
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity studies on [Cp′Fe(μ-I)]2: nitrido-, sulfido- and diselenide iron complexes derived from pseudohalide activation†
Matthias
Reiners
a,
Miyuki
Maekawa
 a,
Constantin G.
Daniliuc
a,
Matthias
Freytag
a,
Peter G.
Jones
a,
Peter S.
White
b,
Johannes
Hohenberger
c,
Jörg
Sutter
c,
Karsten
Meyer
c,
Laurent
Maron
*d and
Marc D.
Walter
*a
a,
Constantin G.
Daniliuc
a,
Matthias
Freytag
a,
Peter G.
Jones
a,
Peter S.
White
b,
Johannes
Hohenberger
c,
Jörg
Sutter
c,
Karsten
Meyer
c,
Laurent
Maron
*d and
Marc D.
Walter
*a
aTechnische Universität Braunschweig, Institut für Anorganische und Analytische Chemie, Hagenring 30, 38106 Braunschweig, Germany. E-mail: mwalter@tu-bs.de
bUniversity of North Carolina at Chapel Hill, Department of Chemistry, Chapel Hill, North Carolina 27599-3290, USA
cUniversity of Erlangen-Nürnberg, Department of Chemistry & Pharmacy, Inorganic Chemistry, Egerlandstr. 1, 91058 Erlangen, Germany
dUniversité de Toulouse, INSA-UPS-LPCNO, CNRS-LPCNO, 135 Avenue de Rangueil, F-31077 Toulouse, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
First published on 11th April 2017
Abstract
The iron half-sandwich [Cp′Fe(μ-I)]2 (Cp′ = 1,2,4-(Me3C)3C5H2, 1) reacts with the pseudohalides NCO−, SCN−, SeCN− and N3− to give [Cp′Fe(μ-NCO)]2 (2), [Cp′Fe(μ-S)]2 (3), [Cp′Fe(μ-Se2)]2 (4) and [Cp′Fe(μ-N)]2 (5), respectively. Various spectroscopic techniques including X-ray diffraction, solid-state magnetic susceptibility studies and 57Fe Mössbauer spectroscopy were employed in the characterization of these species. Mössbauer spectroscopy shows a decreasing isomer shift with increasing formal oxidation state, ranging from Fe(II) to Fe(IV), in complexes 1 to 5. The sulfido-bridged dimer 3 exhibits strong antiferromagnetic coupling between the Fe(III) centers. This leads to temperature-independent paramagnetism (TIP) at low temperature, from which the energy gap between the ground and the excited state can be estimated to be 2J = ca. 700 cm−1. The iron(IV) nitrido complex [Cp′Fe(μ-N)]2 (5) shows no reactivity towards H2 (10 atm), but undergoes clean reactions with CO (5 bar) and XylNC (Xyl = 2,6-Me2C6H3) to form the diamagnetic isocyanate and carbodiimide complexes [Cp′Fe(CO)2(NCO)] (7) and [Cp′Fe(CNXyl)2(NCNXyl)] (8), respectively. All compounds were fully characterized, and density functional theory (DFT) computations provide useful insights into their formation and the electronic structures of complexes 3 and 5.
Introduction
Iron nitrides have been extensively investigated because of their potential in synthetic and biological N2 activation and functionalization.1 In general they can be prepared by direct N2 activation using a low-valent iron species;1h,1l,1n alternatively N-atom transfer reagents such as Li(dbabh) (dbabh = 2,3:5,6-dibenzo-7-azabicyclo[2.2.1]hepta-2,5-diene2 or—more frequently—inorganic azides have been used.1g,1i,3 However, N2 elimination from iron azido precursors usually requires forcing conditions such as reduction or photochemical/thermal activation.1g,1i,3 Nevertheless, surprisingly facile conversion of [Bu4N]N3 to the corresponding iron nitride derivative has been observed in the context of trinuclear iron compounds such as [(tbsL)Fe3(thf)] (tbsL = [1,3,5-C6H9(NPh-o-NSitBuMe2)3]6−) to yield [Bu4N][(tbsL)Fe3(μ3-N)].1j,4Our laboratory recently reported the synthesis of [Cp′Fe(μ-I)]2 (Cp′ = η5-1,2,4-(Me3C)3C5H2, 1)5 and its reactivity toward KBH4 to form the metastable, diamagnetic complex [Cp′FeBH4], which releases H2 to form a trimeric iron boryl [Cp′FeBH2]3.6 In contrast, the diiron trihydride and tetrahydride compounds, [Cp′2Fe2(μ-H)3] and [Cp′Fe(μ-H)2]2 can be isolated from the reaction of 1 with KHBEt3.7 Although no N2 activation was achieved with [Cp′Fe(μ-H)2]2, it serves under mild conditions as a synthon for reactive iron(I) Cp′Fe-fragments, as illustrated by its facile reaction with white phosphorus (P4).7 These results suggested that the bulky Cp′Fe-fragment provides sufficient steric protection to kinetically hamper [Cp′2Fe] formation and can also stabilize iron in higher (formal) oxidation states.8 The successful synthesis of the phosphido-bridged complex [Cp′Fe(μ-P)]2 by photochemically induced CO elimination from [Cp′Fe]2(μ-P2)(μ-CO) (Chart 1) also supports this assumption.9 In this contribution we report on some observations regarding the reactivity and redox chemistry of [Cp′Fe(μ-I)]2 (1) with pseudohalides such as NCO−, SCN−, SeCN− and N3−.
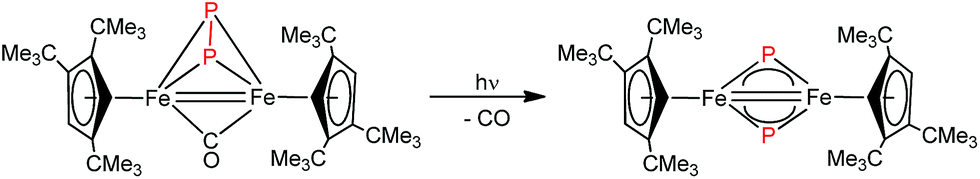 | ||
| Chart 1 Preparation of [Cp′Fe(μ-P)]2.9 | ||
Results and discussion
Reaction with pseudohalides
The products formed by the reaction 1 with the KOCN, KSCN, KSeCN and NaN3 may be divided into three classes; the reactions proceed either with (a) no redox chemistry (OCN−), (b) a 2-electron redox process (SCN− and SeCN−) or (c) a 4-electron redox process (N3−) to give [Cp′Fe(μ-NCO)]2 (2), [Cp′Fe(μ-S)]2 (3) and [Cp′Fe(μ-Se2)]2 (4), or [Cp′Fe(μ-N)]2 (5), respectively (Scheme 1).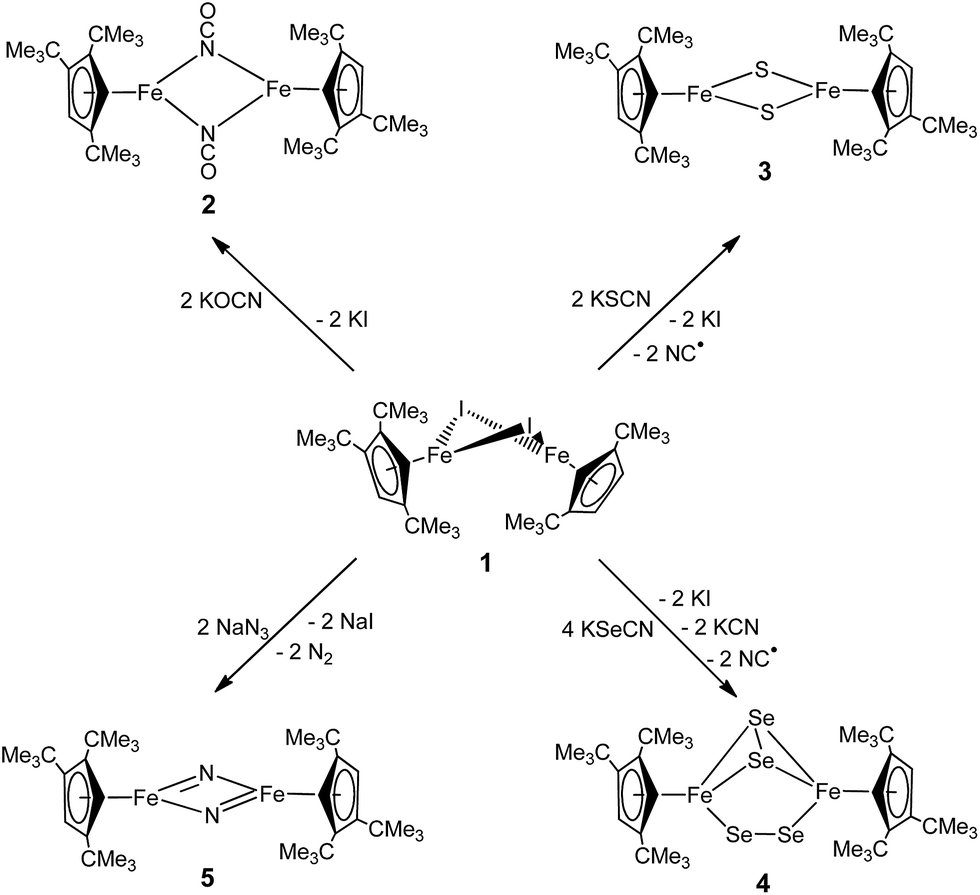 | ||
| Scheme 1 Reaction of 1 with various pseudohalides. | ||
Synthesis of a dimeric μ-NCO iron(II) complex (2)
Salt metathesis of 1 with KOCN yields the dimeric iron(II) isocyanate complex [Cp′Fe(μ-NCO)]2 (2), which is isolated as green crystals in moderate yield (52%). The paramagnetic complex 2 melts intact at 163 °C and shows a molecular ion (662 amu) in the EI-MS spectrum with the correct isotope distribution. The 1H NMR resonances corresponding to the tBu groups Cp′ ligand (δ = −4.05 (18H) and −7.80 (9H)) are in the same range as those observed for the starting material 1 (δ = −7.8 (18H) and −13.5 (9H)).5 Crystals of 2 suitable for an X-ray diffraction experiment were grown from a concentrated pentane solution at −30 °C (see Table S1 in the ESI†) and the molecular structure is shown in Fig. 1. Complex 2 crystallizes in the monoclinic space group P21/n with crystallographically imposed inversion symmetry.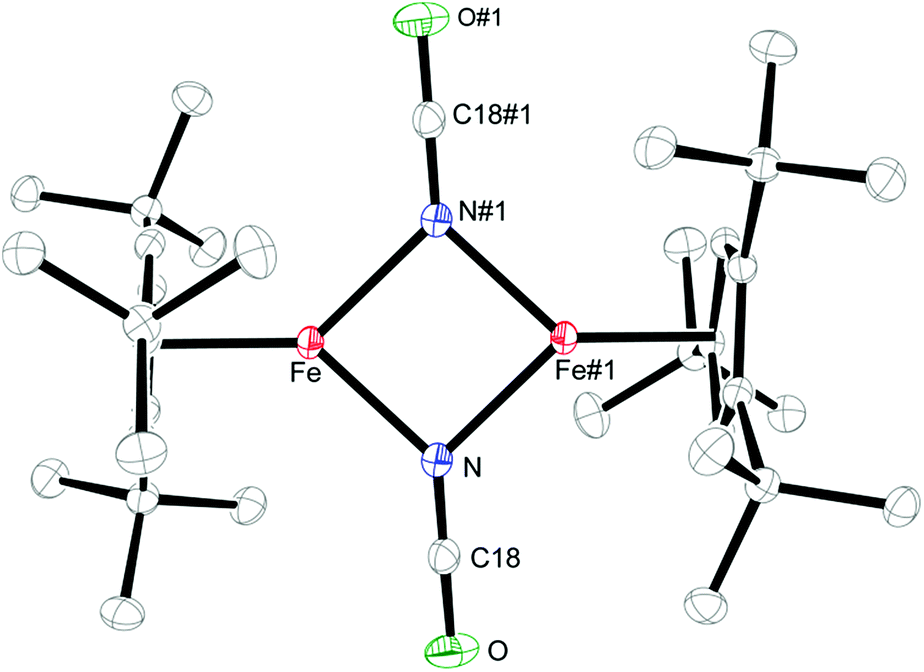 | ||
| Fig. 1 Thermal ellipsoid plot (50% probability) of the molecular structure of 2. H-atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Cp′cent–Fe 1.919, Cp′plane–Fe 1.916, Fe–N 2.0425(12), Fe–N#1 2.0840(12), N–C18 1.1904(19), O–C18 1.1824(19), Fe⋯Fe#1 3.002, N–Fe–N#1 86.65(5), Fe–N–Fe#1 93.35(5), O–C18–N 179.36(18). Symmetry transformations used to generate equivalent atoms: #1 −x + 1, −y + 1, −z + 1. | ||
The X-ray data were of sufficient quality to establish exclusive κN-coordination of the ambidentate OCN−. Furthermore the iron(II) atoms in 1 and 2 adopt a high-spin configuration with 4 unpaired electrons per Fe(II) atom, as can be inferred from the long Cp′cent–Fe bond distances in 1 (1.93 Å),52 (1.92 Å) and [Cp′FeOC6H3(CMe3)2-3,5]2 (1.94 Å.8 For comparison, the Cp′cent–Fe distances in [Cp′2Fe] (S = 0) and [Cp′2Fe][SbF6] (S = 1/2) are significantly shorter at 1.72 Å (ref. 5) and 1.77 Å,10 respectively. The spin state assignment in 2 is further substantiated by solid-state magnetic susceptibility data, which indicate antiferromagnetic coupling between the two Fe(II) (S = 2) centers at low temperature (Fig. 2). The magnetic trace for both complexes may be simulated using a Heisenberg exchange Hamiltonian11 and the respective fit parameters are provided in the figure caption. The extent of antiferromagnetic coupling varies significantly between 1 and 2 (J12 = −6.5 cm−1 and J12 = −13.6 cm−1, respectively) which can be rationalized by the difference in the Fe⋯Fe distances in both complexes of 3.526, 3.002 Å, respectively. Furthermore, there are also significant differences in the respective Fe2X2-cores. Please note that a butterfly structure is adopted for the Fe2I2-core in 1 with afold angle between the two I1–Fe1–I2 and I1–Fe2–I2 planes of 24.49°, whereas a planar Fe2N2-core is realized in 2.5 Another notable feature is a large negative zero-field splitting parameter D that is required to obtain a suitable simulation of the magnetic trace. Although the model employed is not completely sufficient to describe the low-temperature regime found in complex 1 and no significant improvement is achieved when a paramagnetic impurity is assumed. We also want to mention that this feature has been observed in several independently prepared samples prepared over the time frame of several years and measured on different magnetometers and using different sample containers (quartz tubes or KEL-F buckets).
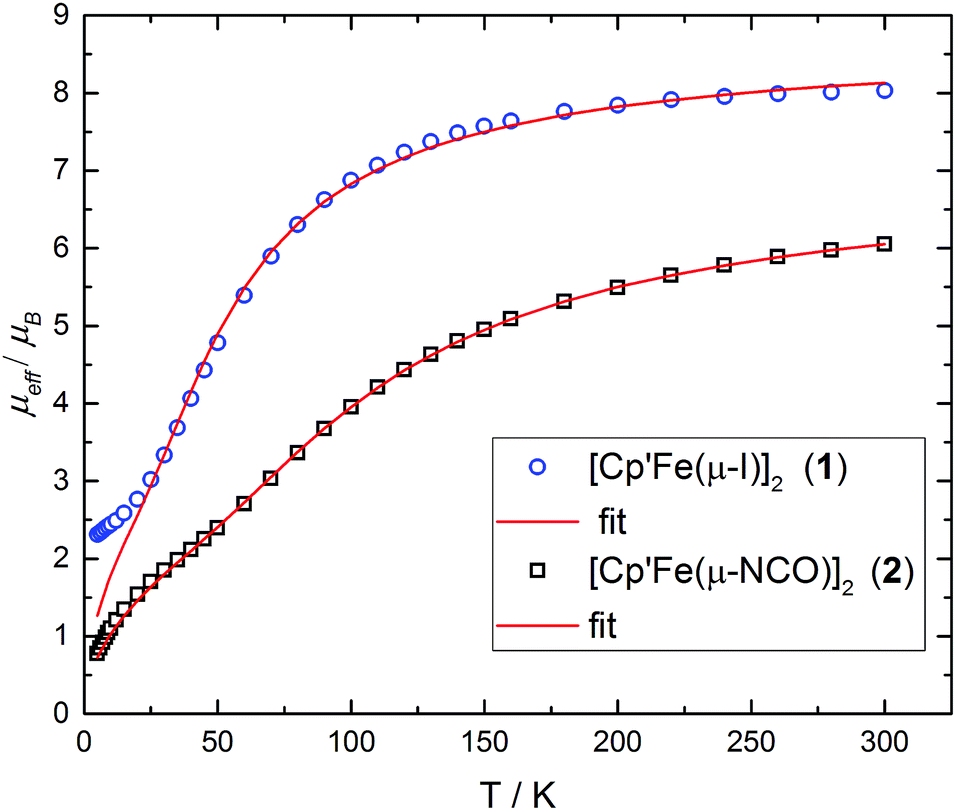 | ||
| Fig. 2 Effective magnetic moment μeffvs. T plots for compounds 1 and 2. Fit parameter for 1: S1 = S2 = 2.0; g1 = g2 = 2.51; J12 = −6.5 cm−1; D1 = D2 = −18.16 cm−1, E/D1 = E/D2 = 0.11. Fit parameter for 2: S1 = S2 = 2.0; g1 = g2 = 2.04; J12 = −13.6 cm−1; D1 = D2 = −32.85 cm−1, E/D1 = E/D2 = 0.10. | ||
While solid-state magnetic susceptibility studies represent a bulk method, the local electronic structure at the individual Fe nucleus may conveniently be probed by 57Fe Mössbauer spectroscopy.12 The isomer shift (δiso) provides information on the s-electron density at the Fe nucleus, which is modulated by the iron oxidation state, the spin state and the covalency in the iron–ligand bonds. However, in strongly covalent compounds unambiguous assignment of the oxidation and spin state exclusively based on δiso is not necessarily straightforward, but some trends have emerged from extensive studies on various iron (coordination) compounds, and are well understood.12 In contrast, Mössbauer investigations on Cp-containing iron complexes, which are more covalent than traditional coordination complexes, have remained relatively rare.13 In zero-field 57Fe Mössbauer spectra (at 77 K), complexes 1 and 2 show well-resolved doublets with isomer shifts δiso = 1.08(1) and 1.05(1) mm s−1 and with a quadrupole splitting ΔEQ = 1.98(1) and 1.49(1) mm s−1, respectively (Fig. 3), which is in the range commonly observed for high-spin (S = 2) Fe(II) species.12a The asymmetry in the quadrupole doublet of 1 indicates a dynamic process, for which the spin relaxation time at 77 K is close to the Mössbauer time scale (ca. 10−7 s) (=intermediate spin relaxation regime).13c
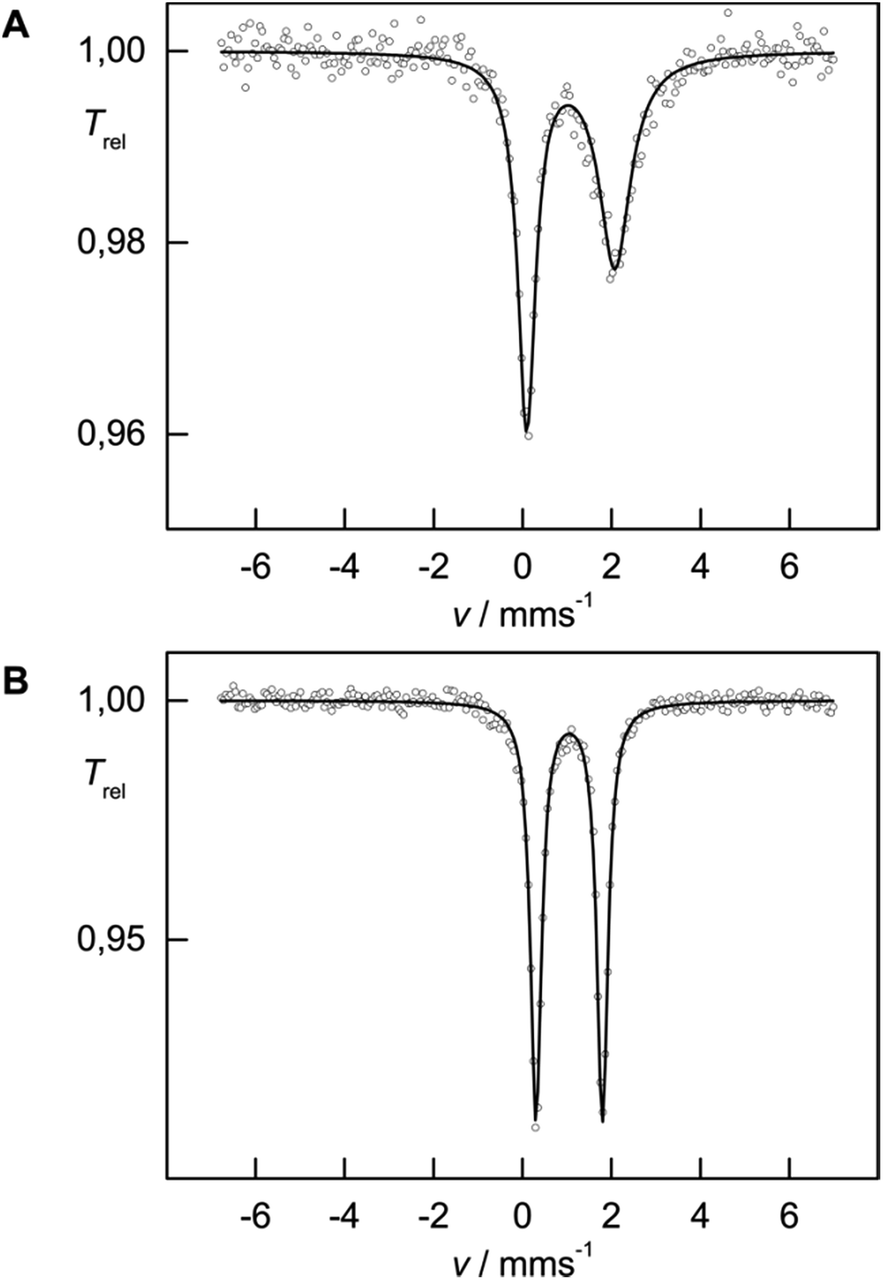 | ||
| Fig. 3 Zero field Mössbauer spectra of 1 (A) and 2 (B) recorded at 77 K. | ||
When coordinating ligands such as Ph3PO and CO are added to 1 and 2, either 16- or 18-valence-electron (VE) complexes are formed (Scheme 2). For example, addition of Ph3PO results in isolation of the paramagnetic, 16-valence-electron (VE) complexes [Cp′Fe(I)(OPPh3)] (see ESI† for details) and [Cp′Fe(NCO)(OPPh3)] (6), respectively. Furthermore, the exchange of free and coordinated Ph3PO is slow on the NMR time scale and no spin state change occurs when the dimeric structure is broken (see ESI† for details). In contrast, exposure of 1 and 2 to CO (1 atm) immediately forms the diamagnetic, 18VE derivatives [Cp′Fe(CO)2(I)]14 and [Cp′Fe(CO)2(NCO)] (7), respectively. The molecular structures of 6 and 7 are shown in Fig. 4 and 5, respectively; selected bond distances and angles are listed in the corresponding figure captions. The most notable difference in the molecular structures of these compounds are the Cp′cent–Fe distances of 1.97 Å vs. 1.73 Å for 6 and 7, which reflect their different electronic ground states of S = 2 and S = 0, respectively.
 | ||
| Scheme 2 Adduct formation of 2 with Ph3PO and CO. | ||
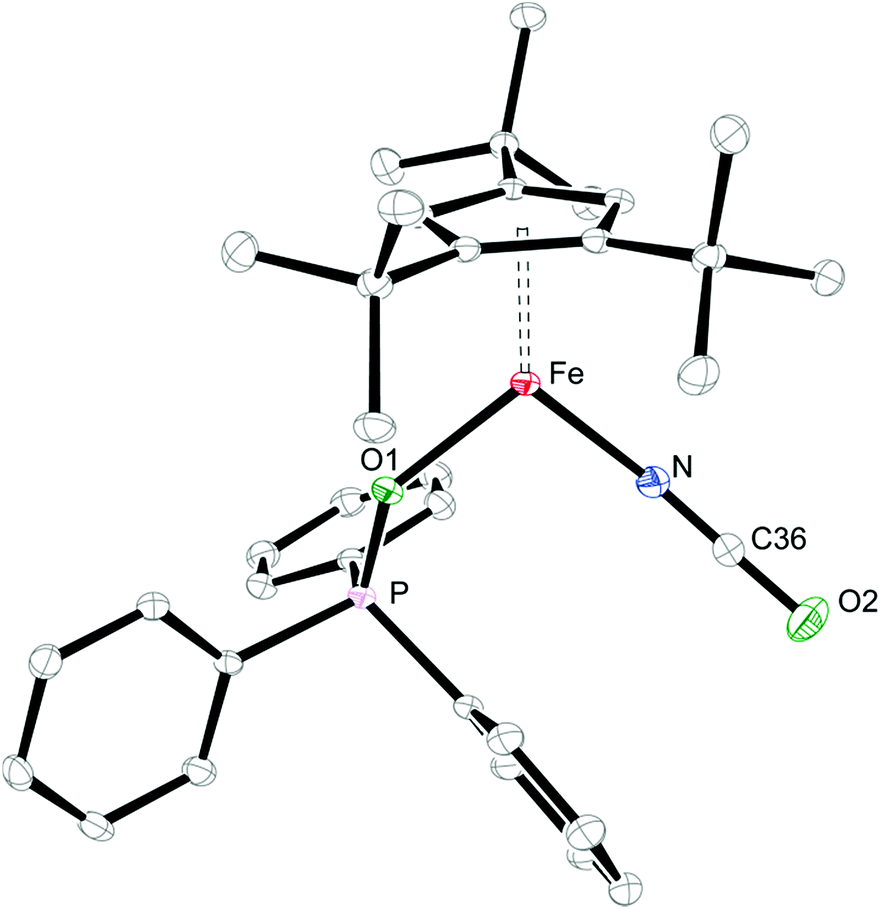 | ||
| Fig. 4 Thermal ellipsoid plot (30% probability) of the molecular structure of 6. H-atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Cp′cent–Fe 1.968, Cp′plane–Fe 1.967, Fe–N 1.9530(17), Fe–O1 2.0164(13), O1–P 1.5074(14), N–C36 1.170(3), C36–O2 1.203(6), O1–Fe–N 101.30(7), Fe–O1–P 137.97(8), Fe–N–C36 154.80(18), N–C36–O2 177.2(2). | ||
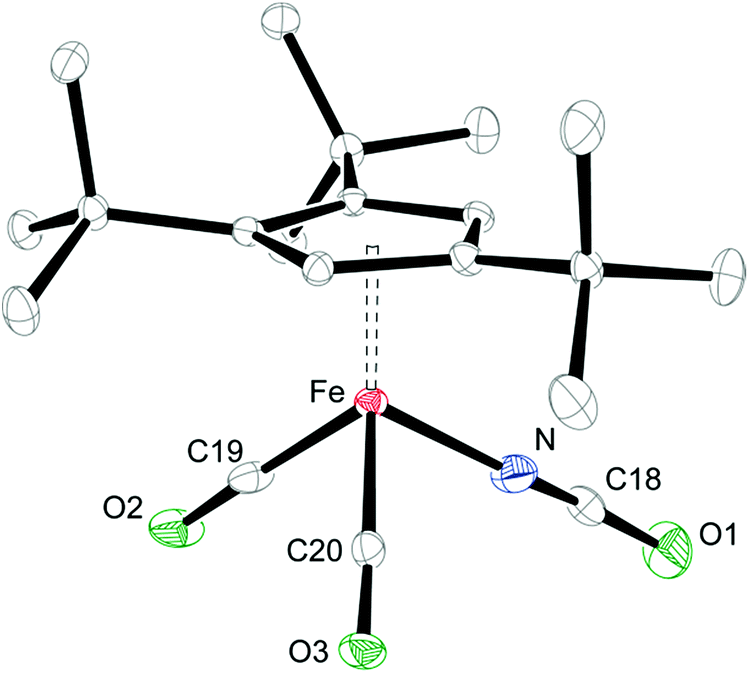 | ||
| Fig. 5 Thermal ellipsoid plot (30% probability) of the molecular structure of 7. H-atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Cp′cent–Fe 1.732, Cp′plane–Fe 1.731, Fe–N 1.952(3), Fe–C19 1.785(3), Fe–C20 1.784(2), N–C18 1.108(4), C18–O1 1.248(4), C19–O2 1.147(3), C20–O3 1.151(3), Fe–N–C18 173.1(2), N–C18-01 173.8(3). | ||
Preparation of sulfido- (3) and diselenido-bridged (4) iron(III) complexes from KSCN and KSeCN
When the heavier homologues KSCN and KSeCN are employed, the Fe(III) compounds 3 and 4 are isolated in low yield (Scheme 1). The formation of these compounds is unexpected, since generally no redox processes are encountered when pseudohalides coordinate to transition metals,15 which implies that the Cp′Fe-fragment is susceptible to redox chemistry. Furthermore, this redox behavior is apparently facilitated by the synergy between two or more Fe atoms, an effect that also accounts for the facile N2 release from [Bu4N]N3 in the presence of [(tbsL)Fe3(thf)].4 Furthermore, S–C bond cleavage of a SCN− ligand is a rare event, but has previously been observed in [Pd3(SCN)(μ3-CO)(μ-dppm)3]+ to form [Pd3(μ3-S)(CN)(μ-dppm)3]+.16 The FeMoco nitrogenase enzyme also transiently binds SCN− before reduction to HCN and H2S occurs; but the mechanism of this biological SCN− reduction remains unknown.17 Cleavage of the S–C and Se–C bond to form 3 and 4 requires that the Fe(II) atoms are oxidized to Fe(III) concomitant with the formation of CN˙ radicals. The remarkable difference between the reactivity of [OCN]− and [XCN]− (X = S, Se) is most likely traced to the inherently weaker C–X bonds for the higher chalcogenide homologues (X = S and Se), which also result in shorter and therefore stronger C–N bonds of 1.15 Å in the free thio- and selenocyanate anions.18 On coordination of [XCN]− (X = S, Se) to two Fe(II) fragments [Cp′Fe]+ (as in complex 2) a redox-process into the σ*-orbitals of the C–X bond can then be initiated leading to the Fe(III) compounds 3 and 4 and CN˙ radicals, which are highly reactive species that readily undergo H atom abstractions from THF to generate HCN,19 or alternatively they may dimerize to cyanogen (CN)2. Unfortunately, we have so far been unable to detect either of these side products spectroscopically. Nevertheless, it is conceivable that the formation of HCN or (CN)2 can induce undesired side reactions, which cause the observed formation of hydrolyzed Cp′ ligand (HCp′) and unidentified paramagnetic species in the reaction mixture. This may also account for the relatively low yields, in which complexes 3 and 4 are isolated as crystalline and analytically pure materials (13% and 15%, respectively). Nevertheless, the formation of the diselenido-bridged complex [Cp′Fe(μ-Se2)]2 (4) can be rationalized by the fact that selenocyanate SeCN− can also act as a Se source.20 The formation of the diselenido-bridged complex 4 suggests that it represents a thermodynamic minimum relative to the selenido-bridged species [Cp′Fe(μ-Se)]2.Iron–sulfur clusters play an important role as biological cofactors involved in electron transfer processes.21 Hence, several bioinorganic model complexes with thiolato ligands have been prepared22 and their electrochemistry studied.23 Despite significant general interest in these systems,24 well-defined [2Fe–2S] derivatives with other than thiolato co-ligands have remained rare.25 From an organometallic point of view, metal chalcogenido clusters are generally prepared by the thermal reaction of elemental chalcogens with transition metal carbonyls such as [CpFe(μ-CO)(CO)]2.26 The outcome of these reactions is not very predictable, but some control can be achieved by changing the steric demand on the Cp-ligands, whereby various iron chalcogenides such as [(η5-C5H5)4Fe4Sn] (n = 4–6),27 [(η5-C5Me5)2Fe2S4],28 [(η5-(Me3Si)C5H4)4Fe4S6],29 [(η5-1,3-(Me3Si)2C5H3)2Fe2S4],29 [(η5-C5H5)2Fe4Se4],30 and [(η5-C5Me5)3Fe3Se8]31 have been isolated. However, the facile and selective formation of [Cp′Fe(μ-S)]2 (3) from 1 and KSCN provides the first organometallic model of the Rieske [2Fe–2S] cluster, whereas thermolysis of [Cp′Fe(CO)(μ-CO)] in the presence of S8 only yields the disulfide-bridged compound [Cp′Fe(μ-S2)]2 (see ESI† for details).
Crystals of 3 and 4 were obtained from concentrated Et2O solutions at −30 °C (see Table S1 in the ESI†) and their molecular structures are shown in Fig. 6 and 7. Selected bond distances and angles are listed in the respective figure captions.
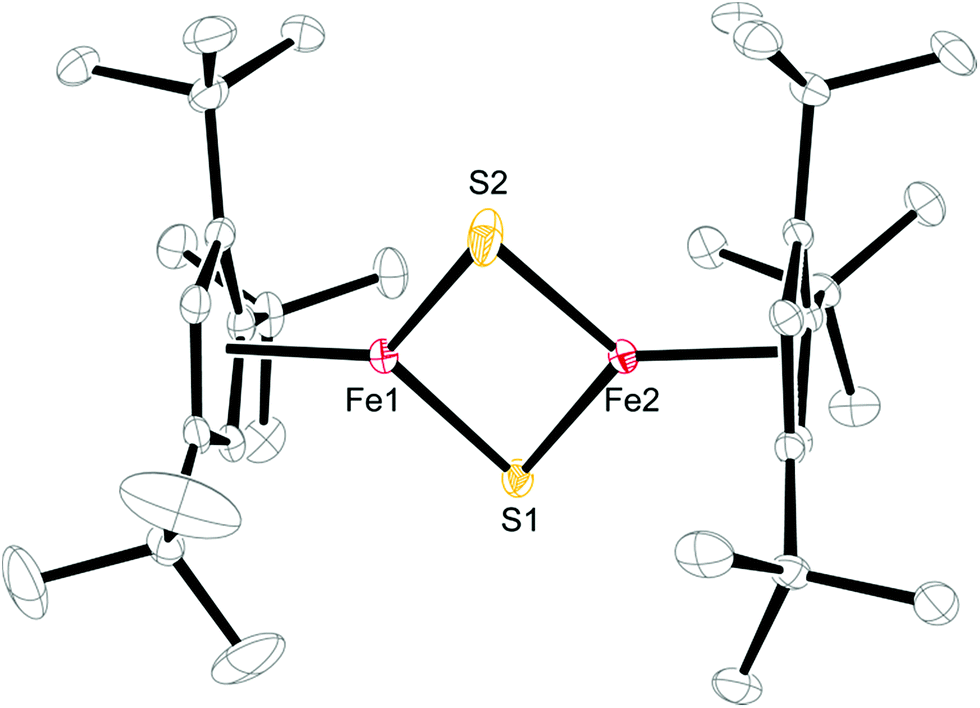 | ||
| Fig. 6 ORTEP of [Cp′Fe(μ-S)]2 (3) (30% probability ellipsoids). H-atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Cp′cent–Fe(ave) 1.833, Cp′plane–Fe(ave) 1.831, Fe1–S1 2.1415(6), Fe1–S2 2.1305(7), Fe2–S1 2.1435(6), Fe2–S2 2.12393(7), Fe1⋯Fe2 2.5873(5), S1⋯S2 2.587, Fe1–S1–Fe2 74.29(2), Fe1–S2–Fe2 74.91(2), S1–Fe1–S2 105.21(3), S1–Fe2–S2 105.37(3). | ||
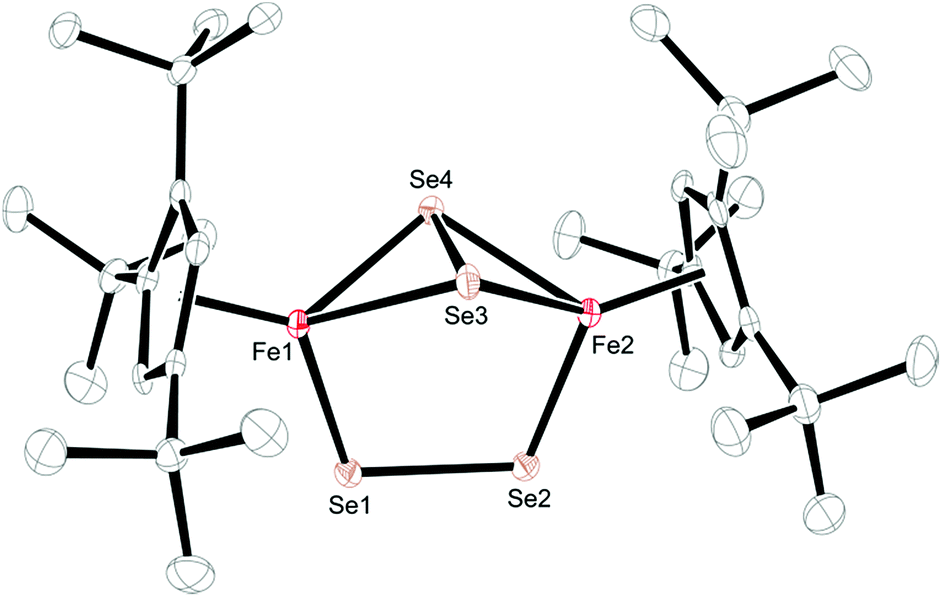 | ||
| Fig. 7 ORTEP of [Cp′Fe(μ-Se2)]2 (4) (50% probability ellipsoids). H-atoms are omitted for clarity. Selected bond distances (Å): Cp′cent–Fe(ave) 1.712, Cp′plane–Fe(ave) 1.710, Fe1–Se1 2.2513(6), Fe1–Se3 2.4055(6), Fe1–Se4 2.4145(6), Fe2–Se2 2.2432(6), Fe2–Se3 2.4213(6), Fe2–Se4 2.4073(6), Se1–Se2 2.2993(5), Se3–Se4 2.3255(5), Fe1⋯Fe2 3.721. | ||
A comparison of the structural parameters for the [2Fe–2S] core in 3 with the biological [2Fe–2S] clusters and their biomimetic models shows some interesting features. Complex 3 has significantly shorter Fe–S bond distances with an average value of 2.1349 ± 0.0093 Å than those found in the structurally characterized biological systems, which range from 2.16–2.23 Å. The latter values are well reproduced in the biomimetic structures (2.185–2.232 Å).25c,32 In addition, the average Fe–S–Fe and S–Fe–S bond angles of 74.60 ± 0.44° and 105.29 ± 0.11° are also at the lower and upper ends of the literature examples.25c,32 Consequently the Fe–Fe distance of 2.5873(5) Å is shorter than that in [(meso-phenyl-dibenzimidazolate)2Fe2(μ-S)]2− (2.7019(5) Å)25c or in [(nacnac)Fe(μ-S)]2 (nacnac = MeC[C(Me)N(2,6-Me2C6H3)]2) (2.669(1) Å).32b The more compact [2Fe–2S] core may manifest itself in the physical properties of 3, e.g., the magnetic exchange interaction between the two Fe(III) atoms (vide infra).
In contrast, the diselenide-bridged compound 4, which displays non-crystallographic twofold symmetry (r.m.s. deviation 0.12 Å), features a rather long Fe⋯Fe distance of 3.721 Å. The Fe–Se distances are 2.2513(6) Å and 2.2432(6) Å for the end-on coordinated [Se2]2− moiety, while those corresponding to the side-on coordinated [Se2]2− moiety range between 2.4055(6) Å and 2.4213(6) Å. This leads to Se–Se distances of 2.2993(5) Å and 2.3255(5) Å for the end-on and side-on coordinated [Se2]2− fragments, respectively. In accord with these different binding modes, two resonances at δ = 2103.2 and 493.2 ppm are found in the 77Se NMR spectrum. Unfortunately, we cannot unambiguously assign these chemical shifts to the different coordination modes. Several transition metal polyselenides,33 are known, such as [{(η5-C5H5Me)V(μ-Se2)}2(μ-Se)],34 but no 77Se NMR data have been provided. Nevertheless, complexes 3 and 4 feature significantly shorter average Cp′cent–Fe distances of 1.83 and 1.71 Å than in their Fe(II) counterparts 1 and 2. This difference of 0.12 Å between 3 and 4 appears unusual, considering that both molecules contain Fe(III) atoms. A d5 system can potentially adopt three different spin states of SFe = 1/2, SFe = 3/2 and SFe = 5/2; and in a dimeric structure these iron centers can either be magnetically independent or anti-/ferromagnetically coupled. A closer inspection of the 1H NMR spectra of 3 and 4 reveals that the resonances of the Cp′ moiety are narrow and occur between δ = 11.0 and 0.4 ppm for 3 and δ = 5.9 and 0.8 ppm for 4. The larger chemical shift window for complex 3 is also found in its 13C{1H} NMR spectrum, which shows very unusual chemical shifts for a diamagnetic compound ranging from δ = 235.3 to 9.7 ppm; the most downfield shifted resonance (δ = 235.3 ppm) is assigned to one of the Cp′ ring-Cipso positions. This indicates that unpaired spin density is transferred from the iron atoms to the Cp′ ligand.35 These observations prompted us to record variable temperature (VT) 1H NMR spectra for 3 and 4. While the 1H NMR resonances in 4 exhibit only a negligible temperature dependence (see ESI† for details), the ring CH resonance in 3 varies from δ = 8.34 to 12.40 ppm in the temperature range 176 K to 376 K, approaching a diamagnetic limit at low temperature (Fig. 8). The different VT behavior can be rationalized by antiferromagnetic coupling between the two Fe(III) atoms in 3 and 4 to form an open-shell singlet ground state, whereby the degree of exchange coupling is significantly different in both materials. While in 4 the Fe(III) atoms undergo strong antiferromagnetic coupling, a smaller energy gap between ground and excited state in 3 allows the thermal population of the (uncoupled) excited state. Based on the underlying Boltzmann distribution the energy gap (2J) between these states can be estimated (Fig. 8).
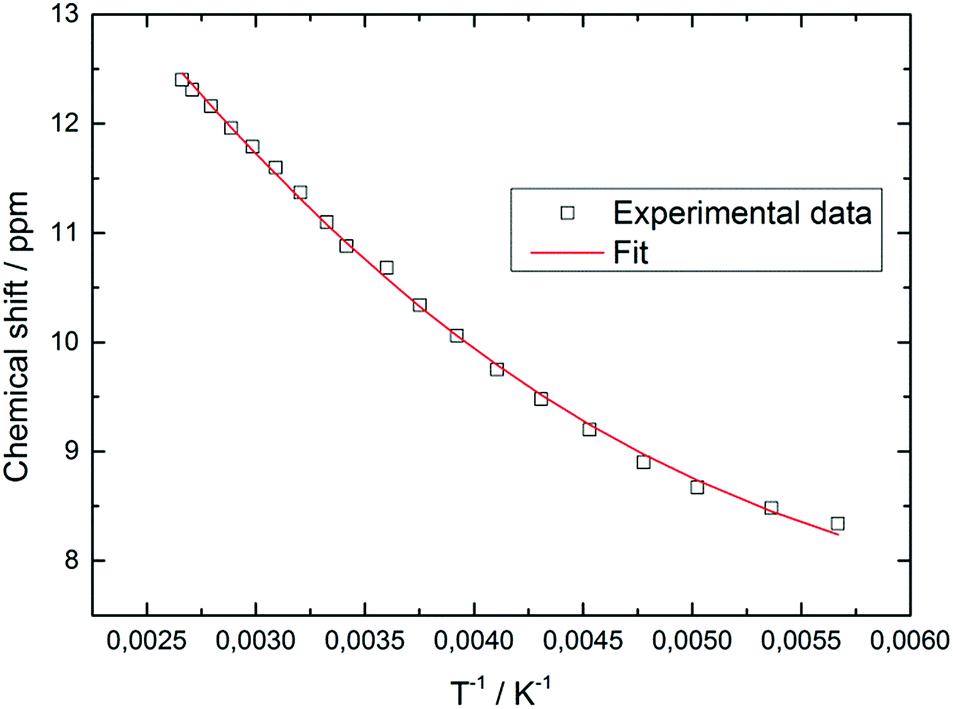 | ||
| Fig. 8 Temperature dependence of the 1H NMR Cp′ ring-CH resonance in 3 and the associated fit. | ||
The fit of the temperature dependence of the 1H NMR chemical shifts of Cp′ ring CH resonance yields an energy separation of |2J| = 654 cm−1 (0.082 eV or 1.87 kcal mol−1). Similar analyses have previously been applied to model spin-equilibria in [(C5Me5)Ni(acac)]36 or [(PNP)RuCl] (PNP = N(CH2CH2PtBu2)2).37
The effective magnetic moment of 3 increases gradually from 0.95 μB at 5 K to 1.98 μB at 360 K, consistent with strong antiferromagnetic coupling between the Fe(III) atoms. Fig. 9 shows the experimental data and the fit to the effective spin Hamilitonian Ĥ = −2J(Ŝ1Ŝ2) + gβ(ŜB), where J is the coupling constant and g represents the average electronic g value. Since the effective magnetic moment is still increasing at 360 K, we cannot unambiguously establish the spin state of the individual Fe(III) atoms, i.e. SFe = 1/2, SFe = 3/2, and SFe = 5/2. While we prefer the intermediate spin description (SFe = 3/2) based on the Cp′cent–Fe(ave) 1.833 Å, the other possibilities were also evaluated (see ESI† for details). Gratifyingly, the coupling constant J remains almost unaffected by the model employed for the simulation of the experimental data; Fig. 9 shows the results obtained for SFe = 3/2. Importantly, the J values determined from solution (NMR, J = −327 cm−1) and solid-state (magnetic susceptibility, J = −354 cm−1) data of 3 are in good agreement with each other. In addition, the antiferromagnetic exchange coupling in 3 is also significantly stronger than in the biomimetic model [(meso-phenyl-dibenzimidazolate)2Fe2(μ2-S)]2− (J = −179 cm−1).25c This increase may be caused by the shorter Fe–Fe and Fe–S bond distances in 3 facilitating direct and super-exchange coupling.
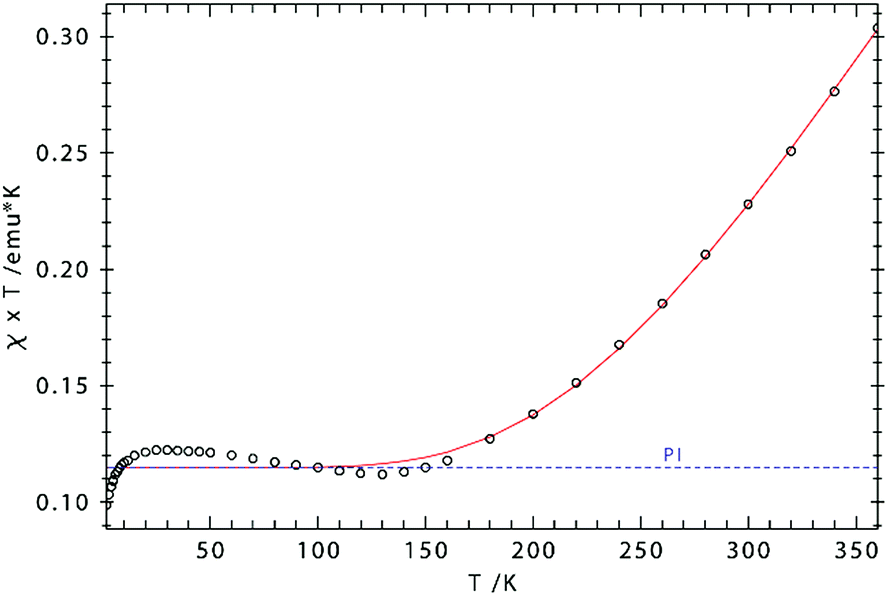 | ||
| Fig. 9 χT vs. T plot for 3. Simulation: S1 = S2 = 3/2; g1 = g2 = 2.244; J = −354 cm−1 [2J = −708 cm−1], paramagnetic impurity (PI, S = 2) = 2.6%, TIP = 1087 × 10−6 emu). | ||
The 57Fe Mössbauer spectra recorded at 77 K for complexes 3 and 4 are shown in Fig. 10. In contrast to compounds 1 and 2, the isomer shifts in 3 and 4 are significantly reduced with δiso = 0.45(1) mm s−1 [ΔEQ = 1.85(1) mm s−1] and δiso = 0.49(1) mm s−1 [ΔEQ = 1.26(1) mm s−1], respectively. This reduction is consistent with a change in formal oxidation state from Fe(II) to Fe(III). DFT computations further provide deeper insights into the electronic structure of 3 (vide infra).
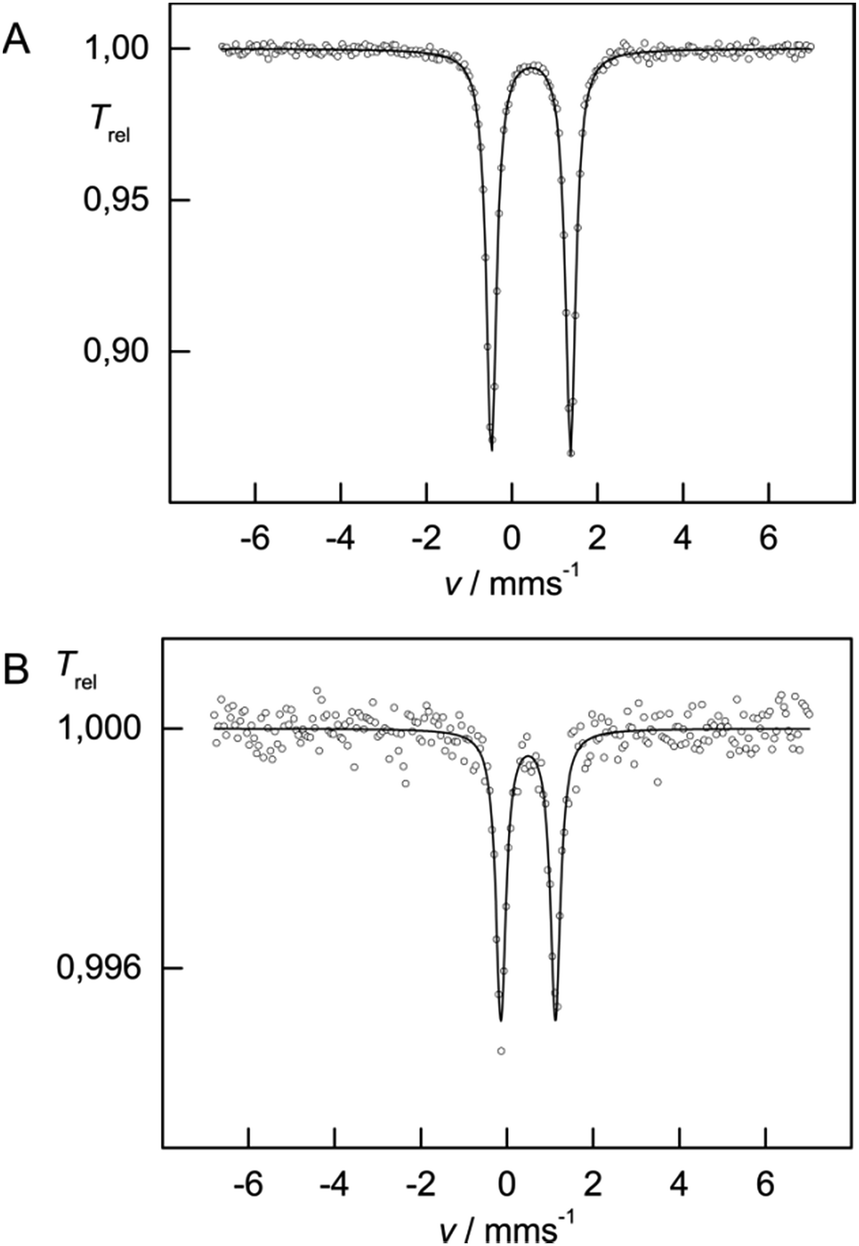 | ||
| Fig. 10 Zero field Mössbauer spectra of 3 (A) and 4 (B) recorded at 77 K. The selenium atoms in 4 act as strong absorber of the Mössbauer γ-radiation, so that only small effects (1.000 to 0.995) could be achieved despite collecting data for 12 days. | ||
Preparation of nitrido- bridged iron(IV) complex (5) from NaN3
Equally interesting is the reaction of 1 with NaN3, from which the dinuclear iron nitrido complex [Cp′Fe(μ-N)]2 (5) is isolated. The formation of 5 was investigated in more detail by 1H NMR spectroscopy in THF-d8. When 1 is dissolved in THF-d8, a yellow solution is formed that shows significantly upfield shifted tBu-resonances in the 1H NMR spectrum at δ = −22.4 and −27.7 ppm, corresponding to 18H and 9H, respectively. We rationalize this change in chemical shifts relative to those recorded in C6D6 solutions of 1 with the formation of a labile mono-THF adduct [Cp′Fe(I)(thf-d8)].5 On addition of NaN3 the THF-d8 solution colour gradually changes from yellow to orange and the 1H NMR resonances of the tBu-groups shift downfield to δ = −15.8 and −22.3 ppm, respectively. Furthermore, a strong IR stretch at 2059 cm−1 indicates the formation of an iron azido intermediate. When the THF solvent is removed under dynamic vacuum, an orange oily residue is initially formed that solidifies and turns olive-green on further exposure to dynamic vacuum. When this olive-green residue is extracted into C6D6, only the resonances corresponding to 5 are observed in the 1H NMR spectrum (see Experimental section for details). Consistent with these observations we propose the following mechanism: initial salt metathesis between 1 and NaN3 forms a solvent-stabilized iron azido intermediate [Cp′Fe(N3)(thf)] (A) that loses the coordinated THF on exposure to dynamic vacuum and generates a dimeric iron azido intermediate [Cp′Fe(μ-N3)]2 (B). Nevertheless, intermediate B is unstable and readily eliminates N2 at ambient temperature (even in the absence of light) (Scheme 3) to yield the dimeric iron nitrido species 5. Although our spectroscopic data support this mechanistic proposal, we have so far been unable to provide structural evidence for intermediates A and B. Nevertheless, the isolation of the 2 and its Ph3PO adduct [Cp′FeI(OPPh3)] is consistent with this proposal (see ESI† for details).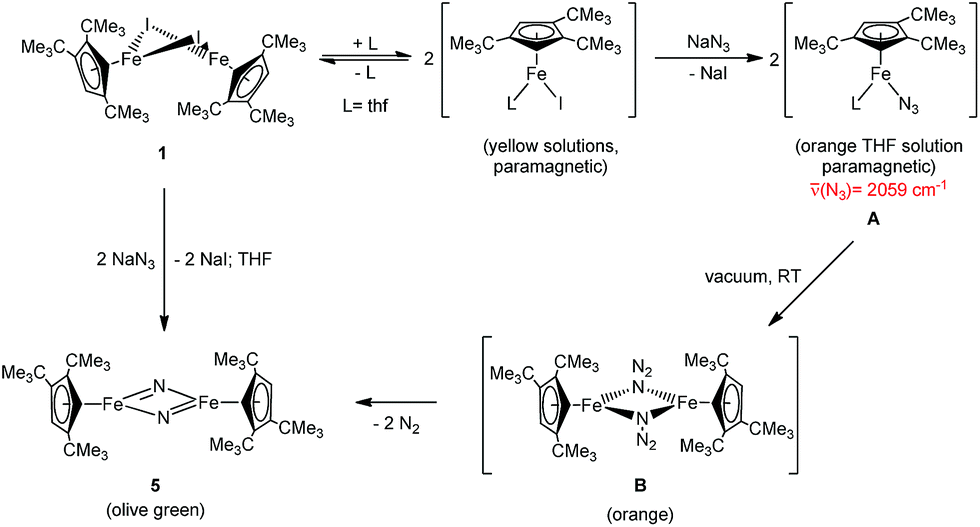 | ||
| Scheme 3 Proposed mechanism for the formation of [Cp′Fe(μ-N)]2 (5). Intermediates given in parenthesis were not isolated. | ||
Complex 5 may be crystallized from concentrated Et2O solution to yield olive-green crystals that decompose at 191–193 °C with gas evolution. In the EI-MS spectrum a molecular ion with the correct isotope distribution is observed, which shows that the Fe2N2 core stays intact under EI-MS conditions. When 15N-enriched sodium azide, Na15N(14N)2 is used, 5 is isolated with a statistical isotopic distribution, that is, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 mixture of [Cp′Fe(μ-15N)]2, [Cp′Fe(μ-14N)]2 and [Cp′2Fe2(μ-15N)(μ-14N)], respectively. In an attempt to probe the stability of the Fe2N2-moiety, unlabeled [Cp′Fe(μ-N)]2 was added to the 15N-enriched statistical mixture in an 1:1 ratio. This mixture was then dissolved in diethyl ether, crystallized and subjected to an EI-MS analysis. However, no enrichment of the mixed [Cp′2Fe2(μ-15N)(μ-14N)] compound was observed, suggesting that under the chosen conditions no dissociation into monomeric [Cp′FeN]-fragments occurred.
:1:2 mixture of [Cp′Fe(μ-15N)]2, [Cp′Fe(μ-14N)]2 and [Cp′2Fe2(μ-15N)(μ-14N)], respectively. In an attempt to probe the stability of the Fe2N2-moiety, unlabeled [Cp′Fe(μ-N)]2 was added to the 15N-enriched statistical mixture in an 1:1 ratio. This mixture was then dissolved in diethyl ether, crystallized and subjected to an EI-MS analysis. However, no enrichment of the mixed [Cp′2Fe2(μ-15N)(μ-14N)] compound was observed, suggesting that under the chosen conditions no dissociation into monomeric [Cp′FeN]-fragments occurred.
The 1H and 13C{1H} NMR spectra of 5 show narrow resonances in the diamagnetic region and the 1H NMR resonances exhibit only negligible temperature dependence (see Experimental section and ESI† for details), which also argues against a dissociation into monomeric species in solution. The 15N NMR spectrum recorded for 15N-enriched 5 features a significantly downfield shifted resonance at δ(15N) = 1118 ppm (relative to NH3(liq)). Interestingly, the related iron phosphido compound [Cp′Fe(μ-P)]2 also exhibits a strongly downfield shifted 31P NMR resonance at δ(31P) = 1406.9 ppm.9
Solid-state structural information on 5 was provided by X-ray diffraction. Fig. 11 shows the molecular structure of 5, which crystallizes in the tetragonal space group P41212 (see ESI† for details); and selected bond distances and angles are provided in the figure caption. The planar Fe2N2–core in 5 has pseudo-D2h symmetry and features nearly equal Fe1–N1 and Fe1–N2 bond distances of 1.744(2) and 1.734(2) Å, respectively. This is surprising, since the M–N bond distances in neutral M2N2 complexes commonly follow a distinct short-long-short-long pattern.38 The situation realized in 5 more closely resembles that typically observed in Fe2O2 complexes with their well-known diamond-core structural motifs.39 This is the first example of a d4 system adopting a M2N2 core and previous examples of neutral M2N2 species are limited to compounds with a formal d0 or d1 electron configuration such as [(η5-C5Me5)V(Cl)(μ-N)]2,40 [(η5-C5Me5)V(Cl)(μ-N)]2,41 {[(Me3SiN{CH2CH2NSiMe3}2)V(μ-N)]2}n (n = 0, −1),38b [(iPr2N)2Cr(μ-N)]2,38a, [{(anti-O3)Nb(μ-N)}2],42, [{(η5-C5Me5)(η3-iPrNC(Me)NiPr)Ta(μ-N)}2]43 [(tBuCH2)(RO)2W(μ-N)]2}n (n = 0, −1)38c and [{(ArO)2Nb(μ-N)}2]n (n = 0, −1).44 Both aspects render 5 and [Cp′Fe(μ-P)]2 rather unusual, with their formal Fe(IV) d4 ground states. The Fe–N bond distances of 1.744(2) and 1.734(2) Å in 5 may also be compared to standard Fe–N single and Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds, which lie in the range 1.9–2.0 Å and 1.6–1.7 Å, respectively.45 For comparison, the Fe–N distances in complex 2 (2.0425(12) and 2.0840(12) Å) are significantly longer than those in 5. Furthermore, the very long N⋯N distance of 2.667 Å precludes any direct interactions between the bridging N-atoms in 5, but the short Fe–Fe distance of 2.2325(7) Å raises questions regarding any direct Fe–Fe interactions, e.g., for FeFe double bonds a Fe–Fe distance of 2.316(1) Å was reported.46 In the related phosphido derivative [Cp′Fe(μ-P)]2 the Fe–Fe distance is as expected longer at 2.5005(4) Å, and to fulfil the 18VE rule the authors originally proposed a FeFe double bond.9 In contrast, for the iron alkylidyne derivative [Cp′Fe(μ-CPh)]2, which also features a short Fe–Fe distance of 2.3558(4) Å, DFT computations and NBO analyses indicate no direct Fe–Fe bonding interaction; and the benzylidyne ligands should instead be considered as deprotonated phenylcarbene fragments coordinated to two [Cp′Fe]+ fragments; thus, forming a low-spin iron(II) complex (Chart 2).47
N double bonds, which lie in the range 1.9–2.0 Å and 1.6–1.7 Å, respectively.45 For comparison, the Fe–N distances in complex 2 (2.0425(12) and 2.0840(12) Å) are significantly longer than those in 5. Furthermore, the very long N⋯N distance of 2.667 Å precludes any direct interactions between the bridging N-atoms in 5, but the short Fe–Fe distance of 2.2325(7) Å raises questions regarding any direct Fe–Fe interactions, e.g., for FeFe double bonds a Fe–Fe distance of 2.316(1) Å was reported.46 In the related phosphido derivative [Cp′Fe(μ-P)]2 the Fe–Fe distance is as expected longer at 2.5005(4) Å, and to fulfil the 18VE rule the authors originally proposed a FeFe double bond.9 In contrast, for the iron alkylidyne derivative [Cp′Fe(μ-CPh)]2, which also features a short Fe–Fe distance of 2.3558(4) Å, DFT computations and NBO analyses indicate no direct Fe–Fe bonding interaction; and the benzylidyne ligands should instead be considered as deprotonated phenylcarbene fragments coordinated to two [Cp′Fe]+ fragments; thus, forming a low-spin iron(II) complex (Chart 2).47
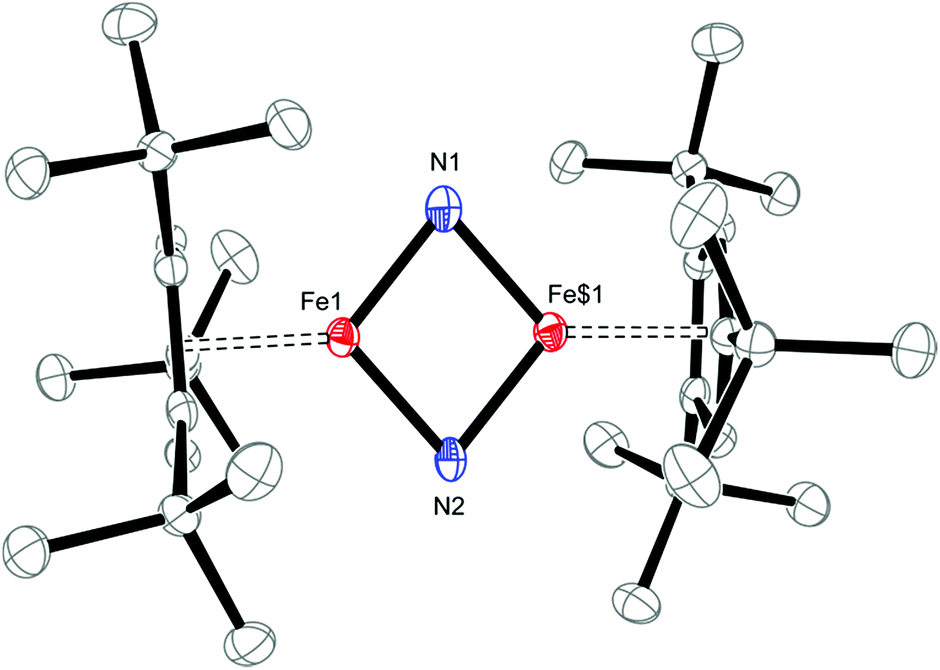 | ||
| Fig. 11 ORTEP of [Cp′Fe(μ-N)]2 (5) (50% probability ellipsoids). H-atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Cp′cent–Fe 1.744, Cp′plane–Fe 1.744, Fe1–N1 1.744(2), Fe1–N2 1.734(2), Fe1$1–N1 1.744(2), Fe1$1–N2 1.734, Fe1⋯Fe1$1 2.2325(7), N1⋯N2 2.667, N1–Fe1–N2 100.14(9), Fe1–N1–Fe1$1 79.59(13), Fe1–N2–Fe$1 80.13(14). Symmetry transformations used to generate equivalent atoms: $1 −y, −x, −z + 0.5. | ||
We will present a more detailed analysis of the bonding in 5, especially in comparison to [Cp′Fe(μ-P)]2 (ref. 9) and [Cp′Fe(μ-CPh)]2,47 in our computational details section. However, the Cp′cent–Fe distance of 1.74 Å in 5 compares well with the values found in [Cp′Fe(μ-P)]2 (1.73 Å)9 and [Cp′Fe(μ-CPh)]2 (1.74 Å);47 and it is also consistent with an S = 0 ground state. To further address the electronic structure, the 57Fe Mössbauer spectrum and solid-state magnetic susceptibility data for 5 were recorded (vide infra). The Mössbauer spectrum of 5 (at 77 K) features a doublet with an isomer shift of δiso = −0.02(1) mm s−1 and a quadrupole splitting ΔEQ = 0.71(1) mm s−1. Within the investigated series of [Cp′Fe(μ-X)]2 compounds this value represents the lowest isomer shift, which is consistent with a reduction of δiso with increasing oxidation state (Fig. 12).
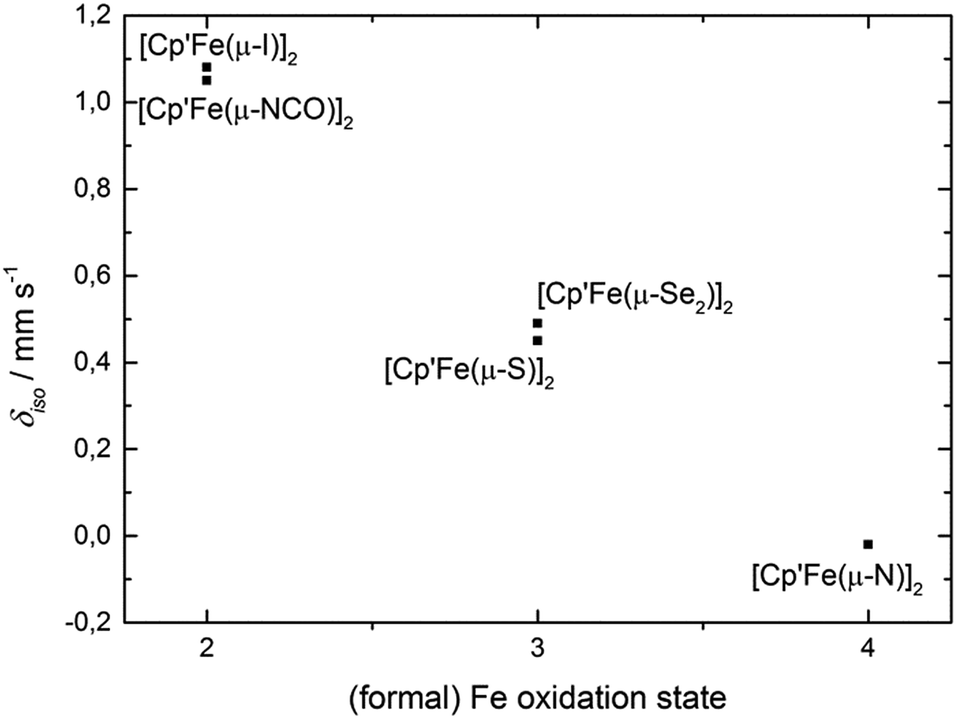 | ||
| Fig. 12 Correlation between the (formal) oxidation state of compounds [Cp′Fe(μ-X)]2 (1–5) and the Mössbauer isomer shift (δiso) at 77 K. | ||
The magnetic susceptibility data show that complex 5 behaves as a temperature-independent paramagnet (TIP) (see ESI† for details) indicating the presence of strong antiferromagnetic coupling between the Fe atoms, mediated either by the nitrido bridges (super-exchange) or an Fe–Fe bond (direct exchange). This is also consistent with the negligible temperature dependence of the 1H NMR chemical shifts.
Reactivity studies on [Cp′Fe(μ-N)]2 (5)
In a preliminary study, we further investigated the reactivity of 5 towards H2, CO, N-heterocyclic carbenes (NHCs) and isonitriles (XylNC, where Xyl = 2,6-Me2C6H3). Complex 5 exhibits no reactivity with H2 when a hexane solution of 5 is exposed to H2 (10 bar) at ambient temperature for one week, but on exposure to CO (5 bar) a rapid conversion occurs to yield [Cp′Fe(CO)2(NCO)] (7) (see Experimental section for details). We assume that in an initial reaction CO reacts with 5 to form the isocyanato-bridged intermediate [Cp′Fe(μ-NCO)]2 (2), which then rapidly reacts with an excess of CO to form 7. This led us also to probe the reactivity with isonitriles such as XylNC and indeed we isolated the carbodiimido complex [Cp′Fe(CNXyl)2(NCNXyl)] (8) (Scheme 4) in good yield. The molecular structure of 8 is shown in Fig. 13, and the selected bond distances and angles are provided in the figure caption. We also monitored this reaction upon addition of substoichiometric quantities of XylNC by 1H NMR spectroscopy. In C6D6 solution we detected starting material 5, product 8 and small quantities of a paramagnetic intermediate, which we assume to be [Cp′Fe(μ-NCNXyl)]2. Unfortunately, every attempt to isolate this intermediate failed, and we attribute this to a slow conversion of 5 to form the proposed [Cp′Fe(μ-NCNXyl)]2 intermediate, followed by a rapid reaction of this species with excess of XylNC.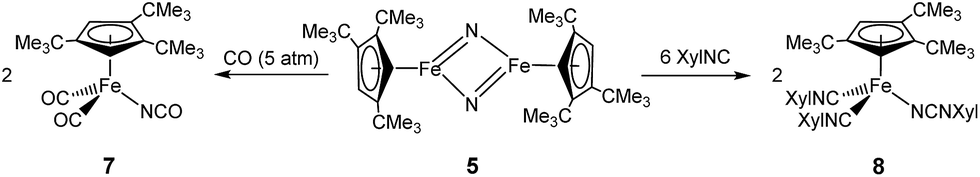 | ||
| Scheme 4 Reactivity of 5 with CO and XylNC. | ||
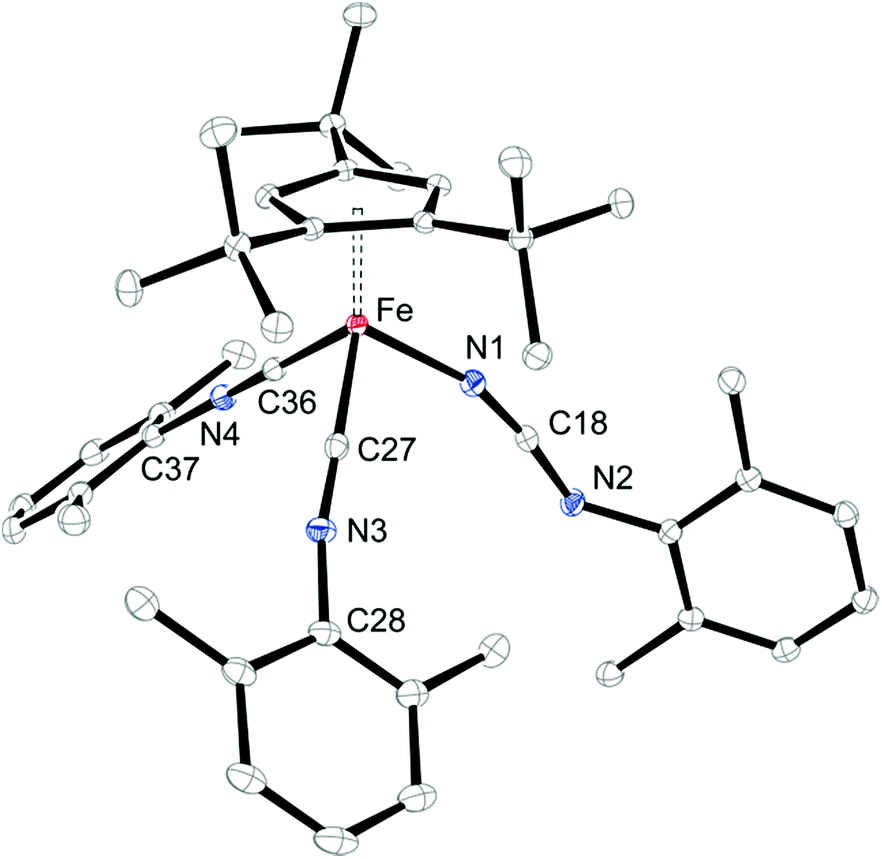 | ||
| Fig. 13 ORTEP of [Cp′Fe(CNXyl)2(NCNXyl)] (8) (30% probability ellipsoids). H-atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Cp′cent–Fe 1.73, Fe–C27 1.8221(13), Fe–C36 1.8265(13), Fe–N1 1.9555(11), N1–C18 1.1640(18), N2–C18 1.2861(17), N2–C19 1.3895(16), N3–C27 1.1769(18), N3–C28 1.3974(17), N4–C36 1.1716(17), N4–C37 1.3964(16), C27–N3–C28 168.29(13), C36–N4–C37 170.48(14), N1–C18–N2 170.44(14). | ||
We then also screened the reactivity of 5 with a series of N-heterocyclic carbenes (NHCs), but only unreacted starting material was recovered. Presumably the sterically hindered nitrido functionalities in 5 are not accessible to bulky substrates.
Computational studies
Computational investigations have become increasingly popular for experimental chemists to rationalize their findings.48 Nevertheless, it is always imperative to calibrate the computational results to experimental data in order to demonstrate the adequacy of the chosen computational method. At the beginning the structure of complex 1 was computed at the B3PW91 level of theory. Gratifyingly, the optimized structure reproduces the experimental data reasonably well (Table 1).| Geometrical data (Å) | Exp | Computed |
|---|---|---|
| Cp(centroid)–Fe | 1.93 | 1.94 |
| Fe1–I1 | 2.6748(5) | 2.79 |
| Fe1–I2 | 2.7088(5) | 2.75 |
| Fe2–I1 | 2.7120(5) | 2.75 |
| Fe2–I2 | 2.6780(5) | 2.79 |
| Fe1⋯Fe2 | 3.5263(8) | 3.83 |
For example, the computed and experimental Cp(cent)–Fe(ave) bond distance is in good agreement, while the computed Fe–I bond distances are elongated by 0.04 - 0.12 Å, which is also reflected in a significant elongation of the computed Fe1 Fe2 (3.83 Å, exp.: 3.5263(8) Å) distance. In a next step, the molecular structures and relative energies of the different spin states of [Cp′Fe(μ-NCO)]2 (2) and of those of the proposed intermediates [Cp′Fe(μ-SCN)]2 and [Cp′Fe(μ-N3)]2 were computed (see ESI† for details). For 2 the computed geometry also compares well with the experimentally determined geometry. In addition, for the cyanato- and isothiocyanato-bridged dimers, the ambiphilic binding modes of these pseudohalides (O vs. N and S vs. N coordination) were also explored and in both cases, the coordination via the N-atom is energetically more favourable (see ESI† for details). While this is indeed consistent with the experiment for compound 2, our computations predict a more complicated situation for the formation of 3, in which μ-κ2-S,N and μ-κ-N coordinated intermediates are suggested to be in equilibrium with each other and may therefore be involved in the formal elimination of CN˙ radicals (see ESI† for details). However, this will require more detailed experimental and computational studies, which are currently ongoing and will be reported in the future.
We then turned our attention to the bonding situation in [Cp′Fe(μ-S)]2 (3) and [Cp′Fe(μ-N)]2 (5). For both molecules a series of different spin states was originally considered using the B3PW91 functional (see ESI† for details). For complex 3 four different spin states were found to be close in energy (i.e., within 0.3 eV) (Fig. 14).
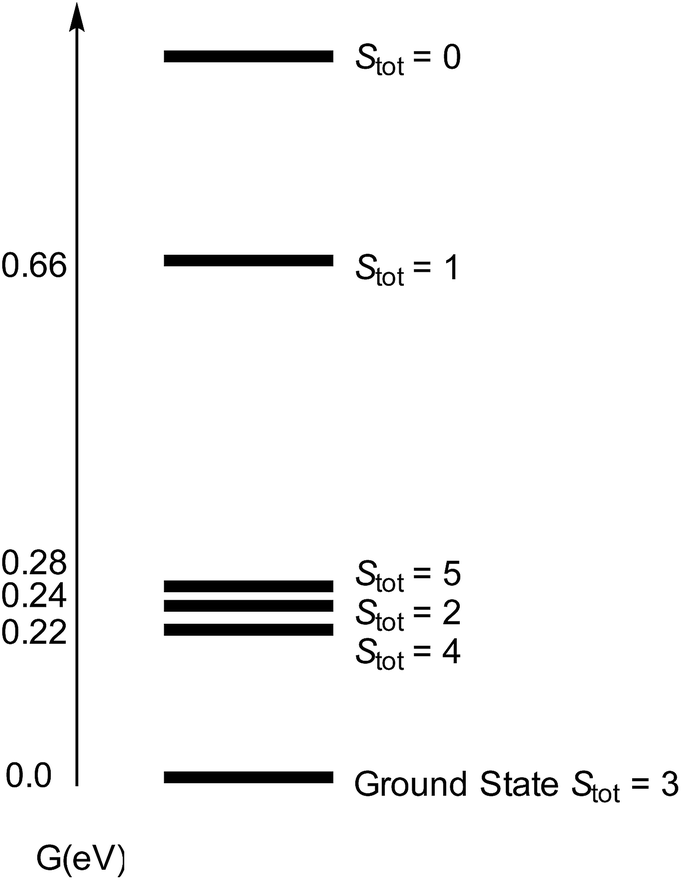 | ||
| Fig. 14 Computed spectrum of electronic spin states (at the B3PW91 level of theory) for [Cp′FeS]2 (3) with Stot = SFe,1 + SFe,2. | ||
However, the ground state configuration of Stot = 3 (computed at the B3PW91 level of theory) is clearly inconsistent with the experimentally observed TIP and indicates unresolved shortcomings of the DFT description of these open shell systems. Therefore, a more sophisticated approach at a higher level of theory is required (vide supra). A similar situation arises when the bonding situation in the iron nitrido compound [Cp′Fe(μ-N)]2 (5) is analysed using DFT methods. The computed molecular structure matches the experimental structure (see ESI† for details), and the potential spin isomers were also evaluated. During these investigations, we found that the quintet and singlet spin states are almost degenerate in energy (their Gibbs free energies (ΔG0) are equal within 0.07 eV). As shown for complex 3, DFT methods can be problematic when applied to open shell systems, especially when different spin states are very close in energy. Complexes 3 and 5 possess d-electron configurations of 3d4.58 and 3d4.33, respectively, which are associated with their formal oxidation states of Fe(III) (3d5) and Fe(IV) (3d4). In 3, the NBO analysis indicates strongly polarized Fe–S bonds with 70% S and 30% Fe composition. Furthermore, at the second order donor–acceptor NBO, another strong donation (73 kcal mol−1) from a π lone pair on sulphur to an empty d orbital on iron is found. The NBO analysis of 5 also suggests strongly polarized Fe–N bonds; a first order analysis reveals four Fe–N single bonds (with 40% Fe–60% N contributions). This strong polarization of the Fe–N bonds is also reflected in their small Wiberg bond indexes of 0.23 and 0.31, respectively. At the second order, a small donation from the nitrogen lone pair to Fe (20 kcal mol−1) is found, in line with the Lewis structure B illustrated in Chart 3.
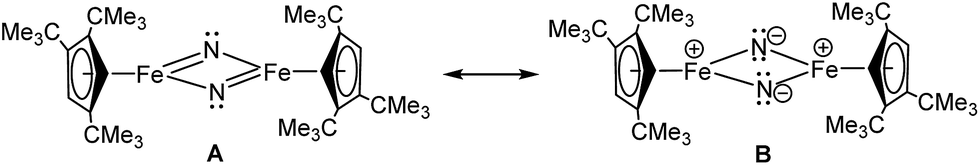 | ||
| Chart 3 Two resonance structures of 5. | ||
To investigate the electronic structure of these compounds in more detail preliminary CASSCF computations were undertaken. For complex 3, six electrons were distributed in 6 orbitals (mainly three 3d orbitals per iron atom that appears as linear combinations between the two iron atoms). Calculations were conducted for the singlet, triplet and quintet spin states. The lowest spin-state is a singlet with a triplet higher in energy (0.14 eV) and a quintet slightly higher (0.28 eV). In the singlet CASSCF wavefunction, two orbitals are doubly occupied whereas two others appear to be singly occupied, yielding an open-shell singlet as ground state. Therefore, in the ground state of 3, the two Fe(III) centres (SFe = 1/2) couple antiferromagnetically, which is consistent with the experiment observation. Applying a similar approach for complex 5, four electrons were distributed into 6 orbitals and singlet, triplet and quintet spin states were computed at the CASSCF level. Alike complex 3, the lowest spin state is found to be a singlet with a quintet higher in energy (0.12 eV) and finally the triplet (0.54 eV). Analysing the singlet wavefunction deeply, it appears that the 4 electrons are populating four different orbitals, leading to an open-shell singlet with an SFe = 1 electron configuration per Fe atom (Ms = ±1). In other words, two Fe(IV, d4) metal atoms with two unpaired electrons on each iron atom are coupled antiferromagnetically with each other (similarly to situation described for 3). At this more sophisticated level of theory it was indeed possible to obtain computational results that match the experimental findings.
The polarized nature of the Fe–N bond (Chart 3, Lewis structure B) is also reflected in the frontier orbitals of 5, in which the nitrogen lone pairs contribute substantially to the highest molecular orbital (HOMO) (Fig. 15). Consistent with [Cp′Fe(μ-CPh)]2,47 a closer inspection of the NBO orbitals confirmed the absence of any (direct) Fe–Fe bonding interactions in complex 5.
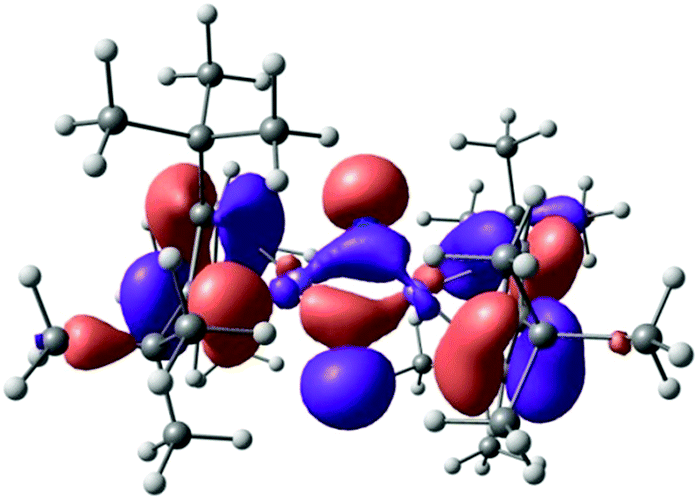 | ||
| Fig. 15 Highest molecular orbital (HOMO) of complex 5. | ||
As we have demonstrated above, the commonly employed DFT methodology can describe the geometries and the overall structural features reasonably well, but fails when the electronic structure of these compounds need to be considered in more detail. However, CASSCF computations on these large dinuclear complexes to provide insights in reaction pathways is too time consuming and therefore not feasible. We therefore decided to probe if the DFT methodology is actually sufficient to adequately describe the observed reactivity patterns of these compounds focusing on the formation of complex 5 and its reaction towards CO was probed computationally (Fig. 16 and 17). Starting from the hypothetical [Cp′Fe(μ-N3)]2 intermediate, DFT methods predict that the initial step involves the cleavage of the N–N bond in one of the azido ligands yielding a transient azido-nitrido species (Fig. 16). The latter undergoes N2 release yielding the product 5 followed by a change of spin state. This reaction pathway was computed at the B3PW91 level of theory with and without the inclusion of dispersion (D3BJ) and solvent corrections (THF). Dispersion and solvent corrections have only a minor influence on the structure of the intermediates and transition states involved, but they dramatically reduce the barriers of the overall reaction and also significantly stabilize the final product 5 (Fig. 17) The overall computed barrier for the conversion of the proposed intermediate [Cp′Fe(μ-N3)]2 (B) to the dinuclear iron nitrido dimer is ΔH‡ = 15.7 kcal mol−1, which agrees with the experimentally observed facile N2 elimination at ambient temperature.
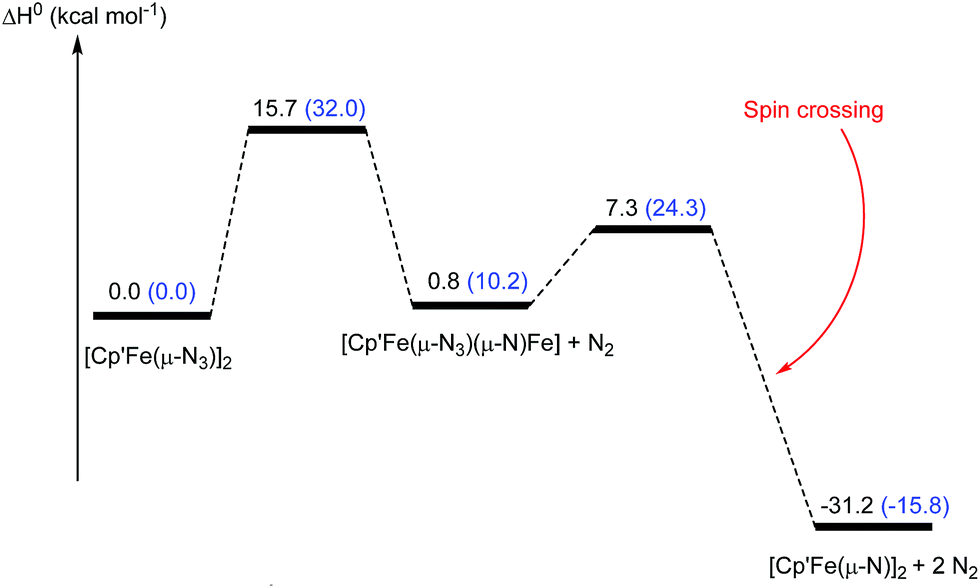 | ||
| Fig. 16 Enthalpy (ΔH0) profile for the formation of [Cp′FeN]2 (5) from the proposed intermediate [Cp′Fe(μ-N3)]2 (B). Values given in parenthesis refer to the values computed at the B3PW91 level of theory without consideration of solvent and dispersion corrections. | ||
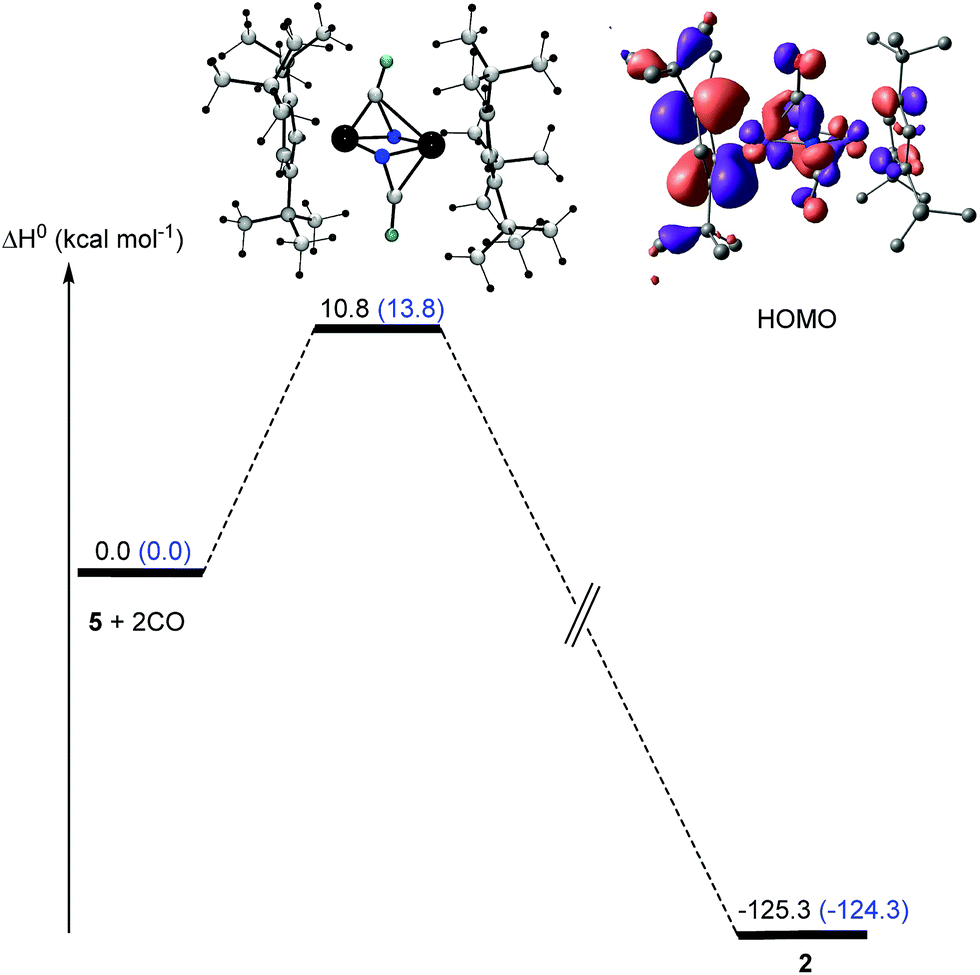 | ||
| Fig. 17 Enthalpy (ΔH0) profile for the formation of [Cp′Fe(μ-NCO)]2 (2) starting [Cp′Fe(μ-N)]2 (5) and CO. Values given in parenthesis refer to the values computed at the B3PW91 level of theory without consideration of solvent and dispersion corrections. | ||
In a next step, the reaction pathway for the reaction of complex 5 with CO was probed. Consistent with the computed HOMO for complex 5 (Fig. 14) the two CO molecules do not attack directly at the lone pair of the N-atoms; instead they approach 5 perpendicularly to the Fe2N2 plane (Fig. 17), which is associated with a low barrier of activation (ΔH‡) of 10.8 kcal mol−1 after consideration of solvent and dispersion corrections. In the transition state electron density is transferred from 5 into the π* orbitals of CO (Fig. 17).
Overall, while the precise electronic structure of compounds 3 and 5 are not adequately described to account for their magnetic properties by standard DFT methods, their molecular structures and the formation and reactivity of 5 can be modeled in sufficient detail yielding reasonable reaction barriers which agree with our experimental observations.
Conclusions
In summary, [Cp′FeI]2 (1) is a valuable starting material for the activation of pseudohalides such as SCN−, SeCN− and N3− yielding sulfido (3), diselenido (4) and nitrido (5) complexes. These compounds were characterized by various spectroscopic techniques including X-ray diffraction, solid-state magnetic susceptibility studies and zero-field 57Fe Mössbauer spectroscopy. Phenomenologically there is a clear correlation between the increasing formal oxidation state in complexes 1 to 5 (ranging from Fe(II) to Fe(IV)) and the decreasing isomer shift observed in their respective Mössbauer spectra. While standard DFT methods reproduce the molecular structures of these complexes reasonably well, their electronic structures require more sophisticated methods to reproduce the data inferred from magnetic susceptibility data of 3 and 5. The iron(IV) nitrido complex 5 shows no reaction towards H2 (even at elevated pressures), but in the presence of CO it converts to the iron(II) isocyanate complex 6. This represents a rare example of successful CO functionalization of a bridging μ-N fragment. In addition, some useful insights into the formation of 5 and its reactivity with CO can be obtained from DFT computations which justify its use to describe the reaction chemistry of these species. Further investigations on the electronic structure of the compounds 3 and 5 using X-ray absorption near edge studies (XANES) and on their intrinsic reactivity towards electrophiles or organic radicals are ongoing and will be reported in due course.Experimental details
General considerations
All reactions and product manipulations were carried out under an atmosphere of dry, oxygen free argon or dinitrogen using standard high-vacuum, Schlenk, or drybox techniques. Argon or dinitrogen were purified by passage through BASF R3-11 catalyst (Chemalog) and 4 Å molecular sieves. Dry, oxygen-free solvents were employed throughout. NMR spectra were recorded on Bruker DRX 500 MHz, a Bruker DRX 400 MHz, or a Bruker 400 MHz AVANCE spectrometer. All chemical shifts are reported in δ units and referenced to the residual protons of the deuterated solvents, which are internal standards, for proton chemical shifts. The elemental analyses were performed by the analytical facilities at the University of California at Berkeley, Robertson Microlit Laboratories of Madison, NJ, or at the TU Braunschweig.Magnetic susceptibility measurements were conducted in a 7 T Quantum Design MPMS magnetometer utilizing a superconducting quantum interference device (SQUID). The samples (10–25 mg) were transferred into a quartz tube and held in place with quartz wool (ca. 5 mg). The quartz tube was flame sealed and transferred into the magnetometer. This method provided a very small and reliable container correction, typically of ca. −2 × 10−5 emu mol−1. The data were also corrected for the overall diamagnetism of the molecule using Pascal constants.49 For a more detailed description see ref. 50. The program package JulX written by Eckhard Bill for exchange coupled systems was used.11
Materials
All solvents were deoxygenated and dried by passage over columns of activated alumina.51 Tetrahydrofuran dried over sodium/benzophenone and freshly distilled prior to use. Deuterated solvents, C6D6, C7D8 and THF-d8 were purchased from Cambridge Laboratories, Inc. or Eurisotope, refluxed for 3 days over sodium metal, vacuum transferred to a Teflon sealable Schlenk flask containing 4 Å molecular sieves, and degassed via three freeze–pump–thaw cycles. [Cp′Fe(μ-I)]2 (1) was prepared as previously reported.5 All other chemicals were purchased from Acros Organics or Sigma Aldrich. KNCO, KSCN, KSeCN and NaN3 were dried overnight under dynamic vacuum at 90 °C.Synthesis
Method A. To a stirred solution of [Cp′Fe(μ-NCO)]2 (2, 100 mg, 0.151 mmol, 1 eq.) in hexane (10 mL), CO (5 bar) was added and the solution stirred for 12 h at ambient temperature. The colour of the solution changed immediately from dark green to red-orange. After removal of the solvent under oil-pump vacuum the red residue was dissolved in a small amount of Et2O (1 mL), filtered and stored at −30 °C to form red crystals. Yield: 104 mg (0.269 mmol, 89%). Mp: 126–127 °C. 1H NMR (300 MHz, C6D6, 300 K): δ = 4.61 (s, 2H, Cp-CH), 1.08 (s, 18H, tBu-H), 1.04 (s, 9H, tBu-H) ppm. 13C{1H} NMR (75 MHz, C6D6, 300 K): δ = 214.7 (CO), 110.0 (C-Cp), 108.0 (C-Cp), 89.2 (CH-Cp), 33.0 (C(CH3)3), 32.5 (C(CH3)3, 31.1 (C(CH3)3), 30.4 (C(CH3)3) ppm. IR (KBr, cm−1): 2228(NCO), 2023(CO), 1978(CO). Elemental analysis calcd (%) for C20H29FeNO3: C 62.02, H 7.55, N 3.62; found: C 61.84, H 7.59, N 3.49.
Method B. To a stirred hexane solution (10 mL) of [Cp′Fe(μ-N)]2 (5, 100 mg, 0.165 mmol, 1 eq.) CO (5 bar) was added and the solution stirred for 12 h at ambient temperature. The colour of the solution changed immediately from dark green to red-orange. The solvent was removed under dynamic vacuum and the red residue was dissolved in a small amount of Et2O (1 mL), filtered and stored at −30 °C to form red crystals. Yield: 116 mg (0.300 mmol, 91%).
Mössbauer spectroscopy
57Fe Mößbauer spectra were recorded on a WissEl Mößbauer spectrometer (MRG-500) at 77 K in constant acceleration mode. 57Co/Rh was used as the radiation source. WinNormos for Igor Pro software has been used for the quantitative evaluation of the spectral parameters (least-squares fitting to Lorentzian peaks). The minimum experimental line widths were 0.20 mm s−1. The temperature of the samples was controlled by an MBBC-HE0106 MÖSSBAUER He/N2 cryostat within an accuracy of ±0.3 K. Isomer shifts were determined relative to α-iron at 298 K.Crystallographic details
X-ray diffraction studies on complex 5 were conducted on a Bruker-AXS SMART APEX-II diffractometer. A suitable crystal was selected and mounted using paratone oil on a MiteGen mylar tip. For all other structures, crystals were mounted on glass fibres in inert oil; data were collected on Oxford Diffraction systems using mirror-focused Cu Kα or monochromatized Mo Kα radiation. Structures were refined anisotropically on F2 using SHELXL-97. Hydrogen atoms were included using rigid methyl groups or a riding model. Special features: For 3, two t-butyl groups were disordered over two positions. Crystal and data collection parameters are presented in Table S1 in the ESI.†Computational details section
All the quantum-chemical calculations were conducted using the Gaussian 09 program suite.52 As functional we used the Becke's 3-parameter hybrid, combined with the non-local correlation functional provided by Perdew/Wang, denoted as B3PW91.53 For Fe and I, the relativistic energy-consistent pseudopotential of the Stuttgart-Köln ECP library was used in combination with its adapted segmented basis.54 For all other atoms, a standard 6-31G** basis set was used.55 All stationary points were identified as minima (number of imaginary frequencies Nimag = 0) or transition states (Nimag = 1).Acknowledgements
We thank the Alexander von Humboldt Foundation for a Feodor-Lynen Fellowship (M. D. W.) and Prof. Maurice Brookhart for providing financial support (through NSF Grant CHE-0615704) and laboratory facilities (M. D. W.) during the initial phase of this research program. Deutsche Forschungsgemeinschaft (DFG) is acknowledged for generous funding by the Emmy Noether and Heisenberg program (WA 2513/2 and WA 2513/6, respectively). LM is grateful to the Humboldt Foundation for a grant of experienced researcher and the Chinese Academy of Science. CalMip is also gratefully acknowledged for a generous computational grant.Notes and references
- (a) C. Ercolani, M. Gardini, G. Pennesi, G. Rossi and U. Russo, Inorg. Chem., 1988, 27, 422–424 CrossRef CAS; (b) S. D. Brown and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 1913–1923 CrossRef CAS PubMed; (c) P. J. Chirik, Angew. Chem., Int. Ed., 2006, 45, 6956–6959 CrossRef CAS PubMed; (d) J. J. Scepaniak, M. D. Fulton, R. P. Bontchev, E. N. Duesler, M. L. Kirk and J. M. Smith, J. Am. Chem. Soc., 2008, 130, 10515–10517 CrossRef CAS PubMed; (e) J. J. Scepaniak, J. A. Young, R. P. Bontchev and J. M. Smith, Angew. Chem., Int. Ed., 2009, 48, 3158–3160 CrossRef CAS PubMed; (f) Y. Nishibayashi, Nat. Chem., 2011, 3, 502–504 CrossRef CAS PubMed; (g) J. J. Scepaniak, C. S. Vogel, M. M. Khusniyarov, F. W. Heinemann, K. Meyer and J. M. Smith, Science, 2011, 331, 1049–1052 CrossRef CAS PubMed; (h) M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780–783 CrossRef CAS PubMed; (i) J. Hohenberger, K. Ray and K. Meyer, Nat. Commun., 2012, 3, 720 CrossRef PubMed; (j) T. M. Powers and T. A. Betley, J. Am. Chem. Soc., 2013, 135, 12289–12296 CrossRef CAS PubMed; (k) J. Torres-Alacan, U. Das, A. C. Filippou and P. Vohringer, Angew. Chem., Int. Ed. Engl., 2013, 52, 12833–12837 CrossRef CAS PubMed; (l) K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559–565 CrossRef CAS PubMed; (m) J. M. Smith, Prog. Inorg. Chem., 2014, 58, 417–471 CrossRef CAS; (n) Y. Lee, F. T. Sloane, G. Blondin, K. A. Abboud, R. García-Serres and L. J. Murray, Angew. Chem., Int. Ed., 2015, 54, 1499–1503 CrossRef CAS PubMed; (o) M. D. Walter, Adv. Organomet. Chem., 2016, 65, 261–377 CrossRef.
- (a) T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2004, 126, 6252–6254 CrossRef CAS PubMed; (b) M. P. Hendrich, W. Gunderson, R. K. Behan, M. T. Green, M. P. Mehn, T. A. Betley, C. C. Lu and J. C. Peters, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17107–17112 CrossRef CAS PubMed.
- (a) J. F. Berry, E. Bill, E. Bothe, S. D. George, B. Mienert, F. Neese and K. Wieghardt, Science, 2006, 312, 1937–1941 CrossRef CAS PubMed; (b) M. P. Mehn and J. C. Peters, J. Inorg. Biochem., 2006, 100, 634–643 CrossRef CAS PubMed; (c) C. Vogel, F. W. Heinemann, J. Sutter, C. Anthon and K. Meyer, Angew. Chem., Int. Ed., 2008, 47, 2681–2684 CrossRef CAS PubMed.
- T. M. Powers, A. R. Fout, S.-L. Zheng and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 3336–3338 CrossRef CAS PubMed.
- M. D. Walter and P. S. White, New J. Chem., 2011, 35, 1842–1854 RSC.
- M. Maekawa, C. G. Daniliuc, P. G. Jones, J. Hohenberger, J. Sutter, K. Meyer and M. D. Walter, Eur. J. Inorg. Chem., 2013, 2013, 4097–4104 CrossRef CAS.
- M. D. Walter, J. Grunenberg and P. S. White, Chem. Sci., 2011, 2, 2120–2130 RSC.
- M. D. Walter and P. S. White, Inorg. Chem., 2012, 51, 11860–11872 CrossRef CAS PubMed.
- C. Eichhorn, O. J. Scherer, T. Sögding and G. Wolmershäuser, Angew. Chem., Int. Ed., 2001, 40, 2859–2861 CrossRef CAS PubMed.
- M. Kreye, D. Baabe, P. Schweyen, M. Freytag, C. G. Daniliuc, P. G. Jones and M. D. Walter, Organometallics, 2013, 32, 5887–5898 CrossRef CAS.
- The program package JulX was used for spin-Hamiltonian simulations and fittings of the data by a full-matrix diagonalization approach, ed. E. Bill, Max-Planck Institute for Chemical Energy Conversion, unpublished results Search PubMed.
- (a) P. Gütlich and J. Ensling, in Inorganic Electronic Structure and Spectroscopy: Methodology, ed. E. I. Solomon and A. B. P. Lever, J. Wiley & Sons, New York, 1999, vol. 1, pp. 161–213 Search PubMed; (b) P. Gütlich, E. Bill and A. X. Trautwein, Mössbauer Spectroscopy and Transition Metal Chemistry, Springer, Heidelberg, 2011 Search PubMed.
- (a) M. L. Good, J. Buttone and D. Foyt, Ann. N. Y. Acad. Sci., 1974, 239, 193–207 CrossRef CAS; (b) J. P. Mariot, F. Varret, P. Michaud, D. Astruc, J. R. Hamon, G. Althoff and P. Batail, J. Phys., Colloq., 1980, 319–320 CAS; (c) M. Reiners, D. Baabe, K. Harms, M. Maekawa, C. G. Daniliuc, M. Freytag, P. G. Jones and M. D. Walter, Inorg. Chem. Front., 2016, 3, 250–262 RSC; (d) M. Malischewski, M. Adelhardt, J. Sutter, K. Meyer and K. Seppelt, Science, 2016, 353, 678–682 CrossRef CAS PubMed.
- M. Maekawa, C. G. Daniliuc, M. Freytag, P. G. Jones and M. D. Walter, Dalton Trans., 2012, 41, 10317–10327 RSC.
- (a) J. S. Thayer and R. West, Adv. Organomet. Chem., 1967, 5, 169–224 CrossRef CAS; (b) R. A. Bailey, S. L. Kozak, T. W. Michelsen and W. N. Mills, Coord. Chem. Rev., 1971, 6, 407–445 CrossRef CAS; (c) Z. Dori and R. F. Ziolo, Chem. Rev., 1973, 73, 247–254 CrossRef CAS; (d) A. M. Golub, H. Köhler and V. V. Skopenko, Chemistry of pseudohalides, Elsevier, Amsterdam, 1986 Search PubMed.
- G. Ferguson, B. R. Lloyd, L. Manojlovic-Muir, K. W. Muir and R. J. Puddephatt, Inorg. Chem., 1986, 25, 4190–4197 CrossRef CAS.
- M. E. Rasche and L. C. Seefeldt, Biochemistry, 1997, 36, 8574–8585 CrossRef CAS PubMed.
- A. F. Hollemann, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, de Gruyter, Berlin, 102 edn, 2007 Search PubMed.
- R. A. Rose, S. J. Greaves, F. Abou-Chahine, D. R. Glowacki, T. A. A. Oliver, M. N. R. Ashfold, I. P. Clark, G. M. Greetham, M. Towrie and A. J. Orr-Ewing, Phys. Chem. Chem. Phys., 2012, 14, 10424–10437 RSC.
- (a) P. Nicpon and D. W. Meek, Inorg. Chem., 1966, 5, 1297–1298 CrossRef CAS; (b) W. J. Stec, G. Zon, W. Egan and B. Stec, J. Am. Chem. Soc., 1984, 106, 6077–6079 CrossRef CAS; (c) A. R. Butler, C. Glidewell and A. E. Waddon, Polyhedron, 1989, 8, 2627–2629 CrossRef CAS.
- H. Beinert, R. H. Holm and E. Münck, Science, 1997, 277, 653–659 CrossRef CAS PubMed.
- P. Venkateswara Rao and R. H. Holm, Chem. Rev., 2004, 104, 527–560 CrossRef CAS PubMed.
- (a) R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS PubMed; (b) E. I. Solomon, X. Xie and A. Dey, Chem. Soc. Rev., 2008, 37, 623–638 RSC.
- W. Yao, P. Gurubasavaraj and P. Holland, Struct. Bonding, 2014, 160, 1–37 CrossRef CAS.
- (a) D. Coucouvanis, A. Salifoglou, M. G. Kanatzidis, A. Simopoulos and V. Papaefthymiou, J. Am. Chem. Soc., 1984, 106, 6081–6082 CrossRef CAS; (b) J. Ballmann, A. Albers, S. Demeshko, S. Dechert, E. Bill, E. Bothe, U. Ryde and F. Meyer, Angew. Chem., Int. Ed., 2008, 47, 9537–9541 CrossRef CAS PubMed; (c) A. Albers, S. Demeshko, S. Dechert, E. Bill, E. Bothe and F. Meyer, Angew. Chem., Int. Ed., 2011, 50, 9191–9194 CrossRef CAS PubMed; (d) Y. Li, Y. Li, B. Wang, Y. Luo, D. Yang, P. Tong, J. Zhao, L. Luo, Y. Zhou, S. Chen, F. Cheng and J. Qu, Nat. Chem., 2013, 5, 320–326 CrossRef CAS PubMed; (e) A. Albers, T. Bayer, S. Demeshko, S. Dechert and F. Meyer, Chem.–Eur. J., 2013, 19, 10101–10106 CrossRef CAS PubMed; (f) A. Albers, S. Demeshko, S. Dechert, C. T. Saouma, J. M. Mayer and F. Meyer, J. Am. Chem. Soc., 2014, 136, 3946–3954 CrossRef CAS PubMed.
- (a) J. Wachter, Angew. Chem., Int. Ed. Engl., 1989, 28, 1613–1626 CrossRef; (b) J. Wachter, J. Coord. Chem., 1987, 15, 219–236 CrossRef CAS.
- (a) P. J. Vergamini and G. J. Kubas, Prog. Inorg. Chem., 1976, 261–282 CrossRef CAS; (b) G. J. Kubas and P. J. Vergamini, Inorg. Chem., 1981, 20, 2667–2676 CrossRef CAS.
- H. Brunner, N. Janietz, W. Meier, G. Sergeson, J. Wachter, T. Zahn and M. L. Ziegler, Angew. Chem., Int. Ed. Engl., 1985, 24, 1060–1061 CrossRef.
- M. Yamada, H. Tobita, S. Inomata and H. Ogino, Bull. Chem. Soc. Jpn., 1996, 69, 861–867 CrossRef CAS.
- H. Ogino, H. Tobita, K. Yanagisawa, M. Shimoi and C. Kabuto, J. Am. Chem. Soc., 1987, 109, 5847–5848 CrossRef CAS.
- S. Inomata, T. Hiruma and H. Ogino, Chem. Lett., 1998, 309–310 CrossRef CAS.
- (a) I. Bertini, H. B. Gray, E. I. Stiefel and J. S. Valentine, Biological Inorganic Chemistry, University Science Books, Sausalito, CA, 2007 Search PubMed; (b) M. E. Reesbeck, M. M. Rodriguez, W. W. Brennessel, B. Q. Mercado, D. Vinyard and P. L. Holland, J. Biol. Inorg Chem., 2015, 20, 875–883 CrossRef CAS PubMed.
- (a) M. A. Ansari and J. A. Ibers, Coord. Chem. Rev., 1990, 100, 223–266 CrossRef CAS; (b) C. Simonnet-Jégat and F. Sécheresse, Chem. Rev., 2001, 101, 2601–2612 CrossRef; (c) M. N. Sokolov, V. P. Fedin and A. G. Sykes, in Comprehensive Coordination Chemistry II, ed. T. J. Meyer, Pergamon, Oxford, 2003, pp. 761–823 Search PubMed; (d) S. M. Dibrov, B. Deng, D. E. Ellis and J. A. Ibers, Inorg. Chem., 2005, 44, 3441–3448 CrossRef CAS PubMed.
- (a) C. M. Bolinger, T. B. Rauchfuss and A. L. Rheingold, Organometallics, 1982, 1, 1551–1553 CrossRef CAS; (b) A. L. Rheingold, C. M. Bolinger and T. B. Rauchfuss, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1878–1880 CrossRef.
- NMR of Paramagnetic Molecules, ed. G. N. La Mar, W. D. Horrocks and R. H.Holm, Academic Press, New York, 1973 Search PubMed.
- M. E. Smith and R. A. Andersen, J. Am. Chem. Soc., 1996, 118, 11119–11128 CrossRef CAS.
- B. Askevold, M. M. Khusniyarov, E. Herdtweck, K. Meyer and S. Schneider, Angew. Chem., Int. Ed. Engl., 2010, 49, 7566–7569 CrossRef CAS PubMed.
- (a) A. L. Odom and C. C. Cummins, Organometallics, 1996, 15, 898–900 CrossRef CAS; (b) G. K. B. Clentsmith, V. M. E. Bates, P. B. Hitchcock and F. G. N. Cloke, J. Am. Chem. Soc., 1999, 121, 10444–10445 CrossRef CAS; (c) Z. J. Tonzetich, R. R. Schrock, K. M. Wampler, B. C. Bailey, C. C. Cummins and P. Müller, Inorg. Chem., 2008, 47, 1560–1567 CrossRef CAS PubMed.
- (a) C. Kim, Y. Dong and L. Que Jr, J. Am. Chem. Soc., 1997, 119, 3635–3636 CrossRef CAS; (b) L. Que Jr, Pure Appl. Chem., 1998, 70, 947–954 Search PubMed; (c) A. J. Skulan, M. A. Hanson, H.-f. Hsu, L. Que Jr and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 7344–7356 CrossRef CAS PubMed.
- T. S. Haddad, A. Aistars, J. W. Ziller and N. M. Doherty, Organometallics, 1993, 12, 2420–2422 CrossRef CAS.
- M. Herberhold, A.-M. Dietel, A. Goller and W. Millius, Z. Anorg. Allg. Chem., 2003, 629, 871–879 CrossRef CAS.
- F. Akagi, T. Matsuo and H. Kawaguchi, Angew. Chem., Int. Ed., 2007, 46, 8778–8781 CrossRef CAS PubMed.
- M. Hirotsu, P. P. Fontaine, A. Epshteyn and L. R. Sita, J. Am. Chem. Soc., 2007, 129, 9284–9285 CrossRef CAS PubMed.
- K. Searles, P. J. Carroll, C.-H. Chen, M. Pink and D. J. Mindiola, Chem. Commun., 2015, 51, 3526–3528 RSC.
- (a) M. M. Olmstead, P. P. Power and S. C. Shoner, Inorg. Chem., 1991, 30, 2547–2551 CrossRef CAS; (b) S. K. Dutta, U. Beckmann, E. Bill, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2000, 39, 3355–3364 CrossRef CAS PubMed; (c) R. E. Cowley, N. J. DeYonker, N. A. Eckert, T. R. Cundari, S. DeBeer, E. Bill, X. Ottenwaelder, C. Flaschenriem and P. L. Holland, Inorg. Chem., 2010, 49, 6172–6187 CrossRef CAS PubMed; (d) R. E. Cowley and P. L. Holland, Inorg. Chem., 2012, 51, 8352–8361 CrossRef CAS PubMed; (e) A. K. Verma, T. N. Nazif, C. Achim and S. C. Lee, J. Am. Chem. Soc., 2000, 122, 11013–11014 CrossRef CAS; (f) S. C. Bart, E. Lobkovsky, E. Bill and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 5302–5303 CrossRef CAS PubMed; (g) E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917–4923 CrossRef CAS PubMed.
- F. A. Cotton, J. D. Jamerson and B. R. Stults, J. Organomet. Chem., 1975, 94, C53–C55 CrossRef CAS.
- G. Y. Vollmer, M. W. Wallasch, D. Saurenz, T. R. Eger, H. Bauer, G. Wolmershäuser, M. H. Prosenc and H. Sitzmann, Organometallics, 2015, 34, 644–652 CrossRef CAS.
- E. R. Davidson, Chem. Rev., 2000, 100, 351–352 CrossRef CAS PubMed , Introduction to a special issue Computational Transition Metal Chemistry.
- C. J. O'Connor, in Progress in Inorganic Chemistry, ed. S. J. Lippard, J. Wiley & Sons, New York, 1982, vol. 29, pp. 203–285 Search PubMed.
- M. D. Walter, M. Schultz and R. A. Andersen, New J. Chem., 2006, 30, 238–246 RSC.
- (a) P. J. Alaimo, D. W. Peters, J. Arnold and R. G. Bergman, J. Chem. Educ., 2001, 78, 64 CrossRef CAS; (b) A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, E. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallington CT, 2009.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS; (b) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic details, additional experimental and structural details, VT 1H NMR studies, magnetic susceptibility studies, 57Fe Mössbauer spectrum of 5, computational details. CCDC 1531255–1531264. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00570a |
| This journal is © The Royal Society of Chemistry 2017 |