
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencep-Doping of graphene in hybrid materials with 3,10-diazapicenium dications†
Alexandra
Roth‡
a,
Tobias A.
Schaub‡
b,
Ute
Meinhardt
b,
Dominik
Thiel
a,
Jan
Storch
c,
Vladimír
Církva
c,
Pavel
Jakubík
c,
Dirk M.
Guldi
*a and
Milan
Kivala
 *b
*b
aInstitute for Physical Chemistry I, Department Chemistry and Pharmacy, University of Erlangen-Nürnberg, Egerlandstrasse 3, D-91058 Erlangen, Germany. E-mail: dirk.guldi@fau.de
bInstitute for Organic Chemistry I, Department of Chemistry and Pharmacy, University of Erlangen-Nürnberg, Henkestrasse 42, D-91054 Erlangen, Germany. E-mail: milan.kivala@fau.de
cInstitute of Chemical Process Fundamentals of the Czech Academy of Sciences, Rozvojová 135/1, CZ-165 02 Prague 6, Czech Republic
First published on 24th February 2017
Abstract
N,N′-Didodecyl-substituted 3,10-diazapicenium salts featuring bromide and hexafluorophosphate counterions have been designed as novel dopants to realize individualized graphene sheets in a series of cutting edge experiments and to intrinsically stabilize them via p-doping. Importantly, electrochemical studies revealed two consecutive irreversible one-electron reductions of the N,N′-didodecyl-substituted 3,10-diazapicenium salts to yield the corresponding radical cation and neutral quinoidal species. Formation of both species was accompanied by characteristic changes in the absorption spectra. The 3,10-diazapicenium bromide was found to be a potent dopant to produce hybrid materials with exfoliated graphene. Microscopy based on AFM and TEM imaging and spectroscopy based on Raman probing corroborated that, upon drying, the hybrid material consists of few layer (5–8 layers) turbostratic graphene sheets that are p-doped. Our findings identify the newly synthesized N,N′-dialkylated 3,10-diazapicenium salts as highly promising candidates for the fabrication of functional graphene materials with tailored properties.
Introduction
Hybrid systems featuring π-conjugated molecular building blocks and exfoliated graphite hold enormous promises for various applications due to the possibility to modulate the electronic nature of the graphene flakes.1 Since its seminal realization, graphene and its production methods have been at the forefront of multidisciplinary studies aiming at its application in, for example, next generation organic electronic devices.2,3 In general, graphene is prepared by either physical4 or chemical processes,5 whereby its quantity and quality decisively impacts the development and performance of prospective applications.3 The liquid phase exfoliation of graphite represents a particularly appealing strategy as it opens up new avenues to construct unprecedented hybrid systems.6 Even though this approach still poses myriads of challenges, hybrid materials consisting of functionalized polycyclic aromatic scaffolds and (nano)graphene came to the forefront in recent years.7–9 Overall, the degree of individualization is controlled by the nature of the molecular exfoliant– the use of planar and rigid scaffolds including porphyrins,7,10 phthalocyanines,11 or perylene-3,4,9,10-tetracarboxylic dianhydrides12 amongst others turned out to be particularly effective for graphite exfoliation. Depending on the electron-deficient or -rich nature of the exfoliant, the liquid phase delamination of graphite represents a versatile methodology to produce p- or n-type doped (nano)graphene hybrids, respectively. The more so, as the careful chemical modification of the molecular exfoliant enables precise control over the degree of doping.13 Through this approach graphene flakes with tailored electronic properties are in reach and the scope of potential applications of graphene in nanoelectronics seems sheer endless.14To the best of our knowledge, apart from the previous report by Stoddart et al.,15 who employed 2,9-diazaperopyrenium di-cation A to directly exfoliate graphite in aqueous media (Fig. 1), positively charged polycyclic aromatic exfoliants have never been utilized. As a matter of fact, it has been documented that the comparatively small size of 2,7-diazapyrenium dication B is insufficient for an intercalation between graphite layers and, in turn, exfoliation of single graphene flakes from graphite.15 A careful consideration of structural prerequisites such as rigidity and planarity of an extended π-conjugation paired with inherent redox activity prompted us to design and realize N,N′-dialkylated 3,10-diazapicenium dication C. As such, dication C combines the features of an exfoliant, a stabilizer, and a p-dopant.
 | ||
| Fig. 1 Chemical structures of dications A and B studied previously by Stoddart et al.15 as exfoliating agents for graphite. Chemical structure of the 3,10-diazapicenium dication C examined in this work. Schematic illustration of a graphene flake stabilized by the diazapicenium dications – counterions are omitted for the sake of simplicity. | ||
Results and discussion
Synthesis and photophysical characterization of N,N′-dialkyl-3,10-diazapicenium salts
The parent 3,10-diazapicene scaffold 1, a member of the hitherto rather underrepresented family of hetero[n]-phenacenes,16 was accessible through microwave-assisted photocyclization of the corresponding diazastilbenes.17 To enhance the solubility of 118 and to introduce electron-accepting properties, n-dodecyl alkyl chains were introduced at the pyridyl nitrogens (for synthetic details, see ESI†). In analogy to the reaction conditions optimized for the dialkylation of related 4,4′-bipyridines, so-called viologens,19 reaction of 1 with 1-bromododecane at 130 °C in N,N-dimethylformamide (DMF) under microwave irradiation furnished the N,N′-didodecyl decorated 3,10-diazapicenium dibromide salt 2 in 89% yield (Scheme 1). To study the impact of the counterion on the photophysical and electrochemical properties, anion exchange reactions were carried out. For that purpose, addition of ammonium hexafluorophosphate to a solution of dibromide 2 in an EtOH/H2O mixture resulted in nearly quantitative precipitation of 3,10-diazapicenium bis(hexafluorophosphate) 3. Compounds 2 and 3 were isolated as air stable, yellow powders, which show sufficient solubility in common organic solvents. | ||
| Scheme 1 Synthesis of the N,N′-dialkylated 3,10-diazapicenium dications. | ||
To gain insights into the characteristics of 3,10-diazapicene 1 and its dialkylated salts 2 and 3, absorption and fluorescence studies were performed in EtOH (Fig. 2). Strong absorptions in the UV region (240–340 nm for 1, 250–350 nm for 2 and 3) followed by weak absorptions centered in the visible region (340–390 nm for 1, 370–440 nm for 2 and 3) were observed due to S0 → S2 and S0 → S1 transitions, respectively.20 The vibrational fine structure with a spacing in the range from 1350 to 1430 cm−1 stems from several in-plane C–H bending and ring breathing modes as previously reported for picene20 and smaller rigid viologens.21 Upon dialkylation of 1, the spectroscopic features are bathochromically shifted by about 42 nm for 2 and 3 and are independent on the nature of the counterion. Energies of the fundamental S0 → S1 transitions were estimated as 3.18, 2.79, and 2.77 eV for 1, 2 and 3, respectively, (Table 1).
 | ||
| Fig. 2 Normalized absorption (solid lines) and fluorescence spectra (dashed lines) of 1 (black, λexc = 324 nm), 2 (blue, λexc = 390 nm), and 3 (red, λexc = 390 nm) in EtOH. | ||
| λ max /nm | E gap /eV | λ em /nm (ϕ/a.u.) | τ /ns | E red,1 (Ered,2)/V | |
|---|---|---|---|---|---|
| a Measured in EtOH. b Band gap calculated from onset of the absorption at 0.1λmax. c Fluorescence quantum yields measured in EtOH with Coumarin 47 as reference (ϕref = 0.73).25 d Fluorescence lifetimes from TCSPC measurements (λexc = 403 nm). e Half-wave potentials recorded by cyclic voltammetry in CH2Cl2 with 0.1 M nBu4NPF6 (scan rate 200 mV s−1, referenced vs. Fc/Fc+). | |||||
| 1 | 383 | 3.18 | 386 | n/a | n/a |
| 2 | 426 | 2.79 | 443 (0.22) | 11.4 | −1.10 (−1.52) |
| 3 | 425 | 2.77 | 442 (0.22) | 10.7 | −1.10 (−1.51) |
Compounds 1, 2, and 3 exhibit intense blue fluorescence in EtOH. For example, the fluorescence of 3,10-diazapicene 1 maximizes at 386, 408, 432, and 459 nm. In contrast, the fluorescence of 2 and 3 is far less resolved with a maximum centered at 442 nm and a shoulder at 464 nm. All compounds 1–3 feature similar fluorescence quantum yields with 0.21–0.22 and Stokes shifts of 910 cm−1. The excitation spectra of 1–3 closely resemble their absorption spectra and, thus, confirm their monomeric nature. These results clearly show that the spectroscopic features of N,N′-dialkylated 3,10-diazapicenium dications are not impacted by the counterion pointing to the presence of solvent separated ion pairs in solution.22 The fluorescence spectra are reasonable mirror images of the absorption spectra including vibrational fine structure indicative of a rigid aromatic fluorophore.21
To determine the fluorescence lifetimes, time-correlated-single-photon-counting (TCSPC) measurements were performed in EtOH with an excitation wavelength of 403 nm and the resulting decays were fit by a monoexponential fitting function. We find that the bromide salt 2 and the hexafluorophosphate salt 3 display comparable fluorescence lifetimes of 11.4 and 10.7 ns (Table 1).
The characterization of diazapicenium bromide 2 was complemented by femtosecond transient absorption spectroscopy to gather a deeper insight into the excited state features and their dynamics. The deconvoluted transient absorption spectra for 2 in EtOH upon excitation at 387 nm taken from a global analysis are shown in Fig. 3 together with the time–concentration profiles. To this end, the singlet excited state features were found in the visible as well as in the near infrared region with maxima at 455, 480, 630, 920, and 1035 nm. All of them are formed instantaneously upon excitation and feature a lifetime of 9.7 ± 1.0 ns. The corresponding intersystem crossing produces a triplet excited state, whose lifetime is 280 ± 30 ns, and whose signature is a 615 nm maximum.
 | ||
| Fig. 3 Top: deconvoluted transient absorption spectra of the singlet (black) and triplet (cyan) excited state of 2 in EtOH obtained by global analysis. Bottom: corresponding concentration–time profiles. | ||
To evaluate the electron accepting strength of 2 and 3, their electrochemical properties were examined by means of cyclic voltammetry in CH2Cl2 with 0.1 M nBu4NPF6 as a supporting electrolyte at a scan rate of 200 mV s−1. Glassy carbon served as a working electrode, a platinum wire as counter electrode, and a Ag/AgCl as pseudo-reference electrode. Overall, the redox properties, which are independent on the nature of the counterions, give rise to two irreversible one-electron reductions at −1.10 V vs. Fc/Fc+ (ELUMO = −3.7 eV)23 and −1.51 V vs. Fc/Fc+ for 2 and 3 (Table 1, see ESI†). Both reductions to afford the corresponding radical cation and subsequently neutral quinoidal form of the N,N′-dialkylated 3,10-diazapicenium salts are comparable to those reported for N,N′-dialkylated 3,8-diazaphenanthrenium viologens and related compounds (Fig. 4).21,24 Notably, the first reduction of 2 and 3 is within the range of electron-deficient aromatics as previously used to fabricate non-covalently p-type doped graphene hybrid materials.7,9,11
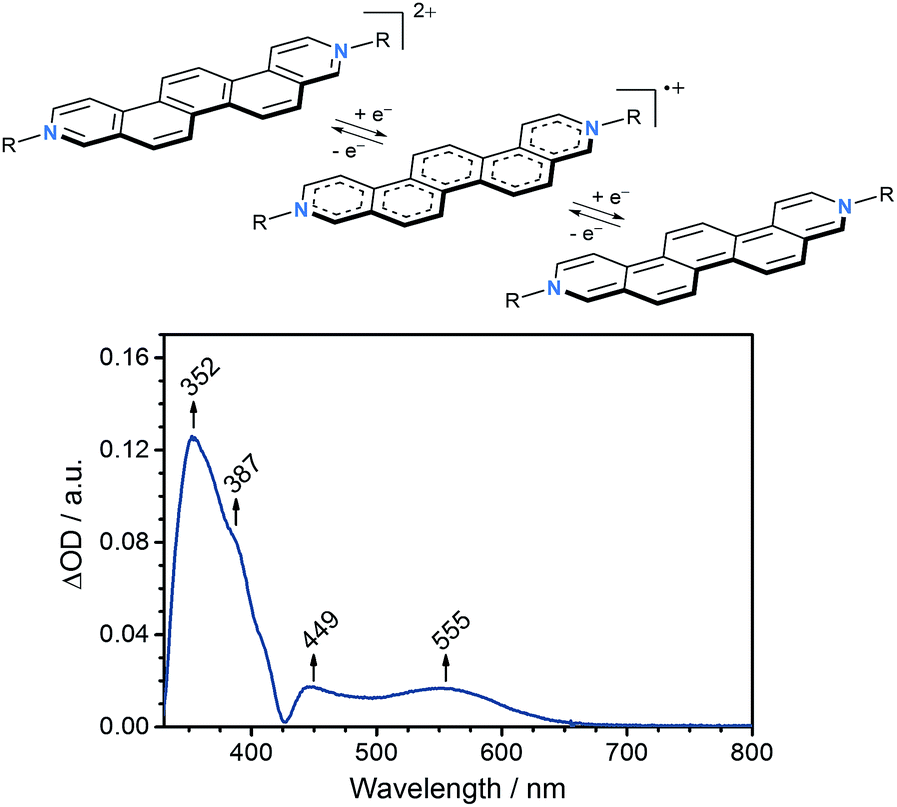 | ||
| Fig. 4 Top: two one-electron reductions of N,N′-dialkylated 3,10-diazapicenium salts (R = alkyl); bottom: differential absorption spectrum of 2 in dry DMF obtained by subtracting the absorption spectrum of 2 recorded at 0.0 V from that recorded at −0.7 V vs. Ag wire. | ||
Based on these results, spectroelectrochemical studies were performed for 2 in dry DMF containing 0.1 M nBu4NPF6 as supporting electrolyte. As working electrode, a platinum mesh was used, while the counter electrode was a platinum plate and a silver wire was used as reference electrode. In agreement with the findings from cyclic voltammetry, the differential absorption spectrum recorded at −0.70 V vs. Ag wire (ca. −1.10 V vs. Fc/Fc+)26 reveals characteristic changes in the absorption, which were ascribed to the one-electron reduction of 2 to the corresponding radical cation (Fig. 4). In agreement with other related viologens,27 absorptions at 350–400 nm as well as at 450–500 nm increase upon reduction and new absorptions evolve between 500–650 nm with a maximum centered at 555 nm.
Exfoliation and characterization of graphene/3,10-diazapicenium hybrid systems
With the key features of the N,N′-dialkylated 3,10-diazapicendiium salt 2 and 328 at hand, we focused on the preparation of the hybrid materials with graphene by adapting a previously reported procedure.11 Initially, a metastable graphene dispersion (EG) was generated in EtOH by ultrasonication for at least 1 h at room temperature and subsequent centrifugation of graphene flakes. This was followed by a stepwise addition of EG to a 3 × 10−6 M solution of 2 in EtOH affording the hybrid system EG/2 which was analyzed by steady state absorption spectroscopy, (for details, see ESI†). Our procedure led to exfoliated graphene flakes, which are stabilized by the diazapicenium cations through π–π interactions. The inherent positive charge of 2 thereby aids in the dispersion of graphene flakes via charge repulsion and in that way minimizes their agglomeration in solution. Thus, as the graphene concentration is increased the absorption features of 2 diminish, indicating strong interaction with the basal plane of graphene (Fig. 5). Notably, the peak at about 270 nm, which steadily increases upon adding various aliquots of the graphene dispersion is due to a van Hove singularity in the density of states of graphene.29 In turn, the fluorescence of 2 is quenched with gradually increasing concentration of EG in the dispersion, which is in agreement with previous reports of closely related positively charged exfoliants by Stoddart et al. (Fig. 5).15 | ||
| Fig. 5 Top: absorption for clarity EG/2 spectra shifted in y-direction to offset light scattering caused by the graphene/graphite flakes; bottom: fluorescence spectra of 2 (black) and EG/2 (blue) upon stepwise addition of EG in EtOH. | ||
A comprehensive tool to disclose ground state interactions between graphene and 2 is based on Raman spectroscopy. The sample preparation involved drop casting of EG/2 onto a Si/SiO2 wafer followed by subsequent drying. To validate our results with EG/2, similar experiments were performed with a reference graphene sample, that is, EG. EG was prepared in analogy to EG/2 without, however, the addition of 2. Statistical analyses of the coated wafer were carried out by measuring mappings consisting of 1000 individual spectra recorded upon an excitation wavelength of 532 nm. From 2D/G-ratios on the order of 0.65 as well as a full width at half maximum (FWHM) of the 2D-band of about 70 cm−1 we conclude that the majority of the spectra resemble turbostratic graphene flakes.30 Restacking of the individual graphene flakes during the drying process is a likely rationale for this finding.
To shed light onto doping effects present in the hybrid material, we focused our attention on the position of the individual Raman bands (Fig. 6). A careful comparison of the histograms regarding the G- and 2D-band positions of EG/2 and reference EG discloses shifts to higher wavenumbers in EG/2. Such an upshift is indicative for p-doping of graphene.31 Such a p-doping is in sound agreement with the electrochemical measurements, in which the low reduction potentials of 2 were documented. In other words, the diazapicenium salt not only acts as a stabilizing agent for exfoliated graphene flakes exploiting the charge repulsion between the cationic scaffolds, but also results in p-doping of the graphene flakes owing to strong electronic coupling in the resulting hybrid material.
 | ||
| Fig. 6 Statistical analysis of the Raman mapping of reference EG (blue bars) as well as of EG/2 (red bars) displaying the Raman shifts of G-band (top) and 2D-band (bottom). The Gaussian fits serve solely as a guide to the eyes. | ||
To further study the nature of the obtained graphene flakes, atomic force microscopy (AFM) and transmission electron microscopy (TEM) were employed. In typical TEM images of EG/2, drop casted from EtOH onto a lacey carbon coated copper grid, few layer graphene with lateral sizes ranging from a few microns as maximum size down to flakes sizes in the nm regime was noted (Fig. 7). Often, the graphene flakes were folded, rolled up, and intertwined. From the height profiles taken from a characteristic AFM image of EG/2, which was also used for the Raman measurements, we infer that next to solvent residues and/or some contaminants thin graphene flakes with average heights of 1.9–2.5 nm are present. In combination with the Raman results, these findings demonstrate that the hybrid material consists of turbostratic graphene flakes with a thickness of 5 to 8 layers when probing solid state samples. It is, however, reasonable to assume that the hybrid material consists in the liquid phase of fewer layers due to the repulsive forces introduced from 2.
 | ||
| Fig. 7 Left: TEM image of EG/2 drop casted onto a lacey carbon grid; for the sample typical intertwined and folded multilayered graphene flakes are depicted; middle: AFM picture of EG/2 drop casted onto a Si/SiO2 wafer; the black dots constitute residual solvent and/or contaminants; right: height profiles extracted from the AFM image as indicated by the graded blue lines. | ||
Next to steady state measurements, also transient absorption studies were carried out to probe EG/2. In the near-infrared region of the spectrum, the usual graphene related bleaching is discernable, for which a lifetime of 0.4 ps was determined. In the visible region of the spectrum, a new transient maximum at 595 nm is discernible for EG/2 (see ESI†). This finding is in sharp contrast to the singlet and triplet excited state features noted for just 2 in EtOH (see Fig. 4). Global analysis of EG/2 afforded a single evolution associated spectrum with a rather short lifetime of 26 ± 3 ps. Tentatively, we assign this species to that of the charge separated state. As derived from Raman experiments, strong interactions between EG and 2 can be regarded as the inception to a doping of EG in the ground state. As such, it is reasonable to expect that a partial shift of charge density in the ground state leads to rapid charge separation kinetics in the excited state. Moreover, the comparison of the deconvoluted transient absorption spectrum with the spectroelectrochemical results for 2 confirms that this newly formed transient relates to the one-electron reduced form of 2 (Fig. 8). In comparison to the absorption spectrum of the electrochemically generated radical cation of 2 the maximum is, however, bathochromically shifted by ca. 40 nm. A possible rationale for this shift is based on the additional stabilization of the radical cation resulting from electronic coupling with the basal plane of the exfoliated graphene flakes in EG/2. Conclusively, the excitation of EG/2 leads to an electron transfer event from graphene to 2 resulting in a hybrid system consisting of p-doped graphene flakes and radical cations of 2.
