
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSemiconducting polymer dots with bright narrow-band emission at 800 nm for biological applications†
Dandan
Chen‡
 ac,
I-Che
Wu‡
b,
Zhihe
Liu
c,
Ying
Tang
c,
Haobin
Chen
c,
Jiangbo
Yu
b,
Changfeng
Wu
*a and
Daniel T.
Chiu
*b
ac,
I-Che
Wu‡
b,
Zhihe
Liu
c,
Ying
Tang
c,
Haobin
Chen
c,
Jiangbo
Yu
b,
Changfeng
Wu
*a and
Daniel T.
Chiu
*b
aDepartment of Biomedical Engineering, Southern University of Science and Technology, Shenzhen, Guangdong 510855, China. E-mail: wucf@sustc.edu.cn
bDepartment of Chemistry and Bioengineering, University of Washington, Seattle, Washington 98195, USA. E-mail: chiu@chem.washington.edu
cState Key Laboratory on Integrated Optoelectronics, College of Electronic Science and Engineering, Jilin University, Changchun, Jilin 130012, China
First published on 1st March 2017
Abstract
The development of near-infrared (NIR) fluorescent probes is critical for in vivo exploration of the fundamental and complex processes in living systems by noninvasive fluorescence imaging techniques. Semiconducting polymer dots (Pdots) are emerging as important probes that exhibit several advantages, such as high brightness and biocompatibility. However, NIR-emitting Pdots are very rare, particularly at the center (∼800 nm) of the first optical window of biological tissues (between 650 nm and 950 nm). In this paper, we describe the synthesis of a semiconducting polymer with bright and narrow-band emission at 800 nm. The polymer was designed by incorporating a NIR porphyrin unit into a conjugated backbone; the polymer used a cascade energy transfer to produce the signal. The resulting Pdots possessed a narrow emission bandwidth (FWHM ∼ 23 nm) and good fluorescence quantum yield (QY = 8%), which is high for a near-IR emitter. The Pdots were bioconjugated with streptavidin for specific labeling of cellular targets, yielding higher staining index when compared with commercial NIR probes, such as PE-Cy7. Moreover, the NIR polymer was combined with a long-wavelength absorbing polymer to make bright Pdots (QY = 15%) for in vivo noninvasive imaging. These NIR Pdots with surface PEGylation led to high-contrast imaging of lymph nodes and tumors in a mouse model. This work highlights the great potential of the NIR Pdots for cellular and in vivo imaging applications.
1. Introduction
Fluorescence imaging techniques are effective tools to noninvasively investigate many fundamental and complex biological processes because they have high signal-to-noise ratios and excellent spatial and temporal resolution.1,2 The potential of a fluorescence technique is significantly dependent on the fluorophore's property. Thus the development of novel fluorescent probes is of vital importance for further improvement of many fluorescence-based techniques. For in vivo biological applications, near-infrared (NIR) fluorescent probes are highly desirable because of minimal auto-fluorescence interference and high tissue-penetration depth in NIR region due to the weak absorption and scattering of biological species.3–5 The most widely used NIR fluorophores are small-molecule organic dyes (e.g., ICG, Cy7, Atto 740 and Alexa 750).6 However, low brightness, small Stokes shift and limited photostability of these NIR dyes are limiting factors for practical biological imaging applications.Among the strides made in nanotechnology, inorganic semiconductor quantum dots (Qdots) with unique size-tunable emission were introduced to biological studies nearly 20 years ago as fluorescent probes.7–9 However, the release of heavy metal ions is a significant concern for living systems. Alternatively, organic dyes can be doped into nanoparticles such as silica and latex beads,10–13 which exhibit improved brightness and photostability when compared with molecular dyes alone. In certain cases, nanoparticles with some size range and surface functional groups are promising for applications, such as for sentinel lymph nodes (SLNs) mapping, because they have a long blood-circulation time.14–18 However, the usefulness of dye-loaded nanoparticles is severely limited by the low fluorophore concentration and possible dye leaching problems.
Semiconducting polymer dots (Pdots) are emerging as organic fluorescent nanoparticles with high brightness, large Stokes shift, and intriguing energy-transfer properties.19–21 These polymeric semiconductors were originally developed for optoelectronic devices,22–24 and later used for highly sensitive chemical sensors.25,26 In the past decade, the adaptation of semiconducting polymers as fluorescent probes has received considerable attention in biology and medicine.27,28 A large variety of Pdots have been demonstrated for applications such as cellular labeling and tracking,29–33 photodynamic and photothermal cancer therapy,34–36 drug delivery,37,38in vivo optical imaging,20,39 photoacoustic imaging,40–45 and molecular detection.46–48 In contrast with inorganic nanoparticles, one prominent advantage of Pdots is that their optical properties are highly tunable because new polymer structures can be synthesized or small molecular fluorophores can be doped into them.49–51 For the latter case, semiconducting polymers as the nanoparticle matrix efficiently absorb light and transfer the energy to the dopant fluorophore, yielding amplified fluorescence when compared with small molecules. Recently, there are major efforts to develop NIR-emitting Pdots for biological applications. Zhang et al. synthesized BODIPY-based Pdots and achieved bright narrow-band emission at ∼720 nm by cascade energy transfer.52 We reported a squaraine-based semiconducting polymer that exhibited efficient NIR narrow-band fluorescence (∼704 nm).21 Chan et al. developed a few Pdot species that extended the emission range to far red and NIR region.53 Pu et al. demonstrated multilayered polymer dots for efficient NIR emission.54 Despite these progresses, the in vivo performance of current Pdots are far from ideal because both the absorption and emission are far away from the center wavelength (∼800 nm) of the first optical window of biological tissue (between ∼650 nm and 950 nm).
This paper describes the design and synthesis of a NIR semiconducting polymer that emits at 800 nm with high quantum yield, narrow emission bandwidth, and a large Stokes shift. The corresponding Pdots possessed a large absorption cross-section at 488 nm, so they had the potential to work as an excellent fluorescent probe for flow cytometry analysis. Quantitative flow-cytometry results indicated the NIR Pdots exhibit a higher stain index when compared with the commercial NIR probe PE-Cy7 in the specific labeling of cellular targets. Moreover, the NIR800 polymer was combined with a semiconducting polymer with a wide absorption band from 500 to 600 nm, yielding highly fluorescent Pdots for in vivo imaging applications. We showed that the PEG-modified Pdots via tail-vein injection in a mouse model had long blood-circulation time and led to high-contrast imaging of lymph nodes and tumors. These results indicated that the NIR800 Pdots are promising for a broad range of in vitro and in vivo bioimaging applications.
2. Results and discussion
2.1. Design and synthesis of NIR800 polymer
For in vivo applications, we were inspired to synthesize a semiconducting polymer that emitted at the center wavelength (∼800 nm) of the first optical window of biological tissue. The polymer was designed as a cascade energy transfer system that comprises of multiple monomeric units. The chemical structures of the five monomers are shown in Scheme 1. Among those, M1 is made from a porphyrin fluorophore (NIR775) that shows favorable absorption and fluorescence in NIR region. NIR775 is a hydrophobic dye and has been doped into various conjugated polymers to prepare NIR-emitting Pdots.55–59 The close packing of the Pdots allows for energy transfer from the polymer donor to the molecular acceptor, yielding bright fluorescence from the NIR775 dye. However, the emission peak of the NIR775 dye (∼775 nm) is still a distance away from the center wavelength of 800 nm. Covering this distance is not trivial for multiplexed detection and in vivo imaging that typically use narrow bandpass filters to maximize the signal-to-noise ratio. Furthermore, as the dye is doped into the polymer matrix via physical association, there can be several issues, such as dye leaching or segregation, that can decrease the fluorescence of the doped Pdots. In addition, the energy transfer from conventional conjugated polymers to NIR 775 is typically restricted by their small spectral overlap.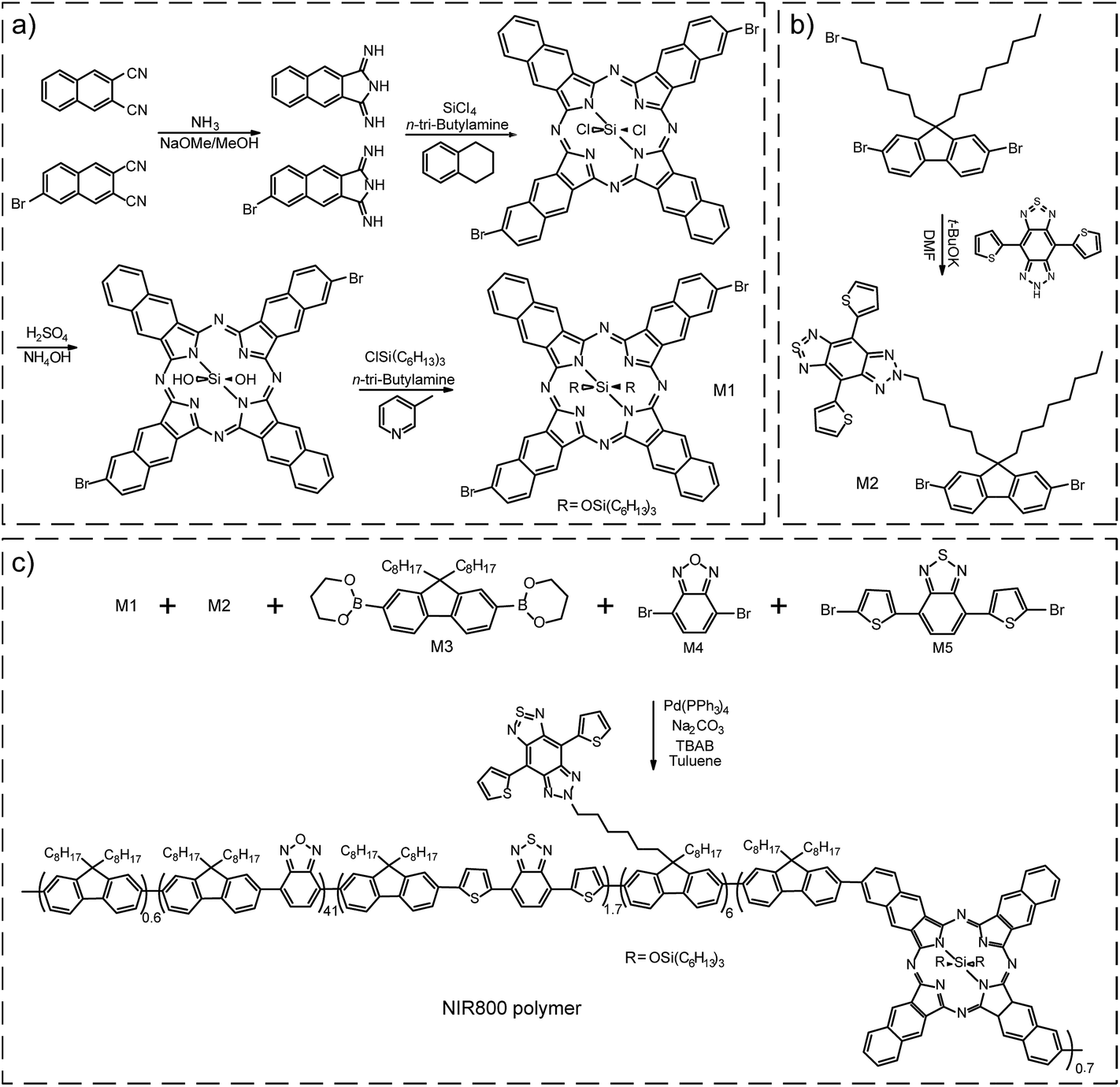 | ||
| Scheme 1 (a) Synthetic route of monomers M1. (b) Synthetic route of monomers M2. (c) Synthetic route of NIR800 polymer by Suzuki coupling reaction. | ||
Here, we intended to design a narrow-band emissive monomer based on NIR775 and then incorporate this unit into conjugated backbone to produce bright and narrow-band NIR emission. First, the conjugation length of NIR775 unit in the conjugated backbone was enlarged in comparison with the size of the molecular NIR775, which shifted the emission to the long wavelength region (∼800 nm). Second, the covalent incorporation overcame the troublesome issues, such as dye leaching and segregation, in the physical doping method. Third, the on-chain energy transfer to the NIR775 unit was more efficient as multiple pathways could occur even when the spectral overlap is negligible.21
For the NIR emissive unit, we synthesized the NIR775-Br monomer (M1) by a five-step reaction (Scheme 1a). According to a previous report,60 we used 1H-benzo[f]isoindole-1,3(2H)-diimine and 6-bromonaphthalene-2,3-dicarbonitrile as starting materials to form a bromo-substituted porphyrin ring and attached tri-hexylsilane chain to increase the solubility of the M1 unit. Nuclear magnetic resonance (NMR) results confirmed the successful synthesis of the M1 molecule (Fig. S1 and S2†). The M1 monomer exhibited an absorption peak at 772 nm and a fluorescence peak at 777 nm (Fig. S3†). The fluorescence quantum yield of M1 monomer in THF was ∼13%, and the full width at half maximum (FWHM) of the emission peak was ∼22 nm. These spectroscopic properties of the M1 monomer were similar to those of the NIR775 dye without bromine groups.
Besides the M1 emissive unit, several other monomers (M2–M5) were used in the polymer synthesis to obtain a large absorption coefficient in the visible region and facilitate the occurrence of cascade energy transfer to the M1 unit. The monomer 6-(6-(2,7-dibromo-9-octyl-9H-fluoren-9-yl)hexyl)-4,8-di(thiophen-2-yl)-6,7-dihydro-5H-[1,2,3]triazolo[4′,5′:4,5]benzo[1,2-c][1,2,5]thiadiazole, called M2, was synthesized according to a reaction previously described (Scheme 1b).61,62 The NMR spectrum of M2 was shown in Fig. S4.† The M2 monomer consisted of a chromophore in the side chain of the fluorene unit that had an absorption around 630 nm and an emission around 680 nm (Fig. S5†). In this polymer design, M2 served as an intermediate unit to maximize the cascade energy transfer.
M1 and M2 were copolymerized with three conventional monomer units, which were 9,9-dioctylfluorene-2,7-diboronic acid bis(1,3-propanediol)ester (M3), 4,7-dibromobenzo[c][1,2,5]oxadiazole (M4), and 4,7-bis(5-bromothiophen-2-yl)benzo[c][1,2,5]thiadiazole (M5). The polymerization was conducted via palladium-catalyzed Suzuki coupling to synthesize the NIR800 polymer (Scheme 1c). The molecular weight of the resulting polymer was determined to be ∼27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 652 daltons with a polydispersity of ∼2.9 by gel permeation chromatography. As shown in Fig. S6,† the absorption of the polymer in THF solution was consistent with the feeding ratio of the five monomers. The NIR800 polymer in THF solution showed a broad fluorescence band in visible region because of its major conjugated backbone structure. In the nanoparticle form, however, the same polymer could yield NIR narrow-band fluorescence because of the efficient intra- and inter-chain exciton diffusion and effective energy funneling to the M1 unit.
652 daltons with a polydispersity of ∼2.9 by gel permeation chromatography. As shown in Fig. S6,† the absorption of the polymer in THF solution was consistent with the feeding ratio of the five monomers. The NIR800 polymer in THF solution showed a broad fluorescence band in visible region because of its major conjugated backbone structure. In the nanoparticle form, however, the same polymer could yield NIR narrow-band fluorescence because of the efficient intra- and inter-chain exciton diffusion and effective energy funneling to the M1 unit.
2.2. Preparation and characterizations of NIR800 Pdots
The NIR800 polymer was used to prepare Pdots by the reprecipitation method.63 During the preparation, the polymer chains folded and collapsed because of hydrophobic interactions, resulting in stable aqueous solutions of nanometer-sized Pdots (Scheme 2). In contrast to the NIR775-doped Pdots, the NIR800 Pdots were directly and reliably produced from the precursor polymer, reducing the batch-to-batch variation that occurs with the dye doping method. This is particularly important because the dye doping concentration was typically low (∼1 wt%) to maintain a high quantum yield.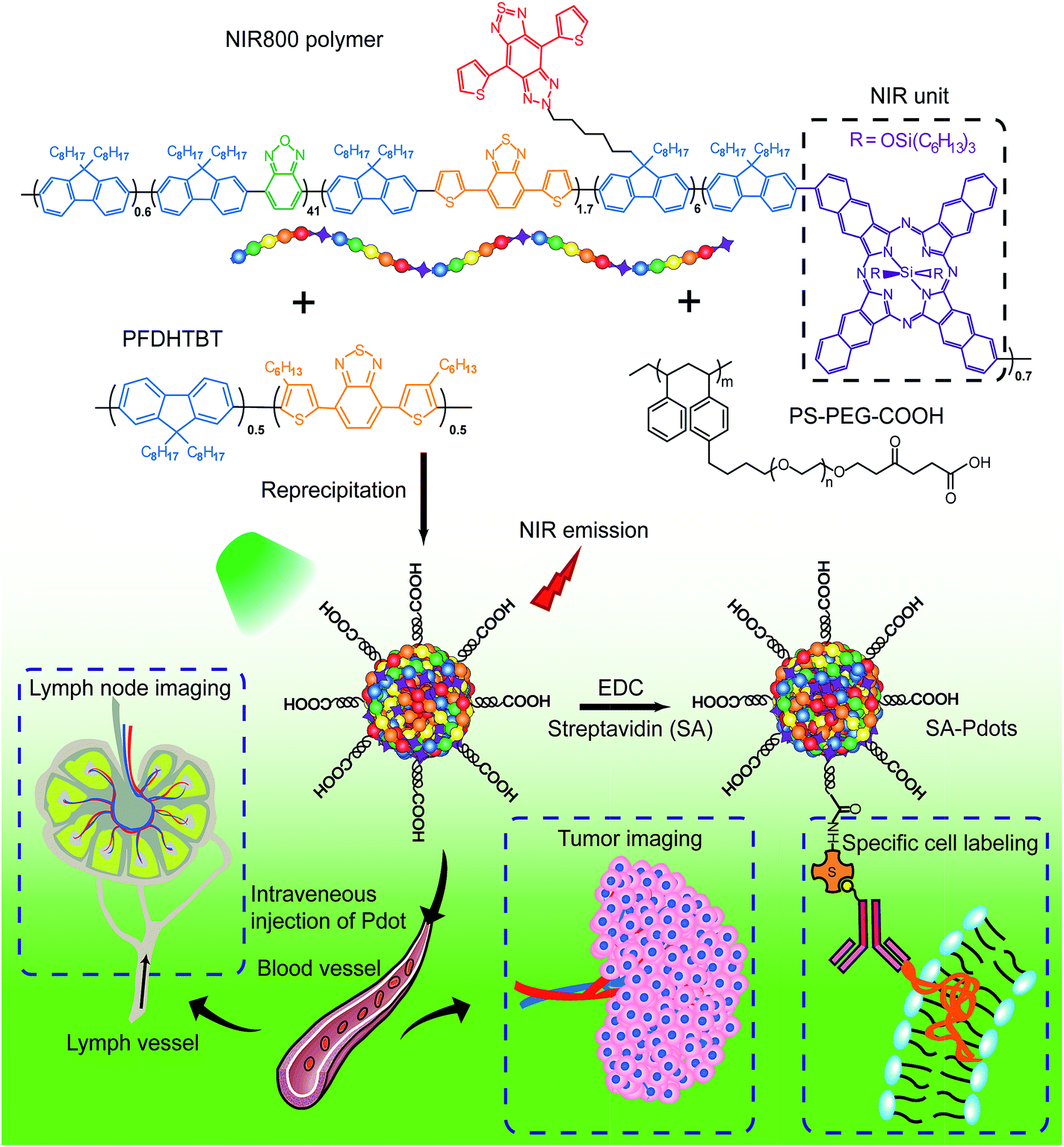 | ||
| Scheme 2 Schematic illustration of the preparation of NIR800 Pdots for specific cellular labeling and blended NIR800 Pdots for tumor and lymph-node imaging. | ||
The NIR800 Pdots exhibited spherical morphology with an average diameter of 16 nm, as determined by dynamic light scattering (DLS) and transmission electron microscopy (TEM) (Fig. 1a). The surface of Pdots was functionalized by blending a small portion (20 wt%) of poly (styrene maleic anhydride) (PSMA), yielding the zeta potential of −36.5 mV and abundant surface carboxyl group for subsequent bioconjugation. As shown by spectroscopic measurements (Fig. 1b), the absorption of the NIR800 Pdots was comparable to that of the precursor polymer, presenting a broad absorption spectrum from 400 nm to 600 nm because of the presence of multiple monomers (M2–M5). Thus, these Pdots were suitable as fluorescent probes for fluorescence imaging and flow cytometry applications. Notably, the Pdots exhibited narrow-band fluorescence with an emission peak at ∼800 nm and a FWHM of 23 nm, a quantum yield of ∼8% and a Stokes shift of ∼330 nm. The quantum yield of the Pdots was comparable to or higher than the values reported for good NIR fluorophores, such as indocyanine green (ICG, ∼2%),6 NIR7.0 (∼2.5%)64 and NIR775 (∼7%).55 Moreover, the emission band width (∼23 nm) was much narrower than those of NIR dyes and inorganic quantum dots. In addition, the per-particle brightness of the Pdots was more than two orders of magnitude higher than those of the small dye molecules because of the extraordinary absorption cross section.
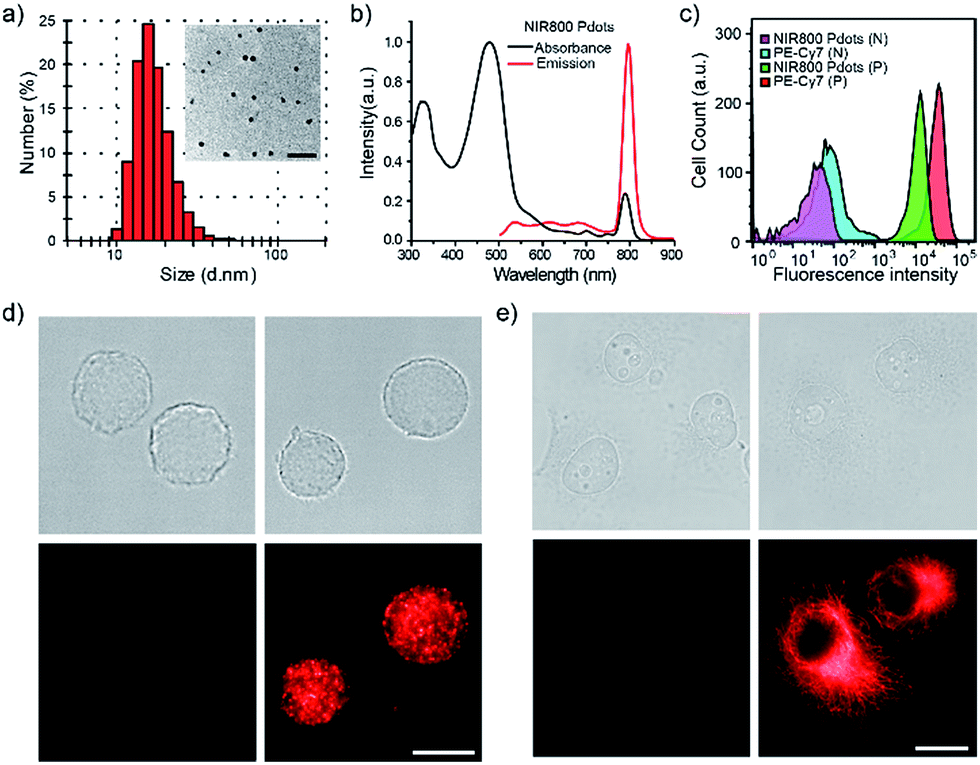 | ||
| Fig. 1 Characterizations of NIR800 Pdots and specific cell labeling by NIR800 Pdots. (a) Hydrodynamic diameter of the NIR800 Pdots. The inset shows a representative TEM image. The scale bar represents 100 nm. (b) Spectral character of the NIR800 Pdots. (c) Flow cytometry of MCF-7 cells labeled with NIR800 Pdots or PE-Cy7 dye via specific antigen–antibody and streptavidin–biotin interaction with nonspecific labeled cells as control. (d) Fluorescence imaging of cell-surface marker (EpCAM) in MCF-7 cells labeled with Pdot–streptavidin. The scale bar represents 20 μm. (e) Imaging of microtubules in MCF-7 cells labeled with Pdot–streptavidin. In d and e, the right panels show MCF-7 cells incubated sequentially with primary antibody and Pdot–streptavidin; the left panels show control samples in which the cells were incubated with Pdot–streptavidin alone (no primary antibody). The scale bar represents 20 μm. | ||
2.3. Bioconjugation and specific cellular labeling with NIR800 Pdots
Next, we explored the use of the NIR Pdots for specific cell-surface and subcellular labeling. The NIR800 Pdots with surface carboxyl groups was conjugated to streptavidin via the EDC-catalyzed coupling method. After conjugation with streptavidin, the size of NIR800 Pdots was increased from 16 nm to 26 nm in diameter and the zeta potential changed from −36 mV to −27 mV (Fig. S7†), indicating successful streptavidin conjugation on the Pdot surface. Then, MCF-7 cells were incubated with biotinylated, monoclonal anti-EpCAM antibodies followed by incubation with Pdot-bioconjugates (Scheme 2). Fig. 1c shows the flow-cytometry results of the MCF-7 cells labeled with Pdots in comparison with the commercial probe PE-Cy7. As seen from the figure, the Pdot-bioconjugates, combined with the primary antibody, were highly specific for the cell labeling in comparison to the control in the absence of the antibody.We evaluated the labeling performance of the NIR800 Pdots and compared it with that of PE-Cy7, which is a commonly used NIR fluorescent probe for flow cytometry. For each probe, the staining index was calculated by the ratio of the mean fluorescence intensity of positive population to the mean fluorescence intensity of negative population. The staining index of NIR800 Pdots was determined to be ∼138, which was significantly higher than the ∼88 obtained for PE-Cy7. However, the cell labeling brightness by Pdots was slightly lower than that of PE-Cy7. Fluorescent imaging further confirmed that the cells were specifically labeled by NIR800 Pdots, as the strong fluorescent signal was detected from the positively labeled cells (Fig. 1d). In addition, fluorescent imaging showed that the NIR800 Pdot–streptavidin could also specifically label microtubule structures in the presence of biotinylated anti-tubulin antibody (Fig. 1e). No fluorescent signal was observed from the negative-control samples in the absence of the antibody, confirming specific and effective cellular labeling without non-specific binding. Taken together, the above results demonstrate that the NIR Pdots were highly specific for labeling cellular and subcellular structures.
2.4. Preparation and characterization of blended NIR800 polymer dots
We use a polymer blending strategy to enhance the light absorption of the Pdots in the long wavelength region. The absorption of the NIR800 polymer is dominant in blue region, which is not suitable for in vivo imaging (Fig. 1b). We synthesized a semiconducting polymer PFDHTBT (chemical structure shown in Scheme 2), which exhibited strong absorption band in green region and broad emission from 600 nm to 800 nm (Fig. 2). By taking advantage of the efficient intra-particle energy transfer inside Pdots,65 we intended to shift the light-absorption wavelength and enhance the fluorescence quantum yield of the NIR800 polymer for in vivo imaging applications. The PFDHTBT as the donor and the NIR800 polymer as the acceptor showed reasonable spectral overlap (Fig. 2a), indicating that the energy transfer could occur via Förster mechanism.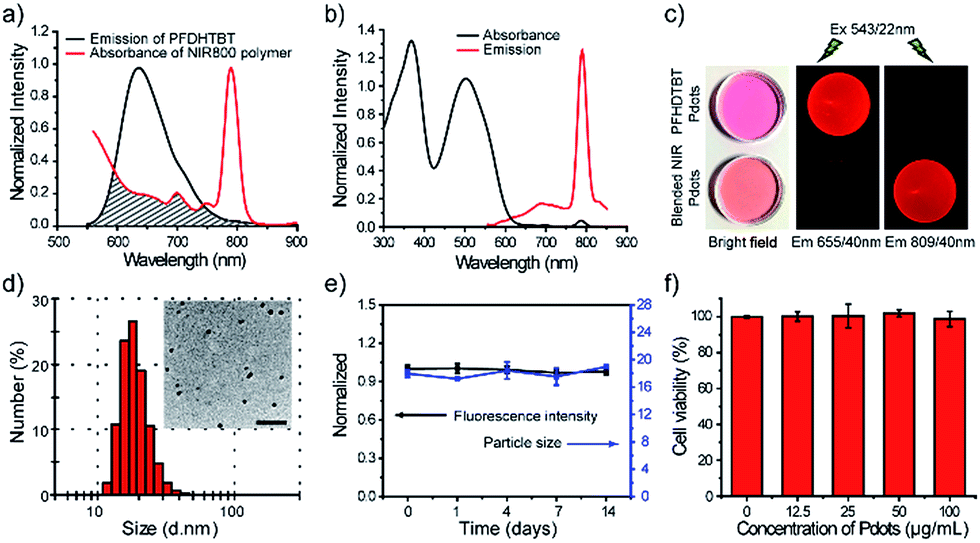 | ||
| Fig. 2 Characterizations of blended NIR800 Pdots. (a) Spectral overlap between the emission of PFDHTBT and the absorption of the NIR800 polymer. (b) Absorption and emission spectra of blended NIR800 Pdots. (c) Ambient-light and fluorescence images of PFDHTBT Pdots and the blended NIR800 Pdots under the same excitation, but different emission, channels using 655 nm and 809 nm band-pass filters, respectively. (d) Hydrodynamic diameter of the blended NIR800 Pdots. The inset shows a representative TEM image. The scale bar represents 100 nm. (e) Fluorescence stability of blended NIR800 Pdots in DMEM medium supplemented with 10% FBS and colloidal stability of blended NIR800 Pdots dispersed in water after continuous incubation at 37 °C. (f) Cell viability of MCF-7 cells incubated with different concentration of blended NIR800 Pdots for 24 hours. | ||
The blended Pdots consisting of the two polymers at variable fractions (0 to 30 wt%) were prepared by using reprecipitation method. As shown by the optical spectroscopy in Fig. S8,† the fluorescence of PFDHTBT was increasingly quenched with the increasing fraction of the NIR800 polymer. At the blending fraction of 25 wt%, the Pdots exhibited minimal fluorescence from the PFDHTBT polymer, but strong fluorescence from the NIR800 polymer, indicating the presence of efficient energy transfer from PFDHTBT to NIR800 polymer (Fig. 2b). As a result, the blended Pdots possessed a broad absorption band from 500 nm to 600 nm in comparison to the pure NIR800 Pdots. In particular, the blended Pdots exhibited a quantum yield (∼15%) for the narrow-band emission, which was apparently enhanced in comparison to the pure NIR800 Pdots (∼8%). The enhanced quantum yield was likely attributable to the reduced chain–chain aggregation of the NIR800 polymer in the blended Pdots. Fig. 2c showed the fluorescent images of aqueous solutions of PFDHTBT Pdots and blended NIR800 Pdots collected by a small-animal imaging system. The two samples at the same concentration were excited by the same light source and their emissions were collected by using 655/40 nm and 809/40 nm band-pass filters. The fluorescent signal from the NIR channel was comparable to the red channel. Single-particle imaging and intensity statistics were performed to compare the per-particle brightness of the blended NIR800 Pdots with Qdot800. As shown in Fig. S9,† the average brightness of Pdots was about two times higher than that of Qdot800.
Blood-circulation time is a key parameter for in vivo targeted delivery of imaging probes and drug molecules. Polyethylene glycol (PEG) groups are typically used for surface functionalization to mask the nanoparticles from the host's immune system and prolong their circulatory time.66–68 A functional polymer consisting of long-chain PEG groups (PS–PEG–COOH) was co-condensed with the polymer blends (PFDHTBT and NIR800) to prepare PEGylated NIR Pdots (Scheme 2). The PEGylated NIR800 Pdots exhibited spherical morphology with an average diameter of 18 nm, as determined by DLS and TEM (Fig. 2d). The zeta potential (−33 mV) of the PEG-Pdots was smaller than that of the PSMA-Pdots (−43 mV), most likely because of the presence of the PEG groups on the Pdot surface. The quantum yields of both the PSMA-Pdots and PEG-Pdots were determined to be ∼15%, indicating that the surface-functional groups did not affect fluorescence brightness of Pdots.
The stability of the NIR Pdots in biological media was evaluated. The Pdots were incubated with DMEM medium supplemented with 10% fetal bovine serum (FBS) at 37 °C. At designated time points, the fluorescence spectra of Pdots in medium and hydrodynamic diameter of Pdots dispersed in water were measured with QE-2100 spectrometer and dynamic light scattering instrument, respectively (Fig. 2e). From the data, we found the Pdots retained more than 95% of its initial fluorescence intensity after two-week storage at 37 °C. The diameter of Pdots is basically unchanged during two-week storage. Therefore, the Pdots is stable in biological environment. The cytotoxicity of NIR Pdots was evaluated on MCF-7 cells by measuring the cell viability with a colorimetric MTT assay. After the MCF-7 cells incubated with different concentrations of NIR 800 Pdots for 24 hours, cytotoxic effects were not observed for high Pdots concentrations, such as 100 μg mL−1 (Fig. 2f), indicating the minimal cellular toxicity of the Pdots in this concentration range. Therefore, the red-shifted absorption, high brightness, the narrow-band emission, and biocompatibility of the blended NIR Pdots will be useful for both fluorescence imaging and multiplexed applications.
2.5. Lymph nodes and tumor imaging by blended NIR800 Pdots
First, in vivo imaging of the PEGylated NIR800 Pdots was examined in health ICR mice that were administered with PEG-NIR Pdots through the tail vein. The mice receiving PSMA-NIR Pdots injection were also presented for comparative studies. Whole-body fluorescence images of the Pdots-injected mice were subsequently collected at designated time intervals. As shown in Fig. 3a, the PSMA-NIR Pdots were quickly accumulated into liver within a few minutes post-injection. The fluorescence intensity of liver reached maximum in 30 minutes post-injection, and then remained relatively steady for the rest of the time during the first day. Because of the high penetration depth of NIR fluorescence, the liver of mouse was clearly mapped across peritoneum. Fluorescent signals were hardly detected from the body of the mouse because of the combined effect of short blood circulation and quick accumulation of the PSMA-NIR Pdots in liver. In contrast, the PEG-NIR Pdots showed distinct blood-circulation kinetics. Fig. 3b shows whole-body fluorescence images of a mouse that received a PEG-NIR Pdots injection. As clearly seen from the figure, the fluorescence of liver reached maximum at a much longer time scale (∼24 hours) as compared to the PSMA-NIR Pdots (∼30 minutes), indicating the apparently prolonged blood circulation of PEG-NIR Pdots. Besides the liver, fluorescence was detected from the body because of the blood circulation as well as the strong fluorescent signal from several spots in the chest and flank. These strong fluorescent spots were attributed to the lymph nodes, as further confirmed by anatomical analysis.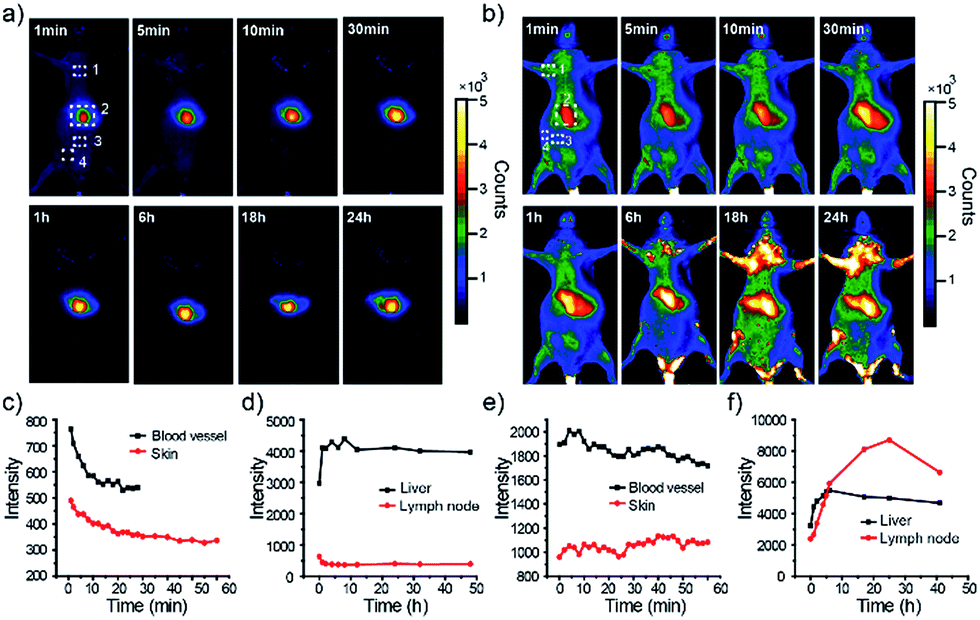 | ||
| Fig. 3 Dynamic imaging of live mice receiving tail-vein injection of PSMA-Pdots or PEG-Pdots. (a) Fluorescence imaging of the mouse intravenously injected with PSMA-NIR Pdots at designated time points. (b) Fluorescence imaging of the mouse intravenously injected with PEG-NIR Pdots at designated time points. The number 1, 2, 3, 4 in (a) and (b) represent lymph nodes, liver, blood vessel and skin ground, respectively. (c) Fluorescence intensity analysis of NIR Pdots in skin background and blood vessel of abdomen from PSMA-NIR Pdots injected mouse. (d) Fluorescence intensity analysis of NIR Pdots in liver and lymph nodes from PSMA-NIR Pdots injected mouse. (e) Fluorescence intensity analysis of NIR Pdots in skin background and blood vessel of abdomen from PEG-NIR Pdots injected mouse. (f) Fluorescence intensity analysis of NIR Pdots in liver and lymph nodes from PEG-NIR Pdots injected mouse. | ||
The whole-body fluorescence imaging enabled quantitative analysis of the pharmacokinetics of the Pdots in live mice. We monitored the fluorescence intensity changes in several locations on the mouse body, which corresponded to blood vessel, skin, lymph node, and liver. Fig. 3c and d show the intensity plots for the mice that received the PSMA-NIR Pdots injection while Fig. 3e and f are for the mice that received the PEG-NIR Pdots injections. As indicated by these results, the fluorescent signal from the skin of the mice injected with PSMA-NIR Pdots quickly decreased to the background level in one hour. However, for the mice injected with PEG-NIR Pdots, the fluorescent signal of skin was essentially unchanged for 2 days. Again, this significant difference is because of the long-chain PEG groups on the Pdots that helped to reduce adsorption of blood protein and prevented the Pdots from rapid clearance by the reticuloendothelial system.
Moreover, we observed strong fluorescent signals from the lymph nodes from the mice receiving the PEG-NIR Pdots injection. The signal increased over 24 hours and then began to decrease. After opening the skin of abdomen, the fluorescent signal of lymph nodes and liver were observed clearly (Fig. 4a). The ratio of the fluorescent signals from lymph nodes to that from the skin was about 10.5; the liver-to-skin fluorescence-signal ratio was about 3.8. The high signal-to-noise ratio makes the NIR Pdots a promising probe for in vivo fluorescence imaging.
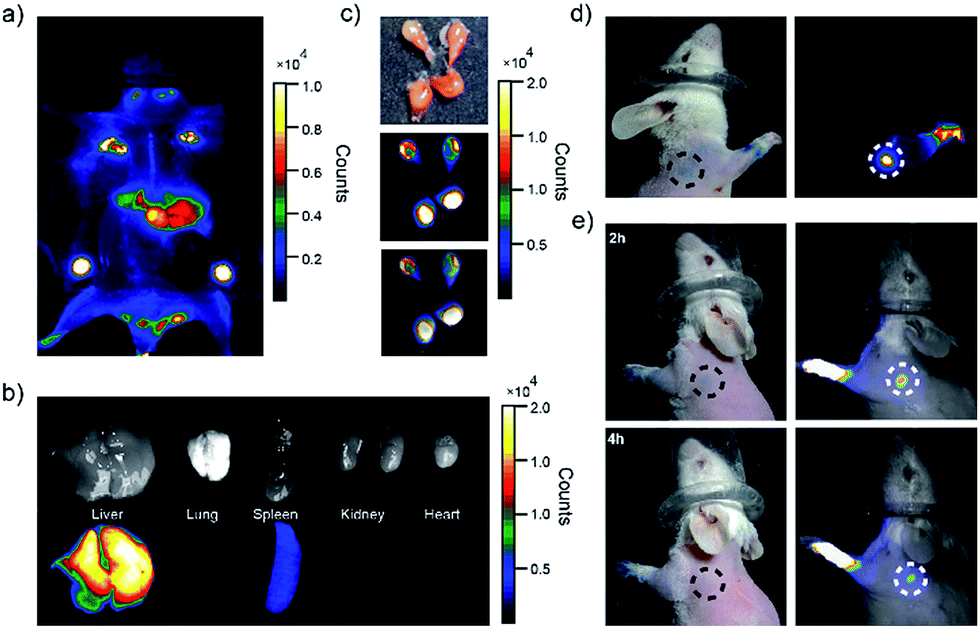 | ||
| Fig. 4 Biodistributions of PEG-NIR Pdots in ICR mice. (a) Whole-body imaging of mouse 24 hours after tail-vein injection of PEG-NIR Pdots. (b) Ambient-light images and fluorescent images of liver, lung, spleen, kidney and heart from the mouse injected with PEG-NIR Pdots. (c) Ambient-light images and fluorescence of lymph nodes from the mouse injected with PEG-NIR Pdots. (d) Images collected 5 min after injections of PEG-NIR Pdot (right) and methylene blue dye (left) into the right paws of the mouse. (e) Fluorescent images and ambient-light images collected 2 h and 4 h injection into the left paws of the mouse, respectively. | ||
The distribution of the PEG-NIR Pdots in different organs of the mice was investigated. As shown by fluorescence imaging of the organs collected from the mice (Fig. 4b and c), the Pdots were accumulated dominantly in liver, lymph nodes, and slightly in spleen. The strong fluorescence of the PEG-NIR Pdots in the lymph nodes reveals a promising application for SLN mapping as well as for precise surgical dissection of the lymph nodes.
A SLN is the first lymph node to receive lymphatic drainage from the site of a tumor. Therefore, a sentinel lymph node biopsy (SLNB) is very important for the detection of cancer metastasis.69–71 The feasibility of NIR Pdots as fluorescent probe for the detection of SLNs was evaluated by a comparative study with a conventional dye staining method. Methylene blue is widely used blue dye for staining SLNs.72 The Pdots and methylene blue dye were intradermally injected into the paws of mice to mimic the administration rout of SLNs contrast agents. The images were acquired by a small-animal imaging system 5 minutes post-injection. The images of the mouse injected PEG-NIR Pdots is shown in Fig. 4d. The high quantum yield and enhanced retention in lymph nodes of PEG-NIR Pdots allowed the lymph node and lymphatic vessels to be clearly seen. The signal-to-noise ratio of the lymph nodes imaged by the PEG-NIR Pdots was clearly superior to that by methylene blue. In addition, when the lymph node mapping was done 2 h and 4 h post-injection of PEG-NIR Pdots and methylene blue dye, the NIR signal from the Pdots was still clearly seen (Fig. 4e). However, the lymph node stained with methylene blue was hardly visible. As small-molecule dyes, methylene blue can quickly flow through the target area, so the time window for imaging is typically short. In contrast, PEG-NIR Pdots with larger diameters have favorable accumulation and retention in lymph nodes. Therefore, the high imaging sensitivity and long retention time in lymph nodes make the PEG-NIR Pdots a promising contrast agent for fluorescence-guided SLN mapping, allowing for real-time guidance throughout a surgery procedure.
Finally, we demonstrated using the PEGylated NIR Pdots for simultaneous tumor imaging and lymph-node mapping in tumor-bearing mice. The whole-body fluorescence imaging was conducted at designated time points in the mice after tail-vein injection of the Pdots. As indicated by the imaging (Fig. 5a), the fluorescent signal was first observed in the liver and lymph nodes, which was consistent with the imaging results of the tumor-free mice receiving PEGylated NIR Pdots. However, due to the long blood-circulation time and the enhanced permeability and retention effect of tumor, the Pdots were gradually accumulated in the tumor; the fluorescent signal reached its maximum at 24 h after the Pdots injection. The region of interest in the images showed that the NIR signal of the tumor, relative to that of the surrounding tissue, was 2.1 ± 0.3 (2 h), 3.1 ± 0.3 (8 h), 4.0 ± 0.3 (12 h) and 5.1 ± 0.6 (24 h) (Fig. 5b). The prominent retention of Pdots at tumor site is beneficial for tumor imaging and therapy. The Pdots have the potential to be used both as imaging probes and drug carriers.
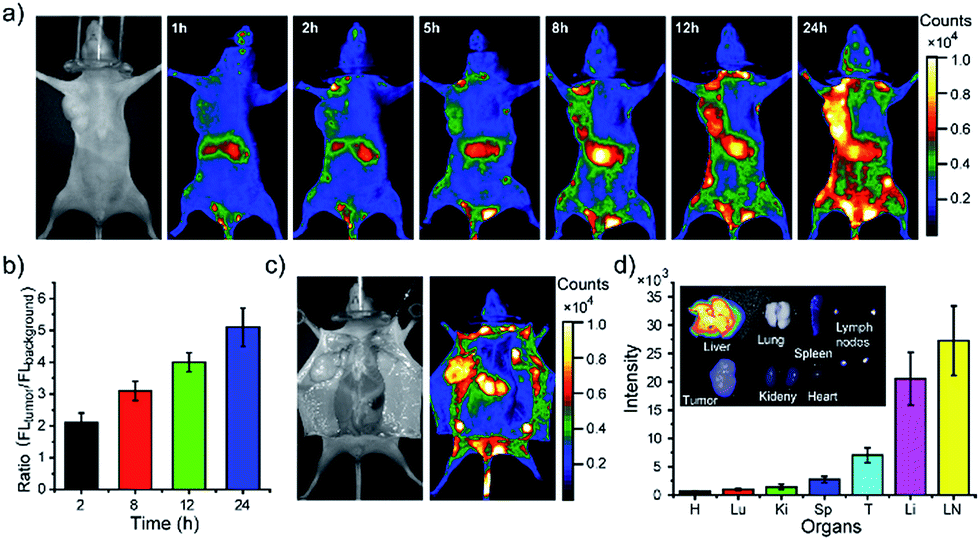 | ||
| Fig. 5 Simultaneous tumor imaging and lymph-node imaging of MCF-7 tumor-bearing nude mice with PEG-NIR Pdots. (a) Fluorescent images of mouse injected with PEG-NIR Pdots collected at designated time points. (b) Fluorescent signal ratio of tumor to background during 24 hour after Pdots injection. (c) Bright-field and fluorescent images of the same mouse opening of abdomen skin 24 hours post Pdot injection. (d) Quantitative fluorescence analysis of the Pdots in liver, lung, spleen, kidney, heart, tumor and lymph nodes (n = 3). Inset shows the overlap image of fluorescence image and bright-field image of liver, lung, spleen, kidney, heart, tumor and lymph nodes. | ||
After opening of the abdominal skin, strong fluorescent signals were detected and the margin of tumor and lymph nodes were clearly seen (Fig. 5c). This observation indicated the potential of the Pdots for image-guided surgery for tumor and SLN resection. The distribution of PEG-NIR Pdots was evaluated at 24 hours post tail-vein injection. The mice were euthanized and the organs, including heart, liver, spleen, lung, kidney, tumor and lymph nodes, were collected. Fluorescent images of Pdots in the organs were obtained by a small-animal imaging system. The fluorescent images showed that the injected PEG-NIR Pdots were mainly distributed to the liver, lymph nodes and tumor (Fig. 5d inset). The quantitative analysis of the Pdots' distribution was performed by analyzing the fluorescent signals of organs from Pdot-injected mice (n = 3). Normally, due to reticuloendothelial system's uptake of Pdots, the fluorescent signal is dominant in liver in comparison to the other organs. However, because of the increased blood-circulation time of the PEG-NIR Pdots in this case, the fluorescent intensity of lymph nodes was significantly higher than that of the liver; the fluorescent signal from the tumor was also enhanced (Fig. 5d). Therefore, the PEG-NIR Pdots, with their suitable size and high brightness in NIR region, have the potential to be important contrast agents for tumor and lymph-node imaging.
3. Conclusion
We synthesized a NIR800 semiconducting polymer by introducing multiple monomeric units into one polymer backbone via Suzuki coupling. The resulting Pdots exhibited a large absorption cross-section, a narrow emission peak (FMHW = 23 nm), and high quantum yields (QY = 8%). The Pdots were effectively excited with a 488 nm laser, making them suitable for flow cytometry and cellular imaging applications. We performed bioconjugation and specific cellular labeling using the streptavidin–Pdot conjugates. Flow cytometry results indicated that the Pdots showed higher staining index in comparison with the commercial probe PE-Cy7. Furthermore, the NIR800 polymer was blended with a long-wavelength absorbing polymer to produce Pdots (QY = 15%) for in vivo applications. The PEGylated Pdots exhibited long blood-circulation time and effectively accumulated to the lymph nodes and tumors, yielding high-contrast fluorescence mapping for precise surgery. These results indicate these NIR Pdots are promising probes for biological assays and contrast agents for clinical imaging applications.Acknowledgements
C.W. thanks the Key Program of National Natural Science Foundation of China (NSFC 61335001, 61222508) and Thousand Young Talents Program for support of this work, and D.T.C. acknowledges support from the National Institute of Health (CA186798) and the University of Washington. J. Yu and D. T. Chiu have financial interest in Lamprogen, which has licensed the described technology from the University of Washington.Notes and references
- S. Falkner, S. Grade, L. Dimou, K. K. Conzelmann, T. Bonhoeffer, M. Gotz and M. Hubener, Nature, 2016, 539, 248–253 CrossRef PubMed.
- Y. Shiba, T. Gomibuchi, T. Seto, Y. Wada, H. Ichimura, Y. Tanaka, T. Ogasawara, K. Okada, N. Shiba, K. Sakamoto, D. Ido, T. Shiina, M. Ohkura, J. Nakai, N. Uno, Y. Kazuki, M. Oshimura, I. Minami and U. Ikeda, Nature, 2016, 538, 388–391 CrossRef CAS PubMed.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- A. L. Vahrmeijer, M. Hutteman, J. R. van der Vorst, C. J. H. van de Velde and J. V. Frangioni, Nat. Rev. Clin. Oncol., 2013, 10, 507–518 CrossRef CAS PubMed.
- L. Yuan, W. Y. Lin, K. B. Zheng, L. W. He and W. M. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- W. C. Chan and S. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS PubMed.
- M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS PubMed.
- S. J. Rosenthal, J. C. Chang, O. Kovtun, J. R. McBride and I. D. Tomlinson, Chem. Biol., 2011, 18, 10–24 CrossRef CAS PubMed.
- C. L. O'Connell, R. Nooney and C. McDonagh, Biosens. Bioelectron., 2016, 91, 190–198 CrossRef PubMed.
- S. W. Bae, W. Tan and J. I. Hong, Chem. Commun., 2012, 48, 2270–2282 RSC.
- M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43, 4243–4268 RSC.
- L. Cerdán, E. Enciso, V. Martin, J. Bañuelos, I. López-Arbeloa, A. Costela and I. García-Moreno, Nat. Photonics, 2012, 6, 621–626 CrossRef.
- W. J. Akers, C. Kim, M. Berezin, K. Guo, R. Fuhrhop, G. M. Lanza, G. M. Fischer, E. Daltrozzo, A. Zumbusch, X. Cai, L. V. Wang and S. Achilefu, ACS Nano, 2010, 5, 173–182 CrossRef PubMed.
- Y. W. Noh, S. H. Kong, D. Y. Choi, H. S. Park, H. K. Yang, H. J. Lee, H. C. Kim, K. W. Kang, M. H. Sung and Y. T. Lim, ACS Nano, 2012, 6, 7820–7831 CrossRef CAS PubMed.
- S. De Koker, J. Cui, N. