
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand-assisted palladium-catalyzed C–H alkenylation of aliphatic amines for the synthesis of functionalized pyrrolidines†
Chuan
He
and
Matthew J.
Gaunt
 *
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: mjg32@cam.ac.uk; Web: http://www-gaunt.ch.cam.ac.uk
First published on 23rd February 2017
Abstract
The development of a ligand-assisted Pd-catalyzed C–H alkenylation of aliphatic amines is reported. Our studies indicated that an amino-acid-derived ligand renders the C–H bond activation step reversible and promotes the traditionally difficult alkenylation process. The C(sp3)–H alkenylation proceeds through a 5-membered-ring cyclopalladation pathway that allows access to complex aliphatic heterocycles that could be useful to practioners of synthetic and medicinal chemistry.
Introduction
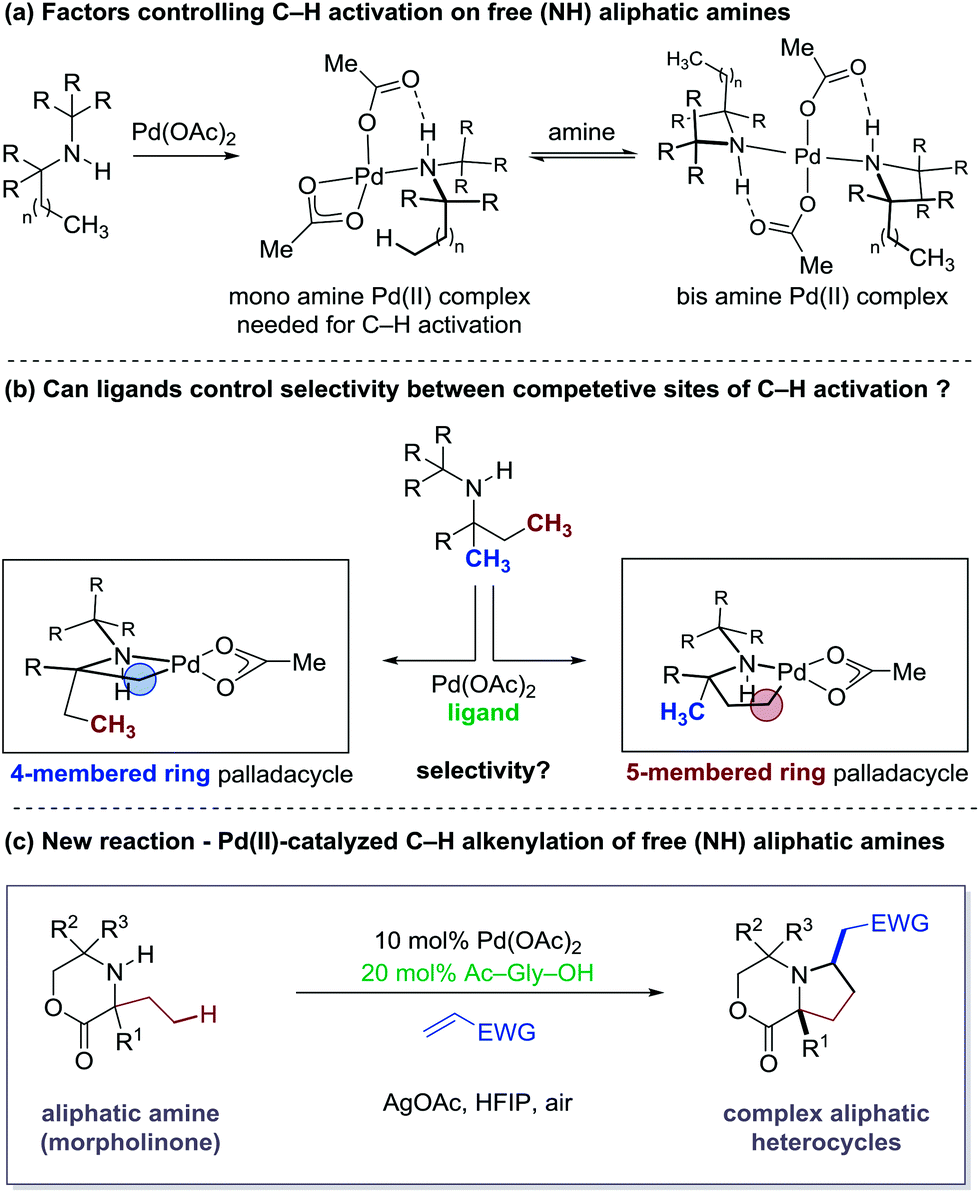
The synthesis of architecturally complex aliphatic amines remains an important challenge to synthetic and medicinal chemists because their structural and functional properties are often fundamental to biological activity in many nitrogen-containing molecules.1 Recently, palladium-catalyzed C–H functionalization of aliphatic amine derivatives has emerged as a potentially powerful tactic for the synthesis of complex variants of these important molecules.2 Central to the continued evolution of these synthetic strategies is the development of new activation modes and transformations on a range of aliphatic amine substrates.Over the last three years, our laboratory has been engaged in the development of C(sp3)–H activation reactions guided by the free (NH)-amine.3 Central to the success of this strategy has been the steric-induced destabilization of rapidly formed bis-amine Pd(II) complexes, which leads to higher concentrations of the putative mono-amine Pd(II) complexes empirically required for C–H bond cleavage. Furthermore, a crucial hydrogen bond between the NH group of the ligated amine and the carbonyl group of the acetate ligand orients the amine such that C–H activation is facilitated (Fig. 1a).3b While we have developed a number of distinct transformation based on discrete 4- and 5-membered cyclopalladation pathways, we began to question whether we could control the selectivity of the C–H activation by the action of an external ligand, which could lead to the development of new transformations on aliphatic amine scaffolds (Fig. 1b). Herein, we describe how an amino acid-derived ligand was found to strongly influence the C–H activation on aliphatic amines with competitive sites of reactivity. By rendering the cyclopalladation step reversible, the amino acid ligand facilitates a productive 5-membered ring cyclopalladation pathway, leading to a new C–H alkenylation reaction to generate complex pyrrolidine-based heterocycles (Fig. 1c). The C–H catalytic alkenylation is operationally simple and works well on a range of substituted morpholinone-derived amines and with a variety of electron-deficient alkenes. Furthermore, the process provides access to previously unexplored aliphatic heterocycles that we believe will be of interest as novel amine scaffolds for drug discovery.
 | ||
| Fig. 1 Evolution of a ligand-controlled C–H alkenylation. | ||
Ligand-controlled reversible C–H activation
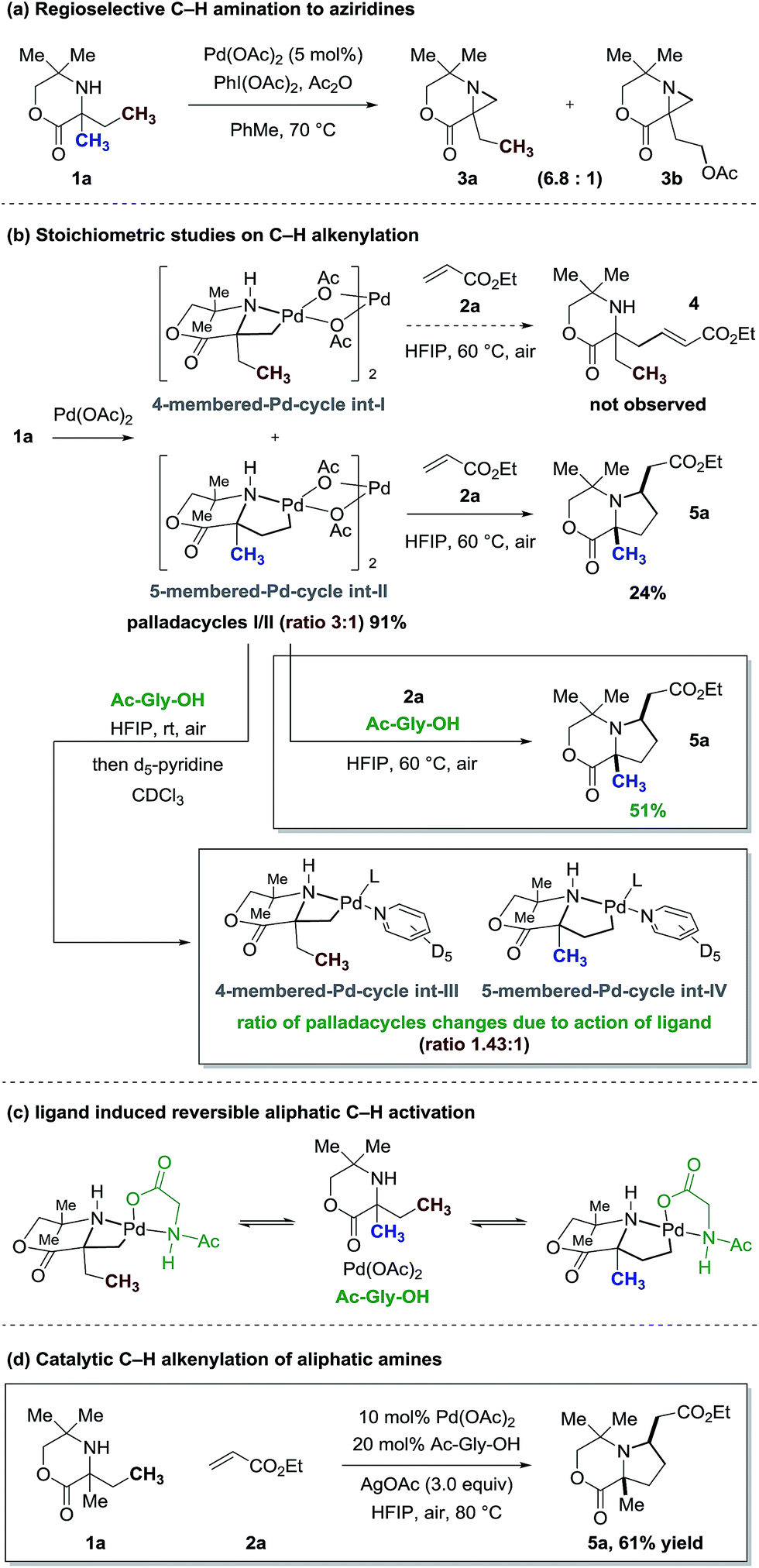
The basis for our studies was an observation made during our original work on Pd-catalyzed C–H amination to form aziridines.3a The signature reaction of this work exploited a 4-membered ring cyclopalladation pathway on a C–H bond at the β-position to the free (NH) amine motif. However, we were surprised to discover that this unusual cyclopalladation mode predominated even when traditionally more favourable C–H activation pathways were accessible; β-C–H activation via a 4-membered ring intermediate was favoured over a more classical 5-membered ring cyclopalladation at a competitive γ-C–H bond (Fig. 3a). Using this scaffold as a starting point for our studies, we elected to investigate this unusual selectivity in the context of a C–H alkenylation reaction.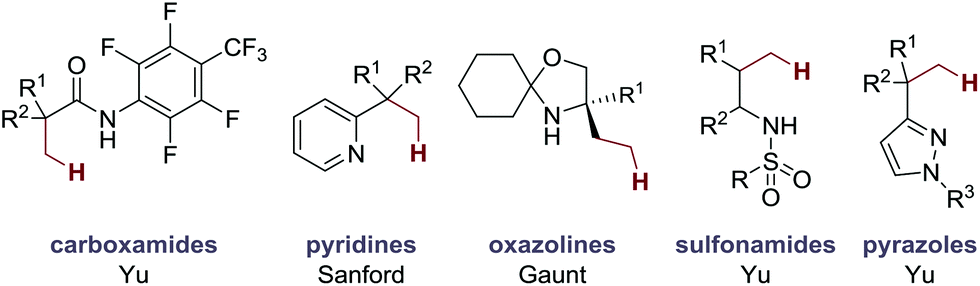 | ||
| Fig. 2 Aliphatic systems capable of C–H alkenylation. | ||
The successful realization of Pd-catalyzed C–H alkenylation reactions has great potential for the synthesis of complex molecules.4 Since the seminal works of Fujiwara and Moritani,5 many different approaches to C–H alkenylation have been developed, in particular on aromatic substrates.6 Central to many of these developments have been the exploitation of Lewis basic directing groups that facilitate the C–H activation step.7 Furthermore, the role of activating ligands has extended the scope of available C–H alkenylation reactions to a variety of substrate classes.6i,8 In contrast, C–H alkenylation in aliphatic systems remains limited to a small number of examples (Fig. 2).3d,9 To date, successful examples of Pd-catalyzed C(sp3)–H alkenylation directly using alkenes include Yu's carboxamide-directed olefination to γ-lactams,9a Sanford's pyridine-directed olefination to pyridinium salts,9b our group's free (NH) amine-directed olefination of amino alcohols derivatives,3d and Yu's sulfonamide-directed olefination9c and pyrazole-directed olefination.9d As a result, the development of new methods for Pd-catalyzed C–H alkenylation remains an ongoing and important goal.
Our studies began with the investigation of a stoichiometric C–H alkenylation process using differentially substituted morpholinone 1a. Treatment of 1a with 1.5 equivalents of Pd(OAc)2 delivered a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of palladacycles (int-I and int-II) in favour of the 4-membered ring complex (Fig. 3b), as expected based on the outcome of the reaction shown in Fig. 3a. When this mixture of palladacycles was treated with acrylate 2a in hexafluoroisopropanol (HFIP) at 60 °C, we did not observe the formation of the expected alkenylation product 4, but found that the bicyclic pyrrolidine product 5a was formed in 24% yield; reaction of only the 5-membered ring palladacycle via sequential carbopalladation of the alkene, β-hydride elimination to a substituted acrylate and aza-Michael addition gave the pyrrolidine-derived heterocycle 5a.
:1 mixture of palladacycles (int-I and int-II) in favour of the 4-membered ring complex (Fig. 3b), as expected based on the outcome of the reaction shown in Fig. 3a. When this mixture of palladacycles was treated with acrylate 2a in hexafluoroisopropanol (HFIP) at 60 °C, we did not observe the formation of the expected alkenylation product 4, but found that the bicyclic pyrrolidine product 5a was formed in 24% yield; reaction of only the 5-membered ring palladacycle via sequential carbopalladation of the alkene, β-hydride elimination to a substituted acrylate and aza-Michael addition gave the pyrrolidine-derived heterocycle 5a.
 | ||
| Fig. 3 Identification of a ligand controlled reversible C–H activation. | ||
Previous work in our group had highlighted an essential role of amino-acid ligands, first introduced by Yu et al.,10 in securing an effective C–H arylation process on the tetramethylpiperidine scaffold.3c Therefore, we treated the mixture of palladacycles formed from morpholinone 1a with acrylate 2a in the presence of two equivalents of glycine-derived ligand (Ac-Gly-OH, 6a, see ESI† for details) and found that the yield of the pyrrolidine product 5a increased to 51%. This result is surprising because, based on the ratio of observed palladacycles, the maximum theoretical yield for this product should be 25%. Therefore, the increased yield of 5a suggested that the amino acid ligand could be reversing the preferred cyclopalladation process to the 4-membered complex and channeling the reaction down the 5-membered ring C–H activation pathway to form 5a (Fig. 3b).
To further investigate this phenomena, we treated the 3:1 mixture of palladacycles int-I/II with ligand Ac-Gly-OH 6a at room temperature, but without the acrylate. Then, we quenched the reaction with d5-pyridine, which enables directly identification of the palladacycles by 1H NMR, and found that the ratio of complexes (4-membered int-IIIvs. 5-membered int-IV) had changed from 3:1 to 1.43:1 (Fig. 3b). This result strongly suggests that the ligand 6a is reversing the cyclopalladation process, such that, in the presence of an alkene, the C–H alkenylation via the 5-membered ring palladacycle becomes a kinetic trap for the equilibrium, selectively forming 5a. To the best of our knowledge, this represents the first example of an amino acid-derived ligand controlling the regioselectivity of C–H activation on aliphatic systems (Fig. 3c). We were able to render this specific transformation catalytic in Pd(OAc)2 by the addition of AgOAc as terminal oxidant, which led to the formation of pyrrolidine 5a in 61% yield (Fig. 3d).
Scope of ligand-assisted C–H alkenylation
While morpholinone 1a had provided an effective scaffold to probe the ligand controlled selectivity between competitive cyclopalladation pathways, variation of substitution at the carbon atom adjacent to the NH motif would be essential in exploring the scope of the process. To test this, we prepared a range of morpholinones, α,α′-morpholinones displaying the ethyl group needed for (5-membered ring) C–H alkenylation alongside a variety of functionalized substituents (Table 1). First, we selected the α,α-diethyl substituted morpholinone 1b and found that the conditions identified for the successful reaction of 1a were effective for this substrate; reaction involving treatment of the amine 1b with ethyl acrylate 2a with 10 mol% Pd(OAc)2, 20 mol% of ligand Ac-Gly-OH 6a, AgOAc and with HFIP as the solvent delivered the desired pyrrolidine product 5b in 86% yield, after isolation by silica gel chromatography. In further assessing the scope of the catalytic reaction, we found that a range of functionalized α,α′-disubstituted morpholinones also underwent smooth ligand assisted C–H alkenylation. Protected hydroxymethyl substituents worked well in 48–78% yields, providing readily manipulable functional groups suitable for down stream modification of varying substitution patterns (5d–5f). Longer alkyl chains with remote functional groups, including esters, protected amines, sulfones even an alkyl boronic ester, proved to be excellent substrates for the C–H alkenylation giving the complex pyrrolidines in good yields (5g–5j). Changing the substituents on the non-reacting side of the morpholinone substrate to a cyclohexyl motif gave 75% yield (5k). Importantly, reducing steric hindrance around the nitrogen atom could also produce the corresponding alkenylation products. By using the chiral morpholinone substrates (1l and 1m), which could be easily assembled from chiral pool amino alcohols, synthetically useful levels of diastereoselective transformation could be achieved to afford the enantiopure pyrrolidine products (5l and 5m). When cyclopropyl substituted morpholinone was subjected to the same reaction conditions, methylene C–H alkenylation took place to form 5n as a single isomer, albeit in a lower yield. When an aromatic group was present close to the NH motif, the reaction unsurprisingly proceeded through the inherently more facile aryl C–H activation pathway to give exclusively ortho sp2 C–H alkenylation to 5o in excellent yields.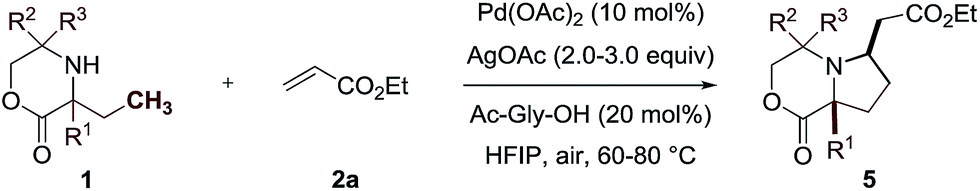
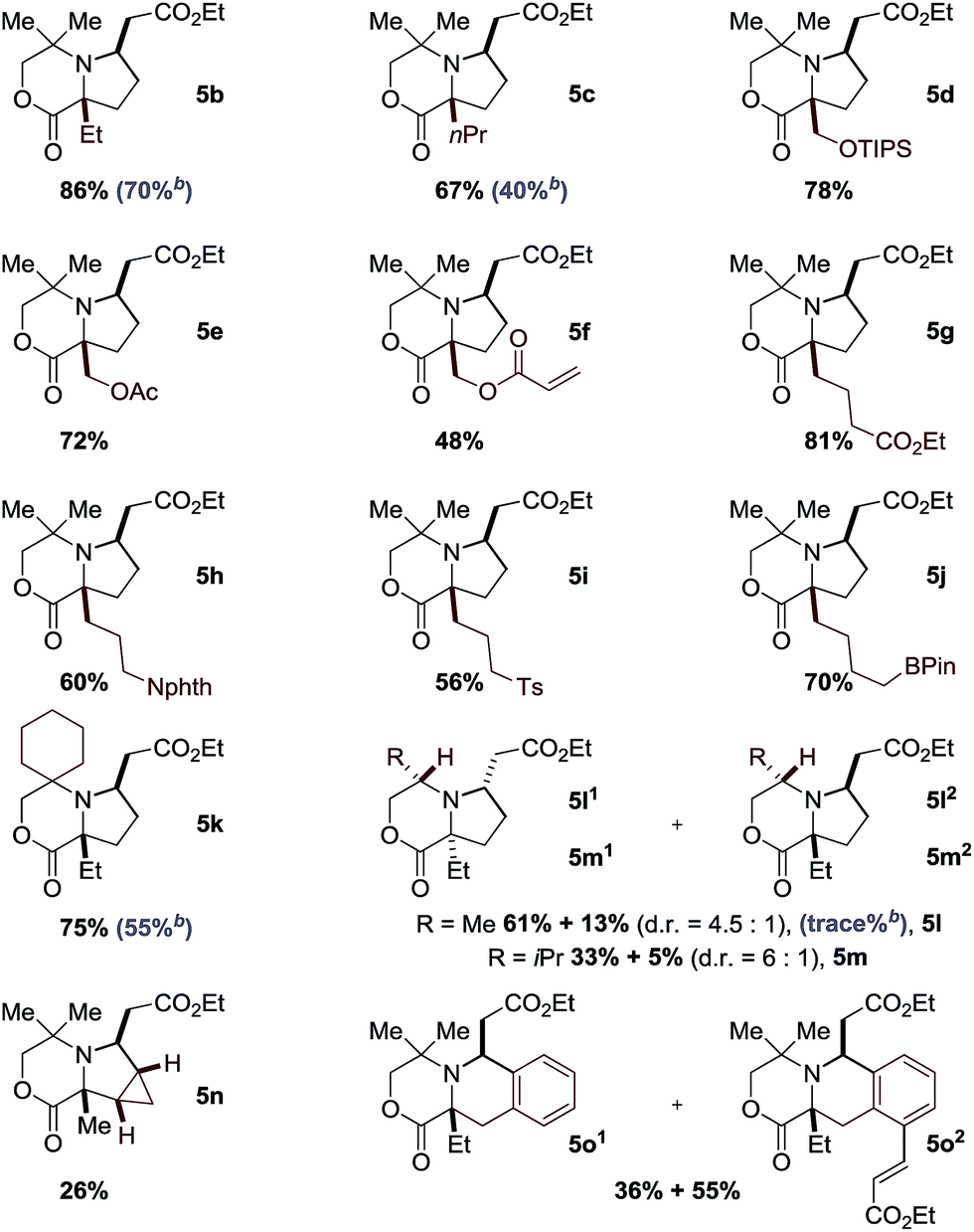
It was interesting to note that even though the reaction worked in some cases in the absence of the amino acid-derived ligand, the yield was significantly increased in its presence. For example, the yield of 5c was increased by almost 30% in the presence of Ac-Gly-OH 6a. More strikingly, C–H alkenylation of the chiral morpholinone 1l to 5l did not proceed in the absence of the ligand 6a. Unfortunately, acyclic amine substrates were unsuccessful when tested under the optimal conditions.11
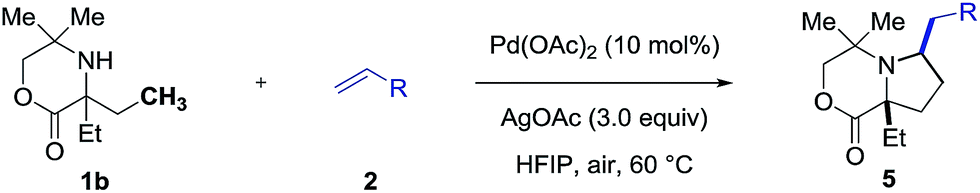
We next explored the range of alkenes that could be transferred as part of this process (Table 2). The reaction was readily extended to a variety of acrylates in excellent yields (5p–5t). α,β-Unsaturated ketones, amides and even acrolein could also be incorporated into the pyrrolidine scaffold (5u–5y). Moreover, vinyl sulfone and vinyl phosphonate also worked well in the C–H alkenylation process, providing useful products (5z and 5aa) with opportunities for further elaboration via well-established methods. Again, we found that ligand Ac-Gly-OH 6a was not necessary for a successful reaction with acrylate coupling partners, but it did have a significant effect on the yield of the reaction with alkene coupling partners such as enones (5v), acrylamides (5x), vinyl sulfones (5z) and vinyl phosphonates (5aa). Unfortunately, simple unfunctionalized alkenes or styrenes were not suitable for this transformation under the current conditions either with or without the use of amino acid ligand.
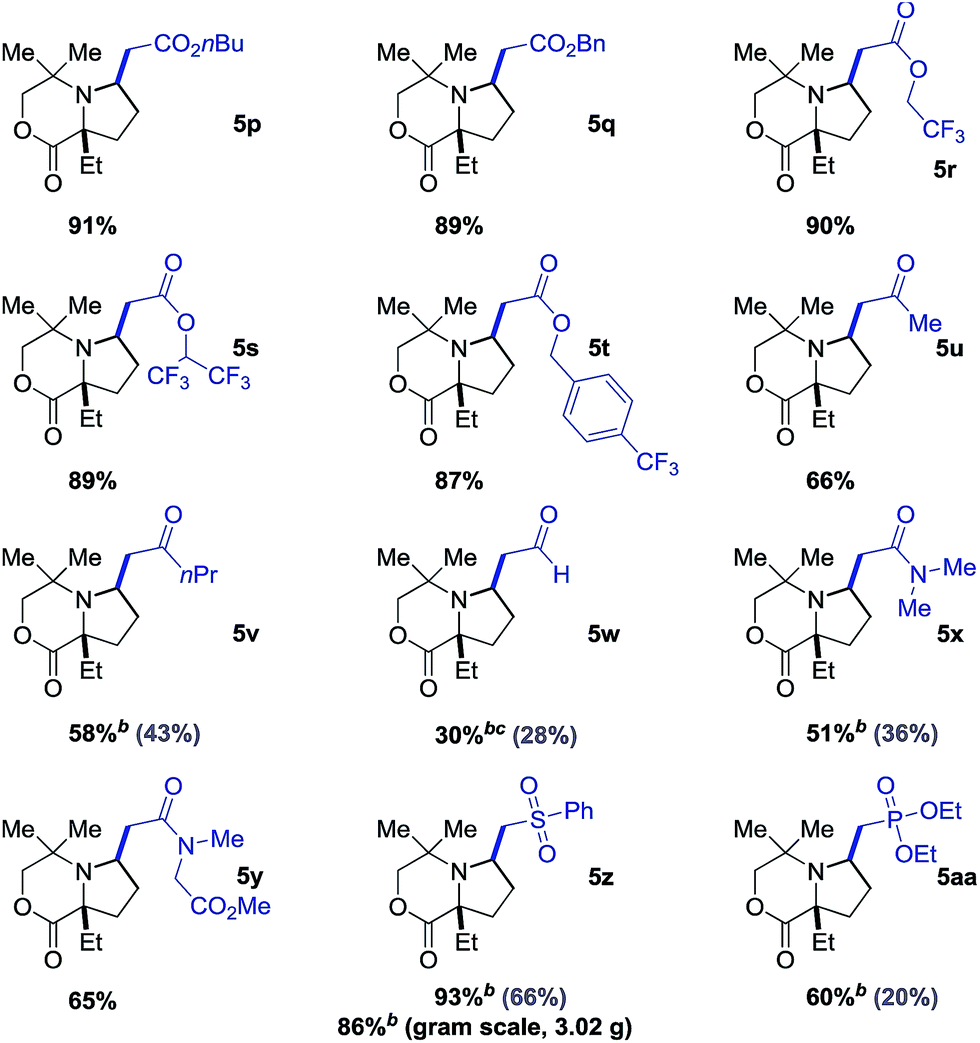
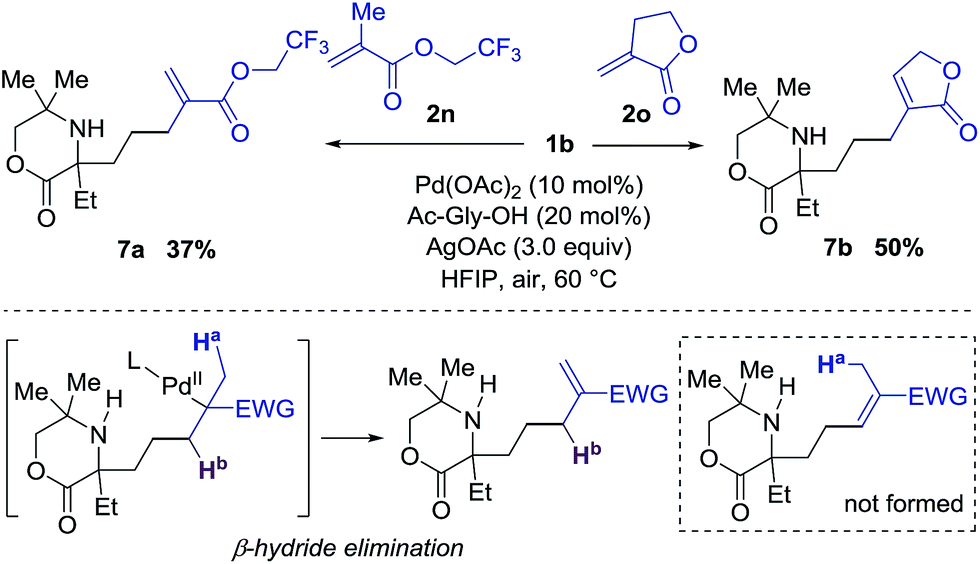
Interestingly, when di-substituted acrylates 2n and 2o were employed in the reaction, the alkenyl products 7a and 7b were obtained in moderate yields with no trace of the corresponding aza-Michael products (Scheme 1). These results infer that the β-hydride elimination step takes place in a different sense to the reaction with the simple acrylates shown in Table 2, and that aza-Michael addition to the corresponding azocene is presumably disfavored in comparison to the stability of the alkene product.
 | ||
| Scheme 1 Reaction of di-substituted acrylates. | ||
Preliminary mechanistic investigations
Although the amino-acid-derived ligand Ac-Gly-OH 6a is not essential in this C–H alkenylation reaction, its presence significantly improves the conversion and yield of product for some examples (5b, 5c, 5k, 5l, 5v, 5x, 5z, and 5aa), which leads to an expanded substrate scope. Given the impact of ligand 6a on the outcome of the catalytic C–H alkenylation reaction and its ability to enable reversible aliphatic C–H bond activation (Fig. 3), we conducted preliminary experiments towards better understanding the mechanism of the new reaction.Firstly, we confirmed that the reaction proceeds through an amine-directed C–H bond activation step. Following the procedure in Fig. 3, treatment of morpholinone 1b with 1.5 equivalents of Pd(OAc)2 delivered the anticipated cyclopalladation complex int-V after stirring at 60 °C with CHCl3 as solvent. The structure of this palladacycle int-V was identified by analysis of the X-ray diffraction pattern of a single crystal, confirming that it was the amine motif that was directing the C–H bond cleavage rather than the lactone function. Further reaction with ethyl acrylate 2a from this palladacycle int-V provided the corresponding pyrrolidine 3b in 65% yield (Scheme 2).
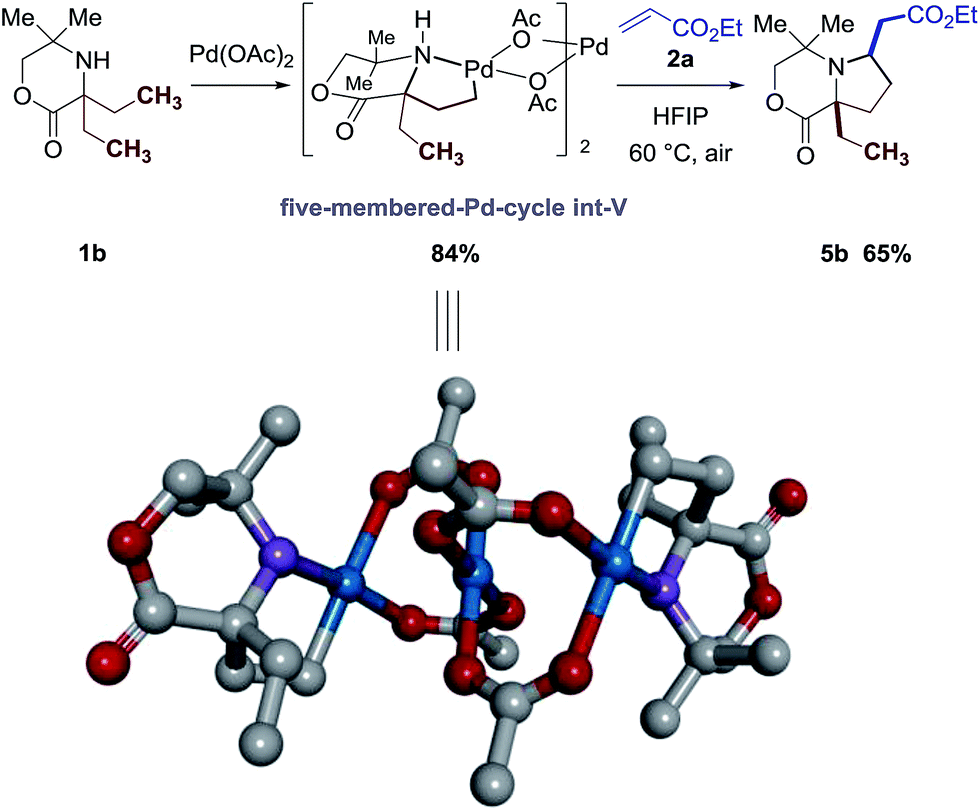 | ||
| Scheme 2 X-ray crystal structure of the amine-palladacycle int-V (hydrogens are removed for clarity) and its stoichiometric reaction with acrylate 2a. | ||
We next investigated some of the basic kinetic parameters of the reaction. Three types of kinetic isotope effect (KIE) experiments were explored with and without the ligand under catalytic conditions.12 First, two parallel reactions were carried out to measure the rate constants, one with 1b fully protiated at the reactive ethyl positions and one with d10-1b fully deuterated (Fig. 4a). In the presence of ligand Ac-Gly-OH 6a, the reaction gave rise to a kH/kD value of 1.02. The lack of a kinetic isotope effect rules out C–H bond cleavage occurring during the turnover-limiting step. Meanwhile, a small isotope effect kH/kD = 1.16 was observed in the absence of ligand, which indicated that C–H bond cleavage is not the turnover-limiting step either. Second, same-flask intermolecular competitive reaction of 1b and d10-1b was investigated (Fig. 4b). In the absence of ligand, a larger primary isotope effect was observed ([PH]/[PD] = 3.00); while in the presence of ligand, a smaller potential isotope effect was observed ([PH]/[PD] = 1.50). Third, reaction of d5-1b containing one protiated ethyl group and one deuterated ethyl group with 2a was also investigated (Fig. 4c). Similar results were obtained that in the absence of ligand, a larger primary isotope effect was observed ([PH]/[PD] = 2.41); while in the presence of ligand, a smaller potential isotope effect was observed ([PH]/[PD] = 1.42). These KIE studies indicate that the C–H bond cleavage step is irreversible without ligand, and occurs after a rate-determining step (possibly the dissociation of the bis-amine Pd(II) complex to the active mono-amine Pd(II) complex int-VI); the loss of AcOH from the Pd(II) complex during the C–H activation step (Fig. 4d) is likely to be the reason why the C–H cleavage step is irreversible. In the presence of ligand, however, the process could begin with amino acid coordination to the Pd(II) catalyst followed by association of one molecule of amine substrate to form int-VII. The N-Ac group serves the role of inner-sphere base need for the C–H bond cleavage step.13 The reversible nature of this step could be explained by the retention of the ligated protonated amide in proxinity to the carbon–Pd bond, facilitating re-protonation (Fig. 4d, see ESI Fig. S2† for a detailed mechanism with and without ligand). Taken together, these results are consistent with the observations of the selective C–H alkenylation of 1a and stoichiometric studies highlighted in Fig. 3.
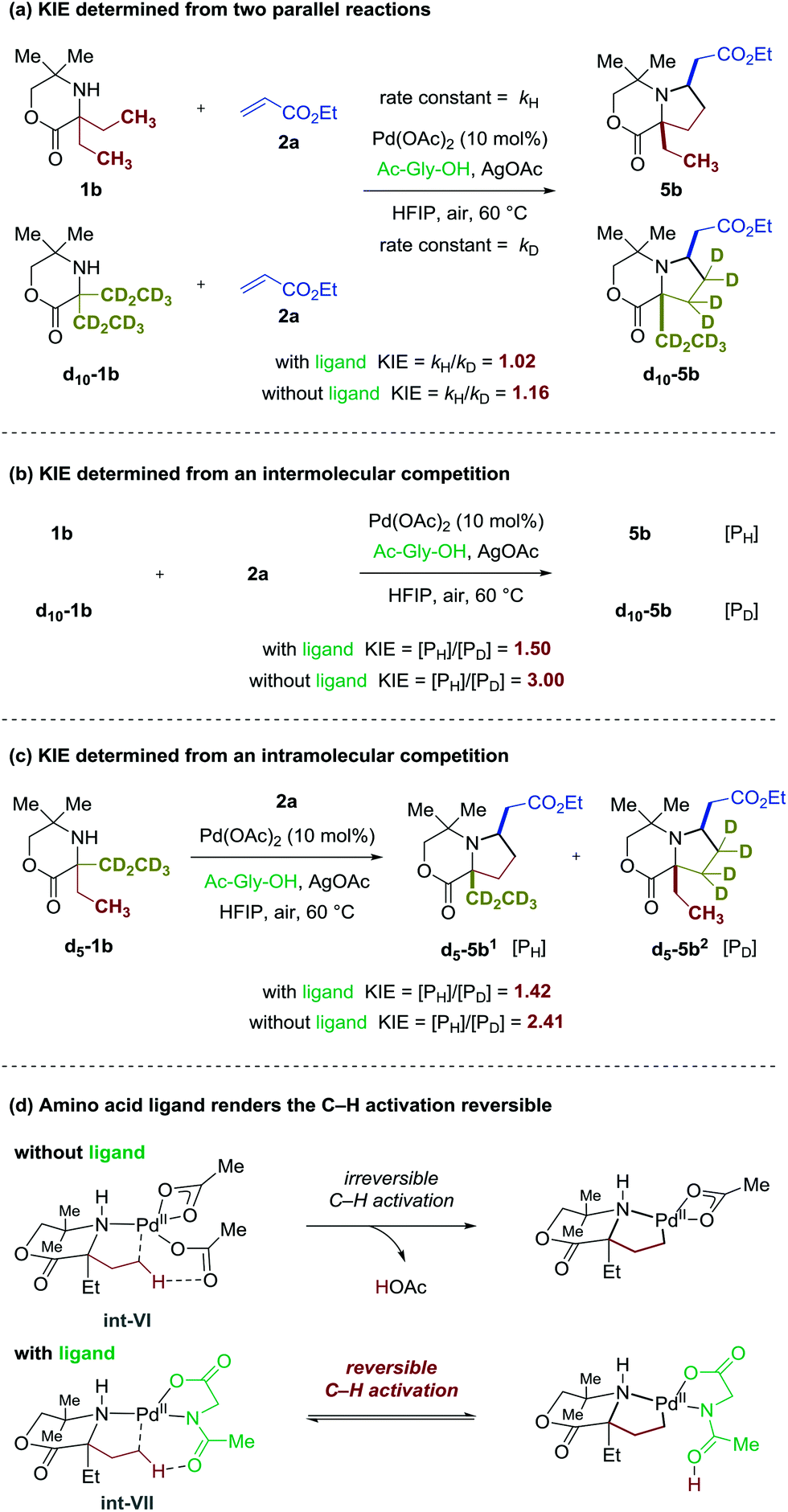 | ||
| Fig. 4 KIE studies of the catalytic C–H alkenylation. | ||
Derivatization of the C–H alkenylation products
Finally, we sought to explore the scaffold elaboration of the products from this new C–H alkenylation process. To demonstrate the efficacy of sequential C–H bond functionalization strategies, we showed that 1b can be selectively functionalized using three step process involving two C–H activation steps. Firstly, catalytic C–H amination through a 4-membered-ring cyclopalladation pathway forms the aziridine 3a; secondly, ring opening of the aziridine provides a functionalized secondary amine 1d;3a and finally, C–H alkenylation through a 5-membered-ring cyclopalladation pathway gives the highly functionalized pyrrolidine derivative 8 (Fig. 5a). To further demonstrate the synthetic versatility of these products, the bicyclic aliphatic heterocycles can be transferred to a series of highly functionalized α-quaternary proline derivatives (Fig. 5b). Reducing the lactone 5z with DIBAL-H followed by the treatment with trifluoroacetic acid (TFA) cleaves the cyclic framework and affords the prolinol product 9 in 91% yield. Further oxidations under appropriate conditions provide the corresponding aldehyde 10 and amino acid proline derivative 11 in good yields.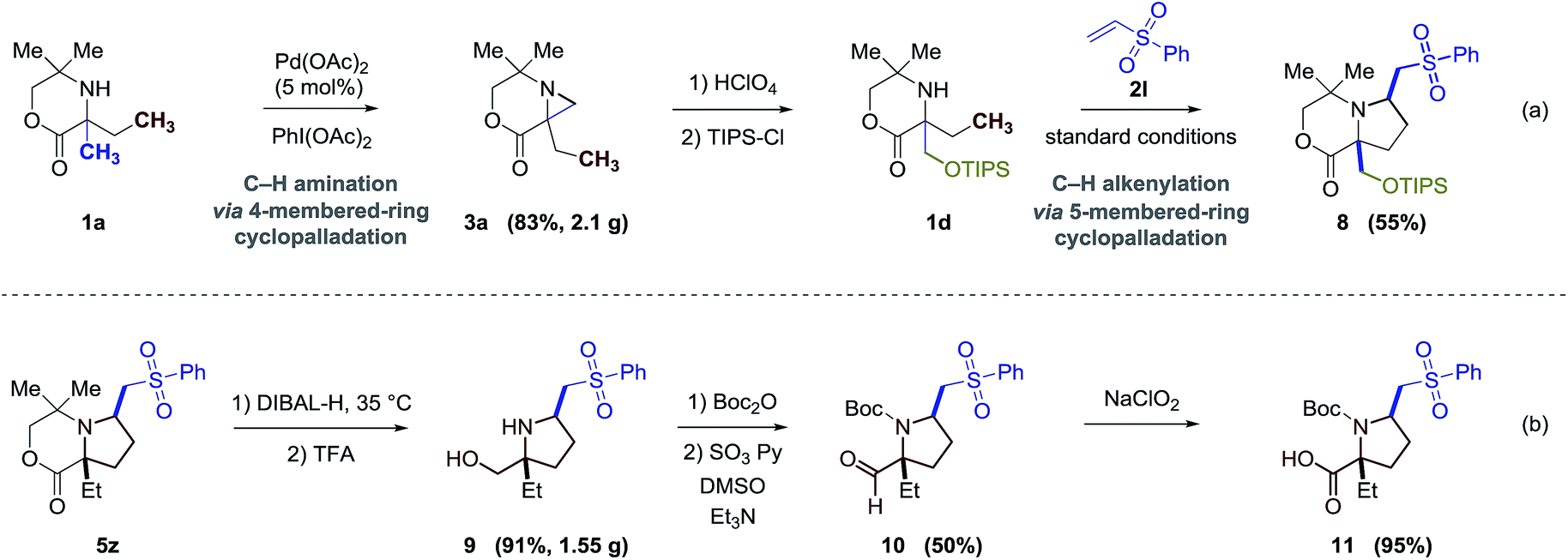 | ||
| Fig. 5 Synthetic applications. | ||
Conclusions
In summary, we have developed a palladium-catalyzed C–H alkenylation of aliphatic amines. This alkenylation reaction proceeds through a 5-membered-ring cyclopalladation pathway that allows alkene insertion and aza-Michael cyclization for the synthesis of various pyrrolidine moieties in good yields with perfect regio- and diastereo-selectivity. The process transforms readily available aliphatic amines into highly functionalized complex aliphatic heterocycles that we believe will prove attractive to practioners of medicinal chemistry as novel scaffolds. We also found that an amino-acid-derived ligand changes the rate-limiting step and enables a reversible aliphatic C–H bond activation, which leads to an expanded substrate scope to be developed on this C–H bond alkenylation. Finally, some non-racemic amino acid-derived ligands were also assessed for the induction of enantioselectivity to this transformation (see ESI Fig. S3†). Although only 12% ee was obtained with Fmoc-(L)-Phe-OH, we believe it is a viable starting point for further development.Acknowledgements
We are grateful to EPSRC (EP/100548X/1 for C. H. & M. J. G.), ERC (ERC-STG-259711 to M. J. G.) and the Marie Curie Foundation (C. H.) for funding. Mass spectrometry data were acquired at the EPSRC UK National Mass Spectrometry Facility at Swansea University. We are also grateful to Dr Andrew Bond for X-ray crystal structure analysis.Notes and references
- (a) R. N. Salvatore, C. H. Yoon and K. W. Jung, Tetrahedron, 2001, 57, 7785–7811 CrossRef CAS; (b) E. W. Baxter and A. B. Reitz, Org. React., 2002, 59, 1–714 CAS; (c) J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC; (d) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed; (e) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed; (f) Y. Yang, S.-L. Shi, D. Niu, P. Liu and S. L. Buchwald, Science, 2015, 349, 62–66 CrossRef CAS PubMed; (g) J. Gui, C.-M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. La Cruz, M. A. Schmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Science, 2015, 348, 886–891 CrossRef CAS PubMed; (h) C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed; (i) F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed.
- (a) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed; (b) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed; (c) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (d) S.-Y. Zhang, G. He, W. A. Nack, Y. Zhao, Q. Li and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124–2127 CrossRef CAS PubMed; (e) N. Rodriguez, J. A. Romero-Revilla, M. A. Fernandez-Ibanez and J. C. Carretero, Chem. Sci., 2013, 4, 175–179 RSC; (f) X. Ye, Z. He, T. Ahmed, K. Weise, N. G. Akhmedov, J. L. Petersen and X. Shi, Chem. Sci., 2013, 4, 3712–3716 RSC; (g) M. Fan and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 12152–12155 CrossRef CAS PubMed; (h) C. Wang, C. Chen, J. Zhang, J. Han, Q. Wang, K. Guo, P. Liu, M. Guan, Y. Yao and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 9884–9888 CrossRef CAS PubMed; (i) K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J.-Q. Yu, Nat. Chem., 2014, 6, 146–150 CrossRef CAS PubMed; (j) J. J. Topczewski, P. J. Cabrera, N. I. Saper and M. S. Sanford, Nature, 2016, 531, 220–224 CrossRef CAS PubMed; (k) J.-W. Xu, Z.-Z. Zhang, W.-H. Rao and B.-F. Shi, J. Am. Chem. Soc., 2016, 138, 10750–10753 CrossRef CAS PubMed; (l) Z. Huang, C. Wang and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 5299–5303 CrossRef CAS PubMed; (m) Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554–14557 CrossRef CAS PubMed; (n) Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26–32 Search PubMed.
- (a) A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129–133 CrossRef CAS PubMed; (b) A. P. Smalley and M. J. Gaunt, J. Am. Chem. Soc., 2015, 137, 10632–10641 CrossRef CAS PubMed; (c) C. He and M. J. Gaunt, Angew. Chem., Int. Ed., 2015, 54, 15840–15844 CrossRef CAS PubMed; (d) J. Calleja, D. Pla, T. W. Gorman, V. Domingo, B. Haffemayer and M. J. Gaunt, Nat. Chem., 2015, 7, 1009–1016 CrossRef CAS PubMed; (e) J. Zakrzewski, A. P. Smalley, M. A. Kabeshov, M. J. Gaunt and A. A. Lapkin, Angew. Chem., Int. Ed., 2016, 55, 8878–8883 CrossRef CAS PubMed; (f) D. Willcox, B. G. N. Chappell, K. F. Hogg, J. Calleja, A. P. Smalley and M. J. Gaunt, Science, 2016, 354, 851–857 CrossRef CAS PubMed.
- (a) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed; (b) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (c) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC; (d) D. Y. K. Chen and S. W. Youn, Chem.–Eur. J., 2012, 18, 9452–9474 CrossRef CAS PubMed.
- (a) I. Moritani and Y. Fujiwara, Tetrahedron Lett., 1967, 8, 1119–1122 CrossRef; (b) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633–639 CrossRef CAS PubMed.
- (a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (b) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318–5365 CrossRef CAS PubMed; (c) E. M. Beck and M. J. Gaunt, Top. Curr. Chem., 2010, 292, 85–121 CrossRef CAS PubMed; (d) M. Miura, T. Tsuda, T. Satoh, S. Pivsa-Art and M. Nomura, J. Org. Chem., 1998, 63, 5211–5215 CrossRef CAS; (e) M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586–1587 CrossRef CAS PubMed; (f) G. Cai, Y. Fu, Y. Li, X. Wan and Z. Shi, J. Am. Chem. Soc., 2007, 129, 7666–7673 CrossRef CAS PubMed; (g) S. H. Cho, S. J. Hwang and S. Chang, J. Am. Chem. Soc., 2008, 130, 9254–9256 CrossRef CAS PubMed; (h) J.-J. Li, T.-S. Mei and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452–6455 CrossRef CAS PubMed; (i) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315–319 CrossRef CAS PubMed; (j) Y. Zhao, G. He, W. A. Nack and G. Chen, Org. Lett., 2012, 14, 2948–2951 CrossRef CAS PubMed; (k) A. Deb, S. Bag, R. Kancherla and D. Maiti, J. Am. Chem. Soc., 2014, 136, 13602–13605 CrossRef CAS PubMed; (l) Q. Wang, J. Han, C. Wang, J. Zhang, Z. Huang, D. Shi and Y. Zhao, Chem. Sci., 2014, 5, 4962–4967 RSC.
- (a) A. D. Ryabov, Chem. Rev., 1990, 90, 403–424 CrossRef CAS; (b) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed; (c) K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (e) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (f) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (g) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (h) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764–7786 RSC; (i) F. Mo, J. R. Tabor and G. Dong, Chem. Lett., 2014, 43, 264–271 CrossRef CAS.
- (a) B.-F. Shi, Y.-H. Zhang, J. K. Lam, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 460–461 CrossRef CAS PubMed; (b) S. Li, G. Chen, C.-G. Feng, W. Gong and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 5267–5270 CrossRef CAS PubMed.
- Examples of C(sp3)–H alkenylation using alkenes: (a) M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3680–3681 CrossRef CAS PubMed; (b) K. J. Stowers, K. C. Fortner and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 6541–6544 CrossRef CAS PubMed; (c) H. Jiang, J. He, T. Liu and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 2055–2059 CrossRef CAS PubMed; (d) W. Yang, S. Ye, Y. Schmidt, D. Stamos and J.-Q. Yu, Chem.–Eur. J., 2016, 22, 7059–7062 CrossRef CAS PubMed; examples of C(sp3)–H alkenylation using vinyl halides: (e) G. He and G. Chen, Angew. Chem., Int. Ed., 2011, 50, 5192–5196 CrossRef CAS PubMed; (f) W. R. Gutekunst, R. Gianatassio and P. S. Baran, Angew. Chem., Int. Ed., 2012, 51, 7507–7510 CrossRef CAS PubMed; (g) B. Wang, C. Lu, S.-Y. Zhang, G. He, W. A. Nack and G. Chen, Org. Lett., 2014, 16, 6260–6263 CrossRef CAS PubMed; (h) Y.-J. Liu, Z.-Z. Zhang, S.-Y. Yan, Y.-H. Liu and B.-F. Shi, Chem. Commun., 2015, 51, 7899–7902 RSC; (i) G. Shan, G. Huang and Y. Rao, Org. Biomol. Chem., 2015, 13, 697–701 RSC.
- (a) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927–8955 CrossRef CAS PubMed; (b) K. M. Engle, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137–14151 CrossRef CAS PubMed; (c) K. M. Engle, P. S. Thuy-Boun, M. Dang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 18183–18193 CrossRef CAS PubMed; (d) D.-W. Gao, Y.-C. Shi, Q. Gu, Z.-L. Zhao and S.-L. You, J. Am. Chem. Soc., 2013, 135, 86–89 CrossRef CAS PubMed; (e) Z.-J. Du, J. Guan, G.-J. Wu, P. Xu, L.-X. Gao and F.-S. Han, J. Am. Chem. Soc., 2015, 137, 632–635 CrossRef CAS PubMed.
- Acyclic substrates (shown below) were unsuccessful when tested under the optimal conditions.
-
. - E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and kinetic details. CCDC 1449203. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00468k |
| This journal is © The Royal Society of Chemistry 2017 |