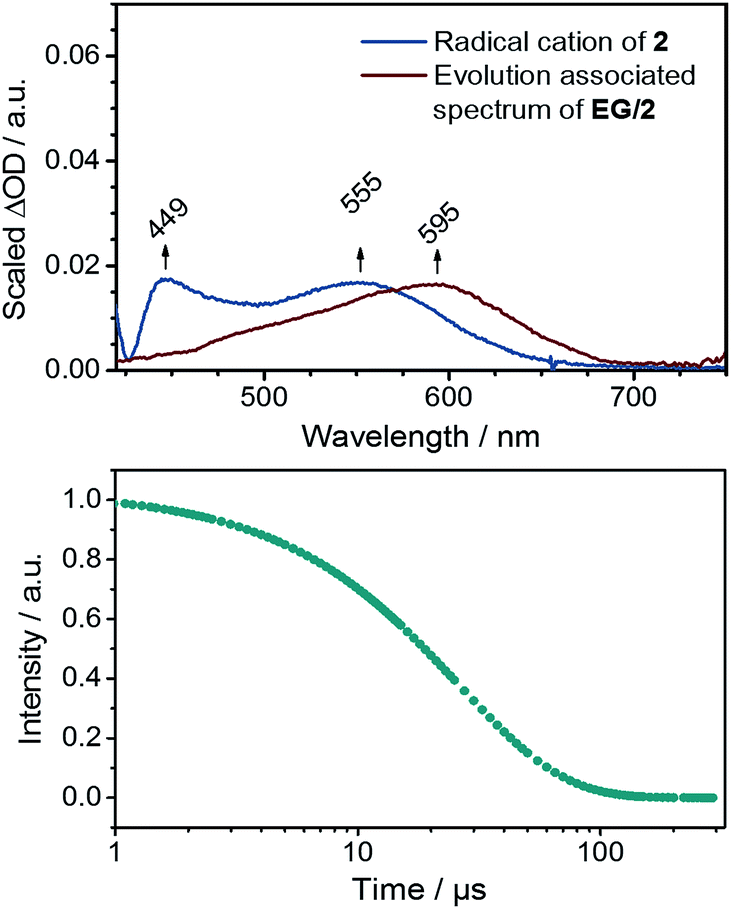 | ||
| Fig. 8 Top: evolution associated spectrum of EG/2 together with the differential spectrum from spectroelectrochemical measurements (see Fig. 4). Bottom: corresponding concentration–time profile. | ||
Conclusions
Two novel N,N′-didodecyl 3,10-diazapicenium salts with bromide and hexafluorophosphate counterions were synthesized as an exfoliant, a stabilizer, and a p-dopant for liquid phase exfoliated graphene. When studied alone, it was demonstrated that the two different counterions of the N,N′-didodecyl 3,10-diazapicenium salts have only negligible influence on the photophysical and electrochemical properties: both N,N′-didodecyl 3,10-diazapicenium salts exhibited comparable absorption, fluorescence, and reduction, which were predominantly dictated by the polycyclic π-conjugated scaffold. Complementary spectroelectrochemical investigations with the 3,10-diazapicenium dibromide revealed that their reductions were accompanied by characteristic changes in the absorption features.The bromide salt was successfully applied to exfoliate graphite and to stabilize the graphene flakes. The resulting hybrids were characterized by an arsenal of steady state and time-resolved spectroscopic as well as microscopic techniques. As such, they corroborate that the ground state of these hybrids is dominated by a shift of charge density from the basal plane of graphene to the positively charged 3,10-diazapicenium: result of this shift is a p-doped graphene. For the excited state, we see an efficient charge transfer from graphene to the positively charged 3,10-diazapicenium. Here, the result is the one-electron reduction of 2 and a hole delocalized on the graphene flakes. Unambiguously, the presence of multilayered turbostratic graphene flakes with modified electronic properties was demonstrated and, hence, the newly synthesized N,N′-dialkylated 3,10-diazapicenium salts open up highly promising new avenues towards the fabrication of hybrid (nano)graphene materials with electronic properties tailored at will.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) as part of SFB 953 “Synthetic Carbon Allotropes”. The Cluster of Excellence “Engineering of Advanced Materials” (EAM) at FAU Erlangen-Nürnberg, and the Graduate School Molecular Science (GSMS) of FAU Erlangen-Nürnberg are gratefully acknowledged for their generous support. V. C., J. S., and P. J. acknowledge the support from the Czech Science Foundation (grant no. 17-02578S).Notes and references
- (a) V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156 CrossRef CAS PubMed; (b) T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2012, 57, 1061 CrossRef CAS; (c) A. Ciesielski and P. Samorì, Adv. Mater., 2016, 28, 6030 CrossRef CAS PubMed.
- (a) A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109 CrossRef CAS; (b) S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217 CrossRef CAS PubMed; (c) D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 9336 CrossRef CAS PubMed; (d) D. A. C. Brownson, D. K. Kampouris and C. E. Banks, J. Power Sources, 2011, 196, 4873 CrossRef CAS.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192 CrossRef CAS PubMed.
- (a) L. Jiao, X. Wang, G. Diankov, H. Wang and H. Dai, Nat. Nanotechnol., 2010, 5, 321 CrossRef CAS PubMed; (b) X. Li, C. W. Magnuson, A. Venugopal, J. An, J. W. Suk, B. Han, M. Borysiak, W. Cai, A. Velamakanni, Y. Zhu, L. Fu, E. M. Vogel, E. Voelkl, L. Colombo and R. S. Ruoff, Nano Lett., 2010, 10, 4328 CrossRef CAS PubMed.
- (a) F. Li, Y. Bao, J. Chai, Q. Zhang, D. Han and L. Niu, Langmuir, 2010, 26, 12314 CrossRef CAS PubMed; (b) Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906 CrossRef CAS PubMed; (c) I. Levchenko, K. K. Ostrikov, J. Zheng, X. Li, M. Keidar and K. B. K. Teo, Nanoscale, 2016, 8, 10511 RSC.
- (a) Y. Si and E. T. Samulski, Chem. Mater., 2008, 20, 6792 CrossRef CAS; (b) M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611 CrossRef CAS PubMed; (c) C. Zhang, W. Lv, X. Xie, D. Tang, C. Liu and Q.-H. Yang, Carbon, 2013, 62, 11 CrossRef CAS.
- D. Kiessling, R. D. Costa, G. Katsukis, J. Malig, F. Lodermeyer, S. Feihl, A. Roth, L. Wibmer, M. Kehrer, M. Volland, P. Wagner, G. G. Wallace, D. L. Officer and D. M. Guldi, Chem. Sci., 2013, 4, 3085 RSC.
- (a) J. Malig, N. Jux, D. Kiessling, J.-J. Cid, P. Vázquez, T. Torres and D. M. Guldi, Angew. Chem., Int. Ed., 2011, 50, 3561 CrossRef CAS PubMed; (b) J. Malig, N. Jux and D. M. Guldi, Acc. Chem. Res., 2013, 46, 53 CrossRef CAS PubMed; (c) D. M. Guldi and R. D. Costa, J. Phys. Chem. Lett., 2013, 4, 1489 CrossRef CAS PubMed.
- L. Wibmer, L. M. O. Lourenço, A. Roth, G. Katsukis, M. G. P. M. S. Neves, J. A. S. Cavaleiro, J. P. C. Tomé, T. Torres and D. M. Guldi, Nanoscale, 2015, 7, 5674 RSC.
- J. Malig, A. W. I. Stephenson, P. Wagner, G. G. Wallace, D. L. Officer and D. M. Guldi, Chem. Commun., 2012, 48, 8745 RSC.
- A. Roth, M.-E. Ragoussi, L. Wibmer, G. Katsukis, G. d. La Torre, T. Torres and D. M. Guldi, Chem. Sci., 2014, 5, 3432 RSC.
- P. Lauffer, K. V. Emtsev, R. Graupner, T. Seyller and L. Ley, Phys. Status Solidi B, 2008, 245, 2064 CrossRef CAS.
- X. Dong, D. Fu, W. Fang, Y. Shi, P. Chen and L.-J. Li, Small, 2009, 5, 1422 CrossRef CAS PubMed.
- (a) S. S. Sabri, J. Guillemette, A. Guermoune, M. Siaj and T. Szkopek, Appl. Phys. Lett., 2012, 100, 113106 CrossRef; (b) H. B. M. Shashikala, C. I. Nicolas and X.-Q. Wang, J. Phys. Chem. C, 2012, 116, 26102 CrossRef CAS PubMed; (c) B. Li, A. V. Klekachev, M. Cantoro, C. Huyghebaert, A. Stesmans, I. Asselberghs, S. de Gendt and S. de Feyter, Nanoscale, 2013, 5, 9640 RSC; (d) T. Hu and I. C. Gerber, Chem. Phys. Lett., 2014, 616–617, 75 CrossRef CAS; (e) L. Zhou, K. Wang, Z. Wu, H. Dong, H. Sun, X. Cheng, H. Zhang, H. Zhou, C. Jia, Q. Jin, H. Mao, J.-L. Coll and J. Zhao, Langmuir, 2016, 32, 12623 CrossRef CAS PubMed.
- S. Sampath, A. N. Basuray, K. J. Hartlieb, T. Aytun, S. I. Stupp and J. F. Stoddart, Adv. Mater., 2013, 25, 2740 CrossRef CAS PubMed.
- (a) V. Calabrese, S. Quici, E. Rossi, E. Cariati, C. Dragonetti, D. Roberto, E. Tordin, F. de Angelis and S. Fantacci, Chem. Commun., 2010, 46, 8374 RSC; (b) C. Mitsui, T. Okamoto, H. Matsui, M. Yamagishi, T. Matsushita, J. Soeda, K. Miwa, H. Sato, A. Yamano, T. Uemura and J. Takeya, Chem. Mater., 2013, 25, 3952 CrossRef CAS; (c) Y. Kubozono, K. Hyodo, H. Mori, S. Hamao, H. Goto and Y. Nishihara, J. Mater. Chem. C, 2015, 3, 2413 RSC.
- (a) V. Církva and S. Relich, Curr. Org. Chem., 2011, 15, 248 CrossRef; (b) J. Storch, V. Církva and M. B. J. Vokál, Czech Republic Pat., CZ 303997, 2013.
- 3,10-Diazapicene was generously provided by J. Storch and V. Církva from the Institute of Chemical Process Fundamentals of the Czech Academy of Sciences, Prague, Czech Republic.
- M. Lamberto, E. E. Rastede, J. Decker and F. M. Raymo, Tetrahedron Lett., 2010, 51, 5618 CrossRef CAS.
- S. Fanetti, M. Citroni, R. Bini, L. Malavasi, G. A. Artioli and P. Postorino, J. Chem. Phys., 2012, 137, 224506 CrossRef PubMed.
- T. Bakupog, E. L. Clennan and X. Zhang, Tetrahedron Lett., 2015, 56, 5591 CrossRef CAS.
- C. D. Geddes, Meas. Sci. Technol., 2001, 12, R53 CrossRef CAS.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bässler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551 CrossRef CAS.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49 RSC.
- G. Jones II, W. R. Jackson, C. Y. Choi and W. R. Bergmark, J. Phys. Chem., 1985, 89, 294 CrossRef.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, 94th edn, 2013 Search PubMed.
- (a) T. M. Bockman and J. K. Kochi, J. Org. Chem., 1990, 55, 4127 CrossRef CAS; (b) C. Reus, M. Stolar, J. Vanderkley, J. Nebauer and T. Baumgartner, J. Am. Chem. Soc., 2015, 137, 11710 CrossRef CAS PubMed.
- Due to the formation of solvent separated ion pairs in solution and similar electrochemical and photophysical characteristics of 2 and 3, we expect comparable results for the electronic interactions between the 3,10-diazapicenium dications and graphene; for further information see the ESI.†.
- V. G. Kravets, A. N. Grigorenko, R. R. Nair, P. Blake, S. Anissimova, K. S. Novoselov and A. K. Geim, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 155413 CrossRef.
- A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed.
- R. Li, Z. Liu, K. Parvez, X. Feng and K. Müllen, J. Mater. Chem. C, 2015, 3, 37 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details and additional spectra. See DOI: 10.1039/c7sc00533d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |