | ND |
For the formation of the C–N bond through mode B (Scheme 3), the water H-shuttle does not help much with only a slightly lower barrier (30.5 kcal mol−1), compared to 31.6 kcal mol−1 without the H-shuttle. The most effective H-shuttle, HO[Si]OCHO, in the case of mode A has a barrier of 27.3 kcal mol−1 in the case of mode B, which is much higher than 18.8 kcal mol−1 through mode A. We thus do not expect that other H-shuttles could aid the stage through the mode B mechanism more efficiently than that through mode A and did not pursue the mode further.
After characterizing the efficiency of these hydrogen transfer mediators in prompting C–N bond formation, we now discuss how the C–N bond could actually be formed. Both eqn (4a) and (4b) are thermodynamically favorable, being exergonic by 9.9 and 6.9 kcal mol−1, respectively. We focus on the kinetics of the reactions using eqn (4a) as an example for simplicity.
It was reported that in the absence of [NHP]H and CO2, [Si](OCHO)2 alone could react with 1a to give 1b. As the efficiency of the reaction was not reported, our energetic results show that the reaction is able to take place, because the barrier for eqn (4a), when using water as a H-shuttle, is 26.4 kcal mol−1, which is somewhat high but in a reasonable range for a reaction to occur. Importantly, when the reaction is initiated to produce silanol, the silanol byproducts can promote the reaction more effectively, with lower barriers (see Table 1). In the presence of [NHP]H and CO2, HCO2− plays the role of initiating the reaction rather than water, because the RDS barrier of 21.8 kcal mol−1 using HCO2− as a catalyst is much lower than 26.4 kcal mol−1 using a water H-shuttle as a promoter. As the reaction proceeds, more and more silanols (HO[Si]OCHO or [Si](OH)2) are produced, thus, silanols take the role of HCO2− to promote C–N bond formation.
3.2 Mechanism for 2a methylation (eqn (2))
Kinjo et al.16 have applied an [NHP]H catalyst to perform formylations of a range of primary and secondary amines. Intriguingly, 2,2,4,4-tetramethylpiperidine (2a) and diisopropylamine (3a) were found to afford N-methylated amines, 2c (eqn (2)) and 3c (eqn (3)), respectively. In general, formamide (the formylation product) was considered to be the intermediate for the methylation of amine with CO2.10,17 The mechanism was also adopted to elucidate the methylation products (2c and 3c). Nevertheless, we reasoned that this could not be true in the present catalytic system (supra infra). Using eqn (2) as an example, we investigate the methylation mechanism.The C–N bond in formylation is formed via the nucleophilic attack of amine (1a) to [Si](OCHO)2 (see TS9− in Fig. 3). Alternatively, we speculated that the hydrides, either [Si]H2 or [NHP]H, may compete with the amine to attack [Si](OCHO)2. Fig. 5 illustrates our computed pathway for 2a methylation, along with key optimized structures. Starting from [Si](OCHO)2, [NHP]H first transfers its Hδ− to a formyloxy carbon of [Si](OCHO)2 with a barrier of 25.1 kcal mol−1 (TS16). Under the catalytic effect of HCO2−, [Si]H2 offers its Hδ− with the higher barrier (27.1 kcal mol−1 at TS16′−). Regardless of which hydride attacks [Si](OCHO)2, the hydride transfer results in an anionic four-membered intermediate IM14−, which corresponds to IM9− in Fig. 3. Subsequently, the [NHP]+ cation attacks an O atom of the four-membered ring viaTS17, breaking the C1–O1 and Si–O2 bonds, resulting in formaldehyde (CH2O) and [NHP]O[Si]OCHO (IM15). The in situ generated CH2O then attacks 2a electrophilically, forming a C–N bond and meanwhile transferring the (N–)H atom of amine to the carbonyl group of the formaldehyde moiety viaTS18, resulting in IM16. The barrier for the process is 26.8 kcal mol−1 (TS18 relative to IM15), which is somewhat high but can be greatly lowered when a H-shuttle is used. For example, a water H-shuttle can lower the barrier to 14.1 kcal mol−1 (TS18′).
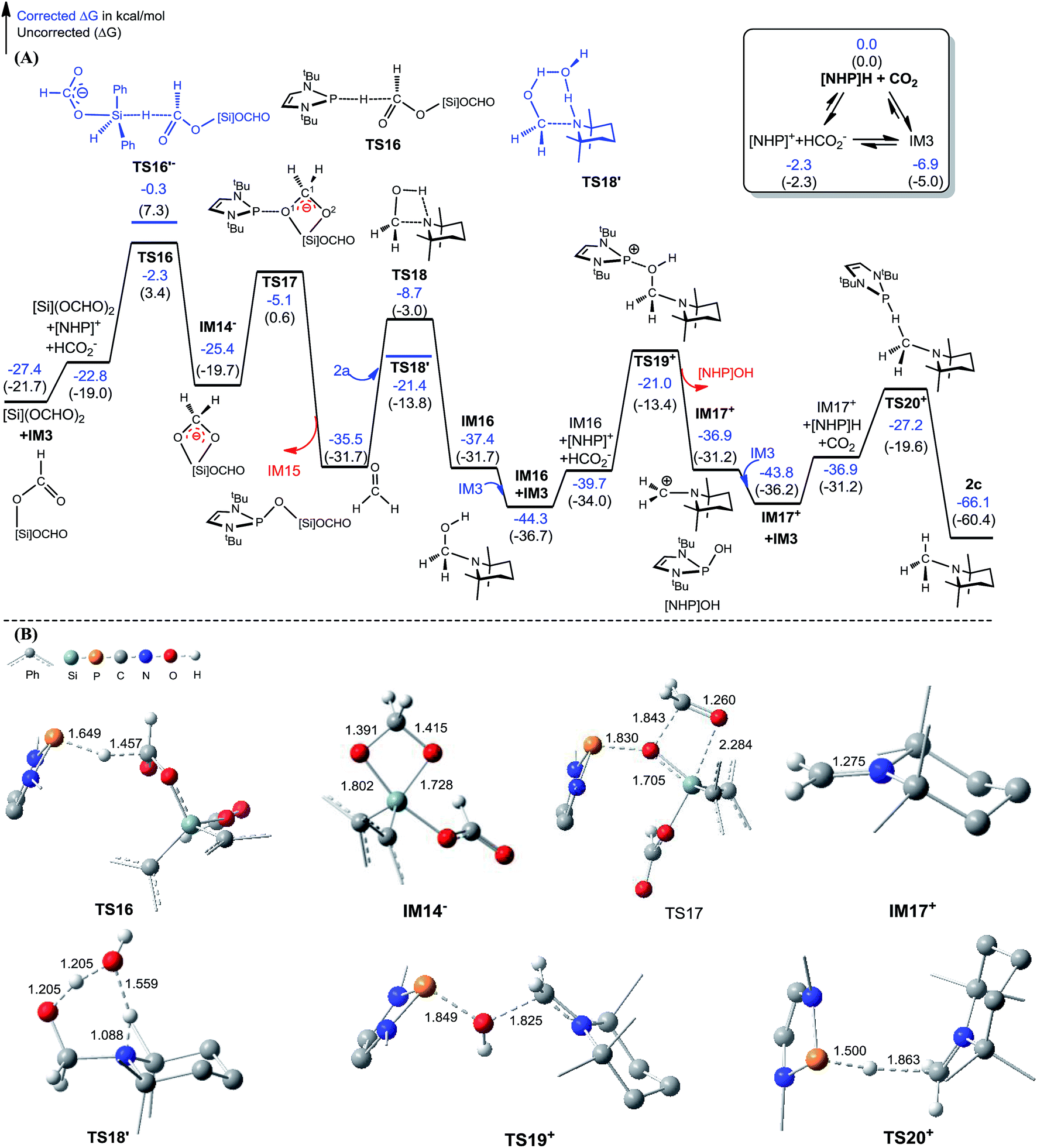 | ||
| Fig. 5 (A) Free energy profile for the methylation of [Si](OCHO)2 + 2a → 2c + H[Si]OCHO. (B) Optimized structures of key stationary points with key bond lengths given in angstroms. Those of others are given in Fig. S11.† Energies are relative to [NHP]H, CO2, 2a, H2O, and [Si]H2 and are mass balanced. | ||
Subsequently, another [NHP]+ attacks the hydroxyl group of IM16viaTS19+, leading to a carbocation species (IM17+) and [NHP]OH with a barrier of 23.3 kcal mol−1 (TS19+ relative to IM16 + IM3). After receiving a Hδ− of [NHP]H or [Si]H2, the carbocation species converts to an N-methylated amine (2c). Our calculations showed that for this step, [NHP]H is a preferred hydride donor with a barrier of 16.6 kcal mol−1 (TS20+ relative to IM17+ + IM3). An attempt using HCO2− to promote the Hδ− transfer of [Si]H2 was not successful, and the geometric optimization to locate the Hδ− transfer TS indicated that the steric effect between the bulky amine and [Si]H2 prevents the hydride transfer.
According to the methylation pathway (Fig. 5A), the reaction seems to consume the catalyst by forming [NHP]O[Si]OCHO (i.e.IM15) and [NHP]OH by-products. However, as detailed in ESI 2,† the two intermediates can be recovered to catalyst [NHP]H feasibly in terms of both kinetics and thermodynamics.
The methylation mechanism involves formaldehyde and a carbocation species IM17+ as the key intermediates. For the viability of formaldehyde, we call attention to the fact that Bontemps, Sabo-Etienne and coworkers experimentally detected formaldehyde in their Ru-catalyzed conversion of CO2 to C2 species with pinacolborane as a reducing reagent.34 Previously, we predicted that formaldehyde could be involved in the NHC- and Ni-catalyzed CO2 conversion to CH3OH.14 The involvement of a carbocation species in CO2 conversion has not ever been reported. For the viability of the carbocation species (IM17+), the cationic species must not form stable species (namely, IM17OCHO) with the anionic HCO2−, because a deep trap would raise the hydrogen transfer barrier from IM17+ + IM3 to TS20+ (Fig. 5A). To estimate the stability of IM17OCHO, we computed the reaction energy of eqn (5). The small endergonicity (1.8 kcal mol−1) of the equation indicates that IM17OCHO is only slightly more stable than IM3.
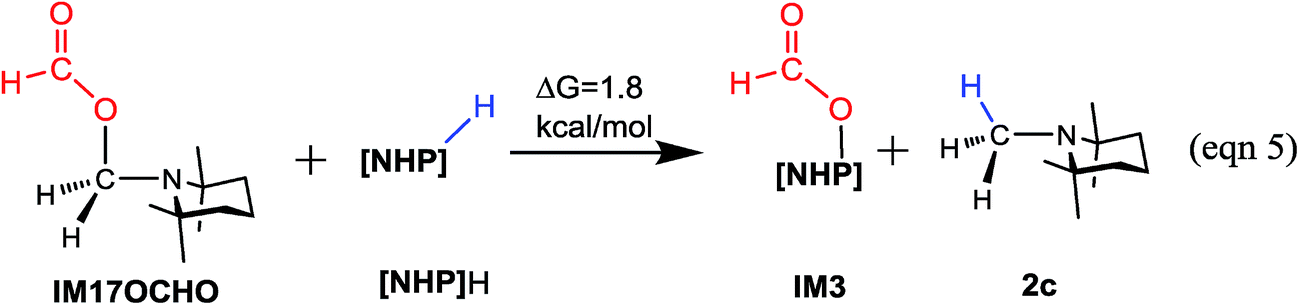
It is interesting to compare the roles of the [NHP]+ and HCO2− ions in formylation and methylation. In 1a formylation (Fig. 3), only the HCO2− component plays the catalytic role and [NHP]+ is a spectator. Differently, in 2a methylation (Fig. 5) the cationic component [NHP]+ plays the catalytic role, and [NHP]+ promotes the generation of CH2O (from IM14− to IM15) and the carbocation species (IM17+) from IM16.
3.3 The origins for chemoselectivities of formylation and methylation
The detailed characterizations of the mechanisms of eqn (1) and (2) facilitate our understanding of the chemoselectivities of the catalytic system. Using the conversion of the first formyloxy group of [Si](OCHO)2 as a representative case, we discuss the origins of the chemoselectivities. Key results for the conversion of the second formyloxy group of [Si](OCHO)2 (i.e. that in HO[Si]OCHO given in Table S1†) support the discussions below. According to the discussion in Section 3.2, the formylation/methylation preference stems from the competition between nucleophilic attacks of amine and hydride (i.e.TS9− in Fig. 3 and TS16 in Fig. 5) to [Si](OCHO)2. Table 2 compares the barriers of the two attacks for different amines. Note that the barrier for methylation is independent of amines. For 1a formylation, the barrier is 21.8 kcal mol−1, which is well below the barrier of 25.1 kcal mol−1 for methylation, thus eqn (1) prefers formylation. In contrast, the barrier (29.3 kcal mol−1, TS9-2a in Fig. 6) for 2a formylation is much higher than the barrier of 25.1 kcal mol−1 for its methylation, rationalizing the production of N-methylated amine (i.e.2c) in eqn (2). The higher formylation barrier of 2a compared to 1a can be attributed to the greater steric effect in TS9−-2a than that in TS9−, as indicated by the shorter H1⋯H2 distance (2.112 Å) than that (2.261 Å) in TS9−. In addition, TS9−-2a suffers steric repulsion between H1 and H3.| Substrate | Formylation | Methylation | Hydride transfer to formamide | ||||
|---|---|---|---|---|---|---|---|
| Hydride source | Hydride source | ||||||
| [NHP]H | HCO2−–[Si]H2 | [NHP]H | HCO2−–[Si]H2 | ||||
| ΔG≠ | ΔG≠ | ΔG≠ | ΔG≠ | ΔG≠ | |||
| a All optimized structures of the transition states are displayed in Fig. S12. b Values in parentheses are the free energy barriers without corrections. | |||||||
| 1a |
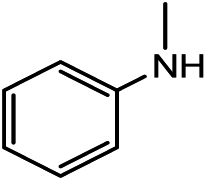
|
21.8(23.7)b | 25.1(25.1) | 27.1(29.0) | 1b | 37.3(37.3) | 36.7(38.6) |
| 2a |
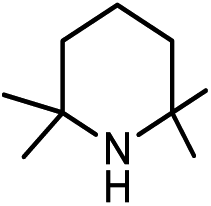
|
29.3(31.2) | 2b | 46.5(46.5) | 41.8(43.7) | ||
| 3a |
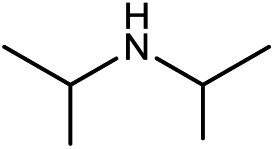
|
20.5(22.4) | 3b | 44.1(44.1) | 43.6(45.5) | ||
| 4a |
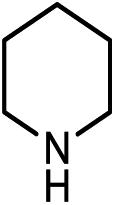
|
18.8(20.7) | 4b | 43.3(43.3) | 43.7(45.6) | ||
| 5a |
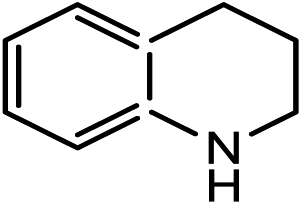
|
20.7(22.6) | 5b | 38.3(38.3) | 36.5(38.4) | ||
| 6a |

|
17.3(19.2) | 6b | 42.5(42.5) | 39.8(41.7) | ||
| 7a |
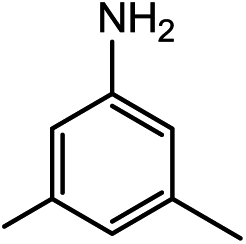
|
22.5(24.4) | 7b | 35.7(35.7) | 34.6(36.5) | ||
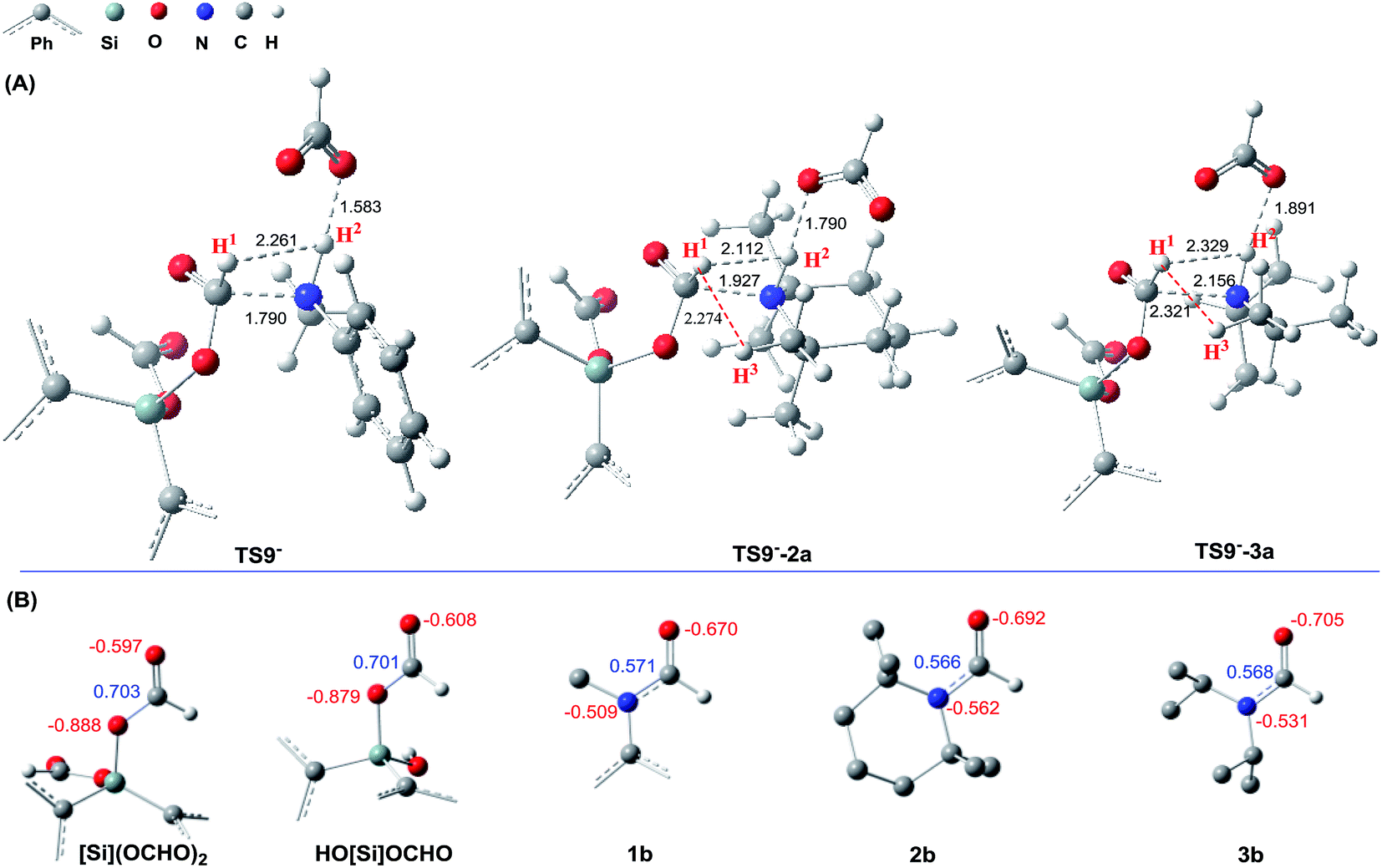 | ||
| Fig. 6 (A) Comparing the structures of the transition states (TS9−, TS9−-2a, and TS9−-3a) resulting in 1a, 2a, and 3a formylations. (B) Comparing the NBO charges (in e) of [Si](OCHO)2 and HO[Si]OCHO with those of formamides (1b–3b). | ||
The competition mechanisms rationalize the chemoselectivities of eqn (1) and (2), but the energetic results disagree with the reported experimental result of eqn (3), affording N-methylated amine 3c. The formylation barrier of 20.5 kcal mol−1 (TS9−-3a in Fig. 6A) for 3a is lower than that (25.1 kcal mol−1) for its methylation. On the other hand, comparing the structures of TS9−-3a and TS9− (the TSs for 3a and 1a formylations respectively), the H1–H2 distance (2.329 Å) in the former is even longer than that (2.261 Å) in the latter, indicating a smaller steric effect in TS9−-3a than in TS9−. In addition, the N atom in 3a bears more negative charge (−0.728e) than that (−0.658e) in 1a, indicating that 3a is more nucleophilic than 1a. Thus both the steric and electronic effect agree with the slightly lower formylation barrier (20.5 kcal mol−1) of 3a than that of 1a (21.8 kcal mol−1). We doubt that eqn (3) might actually produce formamide (3b).
To verify our computed mechanisms and the production of 3b in eqn (3), we performed experiments to study the reactions of 1a–3a (see ESI 3 for experimental details†).35Scheme 4 shows our experimental results. Under the same experimental conditions, we were successful in reproducing the reported results of eqn (1), giving 1a in 96% yield (see eqn (6)). However, our study shows that 3a prefers to undergo formylation, affording formamide (3b) in 56% yield (eqn (8)), rather than N-methylated amine 3c as reported previously (eqn (3)), supporting our computational prediction. For 2a, under the same experimental conditions, we could only obtain traces of 2c. Based on our computed mechanism, we reasoned that the poor performance of the reaction could be due to (a) the barrier for methylation (25.1 kcal mol−1) being higher than that for formylation (e.g. 21.8 kcal mol−1 for 1a formylation) and (b) [NHP]H being required to finally reduce IM17+ to 2c (see Fig. 5), but it could be consumed during the process reaching IM17+. Thus, we modified the experimental conditions as shown in eqn (7) of Scheme 4. Delightedly, under the modified conditions, the methylated amine 2c could be produced in 65% yield. Overall the experimental results corroborate our computational prediction satisfactorily.
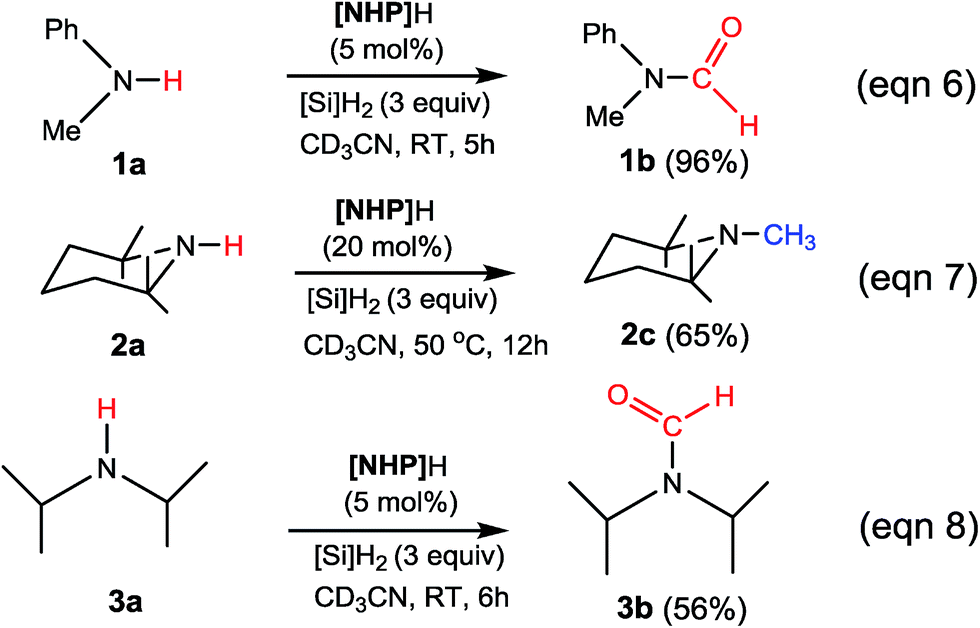 | ||
| Scheme 4 Our experimental results. See ESI 3 for experimental details.† | ||
We have shown that, in the present catalytic system, it is unlikely that methylation passes through formamide as an intermediate. We analyze why this is true. To further reduce formamide, the hydride (either [NHP]H or [Si]H2) should transfer its Hδ− to the carbonyl carbon of formamide, thus the electrophilicity of the carbon should be a factor to determine how favorably the formamide accepts a hydridic hydrogen of a hydride donor. Fig. 6B compares the NBO charges of formamides (1b–3b) with those of [Si](OCHO)2 and HO[Si]OCHO. It can be found that the formyloxy carbon in [Si](OCHO)2 and HO[Si]OCHO bears significantly more positive charge (>0.70e) than that in formamides (<0.58e). Thus [Si](OCHO)2 and HO[Si]OCHO can be reduced more easily than formamides. Consistently, the hydride transfer barriers from [NHP]H to 1b, 2b, and 3b are substantially higher (37.3–44.1 kcal mol−1) than that (25.1 kcal mol−1) to [Si](OCHO)2. This is also true when [Si]H2 is used as the hydride donor with HCO2− as the promoter (see Table 2).
To further corroborate our conclusions, we calculated the RDS barriers for formylation of the other four amines (4a–7a in Table 2) reported in ref. 16. The barriers for formylation of the four amines, ranging from 18.8–22.5 kcal mol−1, are all lower than the barrier for methylation (25.1 kcal mol−1), in excellent agreement with the experimental fact that these amines prefer formylation. Again, the barriers for hydride transfers to their corresponding formamides (4b–5b) are substantially high (>34.6 kcal mol−1). The high reduction barriers of formamides call attention to the sequential mechanism for understanding the methylation of amine with CO2.
4. Conclusions
In this study, we have performed a DFT study to investigate the catalytic mechanisms of the 1,3,2-diazaphospholene ([NHP]H)-mediated formylation/methylation of amines (methylaniline (1a)/2,2,4,4-tetramethylpiperidine (2a)) with CO2 and hydrosilane (Ph2SiH2 = [Si]H2) as a reducing reagent. Formylation of 1a proceeds via three stages, including hydrophosphination of CO2, giving [NHP]OCHO (stage I), reaction of [NHP]OCHO with [Si]H2 to form [Si](OCHO)2 (stage II), and aminolysis of [Si](OCHO)2 to form a C–N bond, finally affording formamide (stage III). Methylation of 2a shares the first two stages of formylation but is different in stage III. After stages I and II, the resultant [Si](OCHO)2 is preferentially subjected to the attack of an [NPH]H hydride, resulting in formaldehyde which then couples with 2a to form a C–N bond in IM16. Subsequently, IM16 converts to a carbocation species. The methyl group is finally formed via hydride transfer of [NHP]H to the carbocation species. Thus, different from the general consideration that methylation passes through formamide as reduced intermediates of CO2, the formylation and methylation in the present catalytic system are two competitive reaction channels. The chemoselectivity originates from the competition between amines and [NHP]H to attack the formyloxy carbon of [Si](OCHO)2. If the attack of an amine (e.g.1a) wins the competition, the transformation affords formamide (1b) and otherwise (e.g.2a) results in N-methylated amine (2c). The reduction of formamides is highly kinetically unfavorable, which calls attention to the sequential mechanism for understanding amine methylation with CO2.On the basis of the detailed pathways, we have the following key findings in terms of reaction modes. The activation of CO2 by [NHP]H establishes a microscopic equilibrium: [NHP]H + CO2 ⇄ [NHP]OCHO ⇄ [NHP]+ + HCO2−. The ions play catalytic roles to facilitate formylation with HCO2− or methylation with [NHP]+. In 1a formylation, HCO2− initially forms a N–H⋯O (of HCO2−) H-bond complex with 1a to attack [Si](OCHO)2. By altering the H-bond pattern between X–H⋯O and X⋯H–O (X = N or O) and shifting the formal charge between HCO2− and the OCO unit in [Si](OCHO)2, HCO2− promotes C–N bond formation and CO2 deoxygenation, finally resulting in formamide. However, it should be noted that, after the formylation is initiated, the silanol byproduct (either HO[Si]OCHO or [Si](OH)2) is more effective than HCO2− to promote the formylation. Formaldehyde and a carbocation (IM17+) were characterized to be two important species to tunnel methylation and the generations of the species require the catalytic action of [NHP]+.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are very grateful to Prof. Dietrich Gudat at the University of Stuttgart for his constructive suggestions on the preparation of the catalyst 1,3,2-diazaphospholene and to the anonymous reviewers for their insightful comments which helped us to improve the paper. We acknowledge the support for this work by the National Natural Science Foundation of China (Grant No. 21373216, 21573233, and 21773240).References
- For recent reviews of CO2 fixation, see: (a) C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS; (b) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kuhn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed; (c) G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed; (d) Y.-N. Li, R. Ma, L.-N. He and Z.-F. Diao, Catal. Sci. Technol., 2014, 4, 1498–1512 RSC; (e) F. J. Fernández-Alvarez, A. M. Aitani and L. A. Oro, Catal. Sci. Technol., 2014, 4, 611–624 RSC; (f) T. Sakakura and K. Kohno, Chem. Commun., 2009, 11, 1312–1330 RSC; (g) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC; (h) A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. Kenis, C. A. Kerfeld, R. H. Morris, C. H. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed; (i) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed; (j) W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC; (k) K. M. Yu, I. Curcic, J. Gabriel and S. C. Tsang, ChemSusChem, 2008, 1, 893–899 CrossRef CAS PubMed; (l) M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC; (m) S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347–3357 RSC; (n) Z.-Z. Yang, L.-N. He, J. Gao, A.-H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602–6639 RSC; (o) M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC; (p) D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765–10780 CrossRef CAS PubMed; (q) A. Dibenedetto, A. Angelini and P. Stufano, J. Chem. Technol. Biotechnol., 2014, 89, 334–353 CrossRef CAS; (r) G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487–498 CrossRef CAS PubMed; (s) L. Wu, Q. Liu, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 6310–6320 CrossRef CAS PubMed; (t) F. J. Fernández-Alvarez, M. Iglesias, L. A. Oro and V. Polo, ChemCatChem, 2013, 5, 3481–3494 CrossRef; (u) C. H. Lim, A. M. Holder, J. T. Hynes and C. B. Musgrave, J. Phys. Chem. Lett., 2015, 6, 5078–5092 CrossRef CAS PubMed; (v) L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395–3403 RSC; (w) I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS; (x) F.-G. Fontaine, M.-A. Courtemanche, M.-A. Légaré and É. Rochette, Coord. Chem. Rev., 2017, 334, 124–135 CrossRef CAS; (y) T. Fan, X. Chen and Z. Lin, Chem. Commun., 2012, 48, 10808–10828 RSC; (z) K. Huang, C. L. Sun and Z. J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC; (a a) T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- For representative experimental papers: (a) M. Khandelwal and R. J. Wehmschulte, Angew. Chem., Int. Ed., 2012, 51, 7323–7326 CrossRef CAS PubMed; (b) F. A. LeBlanc, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2014, 53, 789–792 CrossRef CAS PubMed; (c) A. Berkefeld, W. E. Piers, M. Parvez, L. Castro, L. Maron and O. Eisenstein, Chem. Sci., 2013, 4, 2152–2162 RSC; (d) J. Chen, L. Falivene, L. Caporaso, L. Cavallo and E. Y. Chen, J. Am. Chem. Soc., 2016, 138, 5321–5333 CrossRef CAS PubMed; (e) S. J. Mitton and L. Turculet, Chem.–Eur. J., 2012, 18, 15258–15262 CrossRef CAS PubMed; (f) A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660–10661 CrossRef CAS PubMed; (g) T. Matsuo and H. Kawaguchi, J. Am. Chem. Soc., 2006, 128, 12362–12363 CrossRef CAS PubMed; (h) S. Park, D. Bezier and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11404–11407 CrossRef CAS PubMed; (i) R. Declercq, G. Bouhadir, D. Bourissou, M.-A. Légaré, M.-A. Courtemanche, K. S. Nahi, N. Bouchard, F.-G. Fontaine and L. Maron, ACS Catal., 2015, 5, 2513–2520 CrossRef CAS; (j) S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS PubMed; (k) F. G. Fontaine, M. A. Courtemanche and M. A. Legare, Chem.–Eur. J., 2014, 20, 2990–2996 CrossRef CAS PubMed; (l) M. A. Courtemanche, M. A. Legare, L. Maron and F. G. Fontaine, J. Am. Chem. Soc., 2014, 136, 10708–10717 CrossRef CAS PubMed; (m) N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS PubMed; (n) S. Chakraborty, J. Zhang, J. A. Krause and H. R. Guan, J. Am. Chem. Soc., 2010, 132, 8872–8873 CrossRef CAS PubMed; (o) S. Wesselbaum, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499–7502 CrossRef CAS PubMed; (p) J. Ye and J. K. Johnson, Catal. Sci. Technol., 2016, 6, 8392–8405 RSC; (q) A. Correa and R. Martin, Angew. Chem., Int. Ed., 2009, 48, 6201–6204 CrossRef CAS PubMed; (r) C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS PubMed; (s) T. Fujihara, T. Xu, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2011, 50, 523–527 CrossRef CAS PubMed; (t) T. Ohishi, L. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 8114–8117 CrossRef CAS PubMed; (u) Y. Zhang and S. N. Riduan, Angew. Chem., Int. Ed., 2011, 50, 6210–6212 CrossRef CAS PubMed; (v) K. Ukai, M. Aoki, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2006, 128, 8706–8707 CrossRef CAS PubMed; (w) D. M. Dalton and T. Rovis, Nat. Chem., 2010, 2, 710–711 CrossRef CAS PubMed; (x) C. A. Huff and M. S. Sanford, ACS Catal., 2013, 3, 2412–2416 CrossRef CAS; (y) Z. F. Zhang, E. Xie, W. J. Li, S. Q. Hu, J. L. Song, T. Jiang and B. X. Han, Angew. Chem., Int. Ed., 2008, 47, 1127–1129 CrossRef CAS PubMed; (z) H. Hayashi, S. Ogo and S. Fukuzumi, Chem. Commun., 2004, 2714–2715 RSC; (a a) Y. M. Badiei, W. H. Wang, J. F. Hull, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2013, 52, 12576–12586 CrossRef CAS PubMed; (a b) R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed; (a c) L. González-Sebastián, M. Flores-Alamo and J. J. García, Organometallics, 2013, 32, 7186–7194 CrossRef; (a d) C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, W. Baumann, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701–20704 CrossRef CAS PubMed; (a e) H. Ge, Y. Jing and X. Yang, Inorg. Chem., 2016, 55, 12179–12184 CrossRef CAS PubMed; (a f) I. Knopf and C. C. Cummins, Organometallics, 2015, 34, 1601–1603 CrossRef CAS.
- For representative computational papers of CO2 fixation, see: (a) Q. Zhou and Y. Li, J. Am. Chem. Soc., 2015, 137, 10182–10189 CrossRef CAS PubMed; (b) G. Yang, B. Schäffner, M. Blug, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 800–807 CrossRef CAS; (c) F. Castro-Gomez, G. Salassa, A. W. Kleij and C. Bo, Chem.–Eur. J., 2013, 19, 6289–6298 CrossRef CAS PubMed; (d) A. Uhe, M. Holscher and W. Leitner, Chem.–Eur. J., 2012, 18, 170–177 CrossRef CAS PubMed; (e) W. H. Bernskoetter and N. Hazari, Eur. J. Inorg. Chem., 2013, 2013, 4032–4041 CrossRef CAS; (f) C. H. Lim, A. M. Holder, J. T. Hynes and C. B. Musgrave, Inorg. Chem., 2013, 52, 10062–10066 CrossRef CAS PubMed; (g) P. M. Zimmerman, Z. Zhang and C. B. Musgrave, Inorg. Chem., 2010, 49, 8724–8728 CrossRef CAS PubMed; (h) D. P. Hruszkewycz, J. Wu, N. Hazari and C. D. Incarvito, J. Am. Chem. Soc., 2011, 133, 3280–3283 CrossRef CAS PubMed; (i) T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed; (j) W. Ding, W. Fang, Z. Chai and D. Wang, J. Chem. Theory Comput., 2012, 8, 3605–3617 CrossRef CAS PubMed; (k) H. Batebi, F. Zarkoob, K. Daraei, B. F. Yates and A. Ariafard, J. Organomet. Chem., 2013, 748, 89–97 CrossRef CAS; (l) D. P. Hruszkewycz, J. Wu, J. C. Green, N. Hazari and T. J. Schmeier, Organometallics, 2012, 31, 470–485 CrossRef CAS; (m) H. W. Suh, T. J. Schmeier, N. Hazari, R. A. Kemp and M. K. Takase, Organometallics, 2012, 31, 8225–8236 CrossRef CAS; (n) B. J. Wang and Z. X. Cao, RSC Adv., 2013, 3, 14007–14015 RSC; (o) N. J. Brookes, A. Ariafard, R. Stranger and B. F. Yates, J. Am. Chem. Soc., 2009, 131, 5800–5808 CrossRef CAS PubMed; (p) J. G. Wu, N. Hazari and C. D. Incarvito, Organometallics, 2011, 30, 3142–3150 CrossRef CAS; (q) C. Villiers, J. P. Dognon, R. Pollet, P. Thuery and M. Ephritikhine, Angew. Chem., Int. Ed., 2010, 49, 3465–3468 CrossRef CAS PubMed; (r) Y. Jiang, O. Blacque, T. Fox and H. Berke, J. Am. Chem. Soc., 2013, 135, 7751–7760 CrossRef CAS PubMed; (s) K. W. Huang, J. H. Han, C. B. Musgrave and E. Fujita, Organometallics, 2007, 26, 508–513 CrossRef CAS; (t) R. Lalrempuia, M. Iglesias, V. Polo, P. J. Sanz Miguel, F. J. Fernandez-Alvarez, J. J. Perez-Torrente and L. A. Oro, Angew. Chem., Int. Ed., 2012, 51, 12824–12827 CrossRef CAS PubMed; (u) A. Ariafard, F. Zarkoob, H. Batebi, R. Stranger and B. F. Yates, Organometallics, 2011, 30, 6218–6224 CrossRef CAS; (v) R. Tanaka, M. Yamashita, L. W. Chung, K. Morokuma and K. Nozaki, Organometallics, 2011, 30, 6742–6750 CrossRef CAS; (w) W. Li, D. Huang and Y. Lyu, Org. Biomol. Chem., 2016, 14, 10875–10885 RSC; (x) L. Liu, N. Vankova and T. Heine, Phys. Chem. Chem. Phys., 2016, 18, 3567–3574 RSC.
- (a) Q. Liu, L. P. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6 Search PubMed; (b) A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157–168 RSC; (c) Y. Li, X. Cui, K. Dong, K. Junge and M. Beller, ACS Catal., 2017, 7, 1077–1086 CrossRef CAS.
- S. Schreiner, J. Y. Yu and L. Vaska, J. Chem. Soc., Chem. Commun., 1988, 602–603 RSC.
- (a) O. Krocher, R. A. Koppel and A. Baiker, Chem. Commun., 1997, 453–454 RSC; (b) X. Frogneux, O. Jacquet and T. Cantat, Catal. Sci. Technol., 2014, 4, 1529–1533 RSC; (c) K. Motokura, N. Takahashi, D. Kashiwame, S. Yamaguchi, A. Miyaji and T. Baba, Catal. Sci. Technol., 2013, 3, 2392–2396 RSC; (d) K. Motokura, N. Takahashi, A. Miyaji, Y. Sakamoto, S. Yamaguchi and T. Baba, Tetrahedron, 2014, 70, 6951–6956 CrossRef CAS; (e) X. Cui, Y. Zhang, Y. Deng and F. Shi, Chem. Commun., 2014, 50, 189–191 RSC; (f) L. Zhang, Z. Han, X. Zhao, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2015, 54, 6186–6189 CrossRef CAS PubMed.
- (a) O. Jacquet, C. Das Neves Gomes, M. Ephritikhine and T. Cantat, J. Am. Chem. Soc., 2012, 134, 2934–2937 CrossRef CAS PubMed; (b) S. Das, F. D. Bobbink, S. Bulut, M. Soudani and P. J. Dyson, Chem. Commun., 2016, 52, 2497–2500 RSC.
- C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuery, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef CAS PubMed.
- (a) S. N. Riduan, J. Y. Ying and Y. G. Zhang, J. Catal., 2016, 343, 46–51 CrossRef CAS; (b) O. Jacquet, C. Das Neves Gomes, M. Ephritikhine and T. Cantat, ChemCatChem, 2013, 5, 117–120 CrossRef CAS; (c) L. D. Hao, Y. F. Zhao, B. Yu, Z. Z. Yang, H. Y. Zhang, B. X. Han, X. Gao and Z. M. Liu, ACS Catal., 2015, 5, 4989–4993 CrossRef CAS; (d) H. Lv, Q. Xing, C. Yue, Z. Lei and F. Li, Chem. Commun., 2016, 52, 6545–6548 RSC; (e) T. V. Nguyen, W. J. Yoo and S. Kobayashi, Angew. Chem., Int. Ed., 2015, 54, 9209–9212 CrossRef CAS PubMed.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS PubMed.
- (a) W. C. Chen, J. S. Shen, T. Jurca, C. J. Peng, Y. H. Lin, Y. P. Wang, W. C. Shih, G. P. Yap and T. G. Ong, Angew. Chem., Int. Ed., 2015, 54, 15207–15212 CrossRef CAS PubMed; (b) S. Das, F. D. Bobbink, G. Laurenczy and P. J. Dyson, Angew. Chem., Int. Ed., 2014, 53, 12876–12879 CrossRef CAS PubMed; (c) O. Santoro, F. Lazreg, Y. Minenkov, L. Cavallo and C. S. Cazin, Dalton Trans., 2015, 44, 18138–18144 RSC; (d) Z. Yang, B. Yu, H. Zhang, Y. Zhao, G. Ji, Z. Ma, X. Gao and Z. Liu, Green Chem., 2015, 17, 4189–4193 RSC; (e) L. González-Sebastián, M. Flores-Alamo and J. J. García, Organometallics, 2015, 34, 763–769 CrossRef; (f) X. Cui, X. Dai, Y. Zhang, Y. Deng and F. Shi, Chem. Sci., 2014, 5, 649–655 RSC.
- E. Blondiaux, J. Pouessel and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 12186–12190 CrossRef CAS PubMed.
- (a) A. Tlili, X. Frogneux, E. Blondiaux and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 2543–2545 CrossRef CAS PubMed; (b) X. Cui, Y. Zhang, Y. Deng and F. Shi, Chem. Commun., 2014, 50, 13521–13524 RSC; (c) K. Kon, S. M. Siddiki, W. Onodera and K. Shimizu, Chem.–Eur. J., 2014, 20, 6264–6267 CrossRef CAS PubMed; (d) K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 9554–9557 CrossRef CAS PubMed; (e) Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 12156–12160 CrossRef CAS PubMed.
- (a) F. Huang, G. Lu, L. L. Zhao, H. X. Li and Z. X. Wang, J. Am. Chem. Soc., 2010, 132, 12388–12396 CrossRef CAS PubMed; (b) F. Huang, C. Zhang, J. Jiang, Z. X. Wang and H. Guan, Inorg. Chem., 2011, 50, 3816–3825 CrossRef CAS PubMed.
- M. Wen, F. Huang, G. Lu and Z. X. Wang, Inorg. Chem., 2013, 52, 12098–12107 CrossRef CAS PubMed.
- C. C. Chong and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 12116–12120 ( Angew. Chem. , 2015 , 127 , 12284–12288 ) CrossRef CAS PubMed.
- (a) O. Jacquet, X. Frogneux, C. D. Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC; (b) Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 12156–12160 CrossRef CAS PubMed.
- X. Frogneux, E. Blondiaux, P. Thuery and T. Cantat, ACS Catal., 2015, 5, 3983–3987 CrossRef CAS.
- C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuery, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef CAS PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed; (c) R. Valero, R. Costa, I. D. P. R. Moreira, D. G. Truhlar and F. Illas, J. Chem. Phys., 2008, 128, 114103 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- D. G. Huang, O. V. Makhlynets, L. L. Tan, S. C. Lee, E. V. Rybak-Akimova and R. H. Holm, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1222–1227 CrossRef CAS PubMed.
- Y. Liang, S. Liu, Y. Xia, Y. Li and Z.-X. Yu, Chem.–Eur. J., 2008, 14, 4361–4373 CrossRef CAS PubMed.
- R. L. Martin, P. J. Hay and L. R. Pratt, J. Phys. Chem. A, 1998, 102, 3565–3573 CrossRef CAS.
- For examples: (a) S. Qu, Y. Dang, C. Song, M. Wen, K.-W. Huang and Z.-X. Wang, J. Am. Chem. Soc., 2014, 136, 4974–4991 CrossRef CAS PubMed; (b) S. Qu, H. Dai, Y. Dang, C. Song, Z.-X. Wang and H. Guan, ACS Catal., 2014, 4, 4377–4388 CrossRef CAS; (c) H. Li, M. Wen and Z.-X. Wang, Inorg. Chem., 2012, 51, 5716–5727 CrossRef CAS PubMed; (d) H. Li, X. Wang, F. Huang, G. Lu, J. Jiang and Z.-X. Wang, Organometallics, 2011, 30, 5233–5247 CrossRef CAS.
- (a) H. Li and M. B. Hall, J. Am. Chem. Soc., 2013, 136, 383–395 CrossRef PubMed; (b) C. J. Cramer, Essentials of Computational Chemistry: Theories and Models, John Wiley & Sons, Ltd, New York, 2nd edn, 2004, pp. 378–379 Search PubMed; (c) L. Vigara, M. Z. Ertem, N. Planas, F. Bozoglian, N. Leidel, H. Dau, M. Haumann, L. Gagliardi, C. J. Cramer and A. Llobet, Chem. Sci., 2012, 3, 2576–2586 RSC.
- (a) S. W. Benson, The Foundations of Chemical Kinetics, R. E. Krieger, Malabar, FL, 1982 Search PubMed; (b) Q. Liu, Y. Lan, J. Liu, G. Li, Y.-D. Wu and A. Lei, J. Am. Chem. Soc., 2009, 131, 10201–10210 CrossRef CAS PubMed; (c) F. Schoenebeck and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 2496–2497 CrossRef CAS PubMed; (d) B. Liu, M. Gao, L. Dang, H. Zhao, T. B. Marder and Z. Lin, Organometallics, 2012, 31, 3410–3425 CrossRef CAS.
- (a) S. L. Qu, Y. F. Dang, C. Y. Song, J. D. Guo and Z.-X. Wang, ACS Catal., 2015, 5, 6386–6396 CrossRef CAS; (b) G. Jindal and R. B. Sunoj, J. Am. Chem. Soc., 2014, 136, 15998–16008 CrossRef CAS PubMed; (c) W. Guan, S. Sakaki, T. Kurahashi and S. Matsubara, ACS Catal., 2015, 5, 1–10 CrossRef CAS.
- (a) K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS; (c) F. Weinhold, J. Comput. Chem., 2012, 33, 2363–2379 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision A.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- L. Liu, Y. Wu, P. Chen, C. Chan, J. Xu, J. Zhu and Y. Zhao, Org. Chem. Front., 2016, 3, 423–433 RSC.
- For examples: (a) C.-H. Lim, A. M. Holder, J. T. Hynes and C. B. Musgrave, J. Am. Chem. Soc., 2014, 136, 16081–16095 CrossRef CAS PubMed; (b) C. H. Lim, A. M. Holder and C. B. Musgrave, J. Am. Chem. Soc., 2013, 135, 142–154 CrossRef CAS PubMed; (c) K. S. Sandhya and C. H. Suresh, Organometallics, 2013, 32, 2926–2933 CrossRef CAS; (d) E. Erdtman, E. A. C. Bushnell, J. W. Gauld and L. A. Eriksson, Comput. Theor. Chem., 2011, 963, 479–489 CrossRef CAS.
- L. Zhao, M. Wen and Z.-X. Wang, Eur. J. Org. Chem., 2012, 2012, 3587–3597 CrossRef CAS.
- (a) S. Bontemps, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2014, 136, 4419–4425 CrossRef CAS PubMed; (b) S. Bontemps and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2013, 52, 10253–10255 CrossRef CAS PubMed; (c) G. Jin, C. G. Werncke, Y. Escudie, S. Sabo-Etienne and S. Bontemps, J. Am. Chem. Soc., 2015, 137, 9563–9566 CrossRef CAS PubMed.
- (a) S. Burck, D. Gudat, M. Nieger and W.-W. du Mont, J. Am. Chem. Soc., 2006, 128, 3946–3955 CrossRef CAS PubMed; (b) D. Gudat, A. Haghverdi and M. Nieger, Angew. Chem., Int. Ed., 2000, 39, 3084–3086 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional computational results, energies, and Cartesian coordinates of the optimized structures. See DOI: 10.1039/c7sc00824d |
| ‡ Y. L. and Z.-H. G. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |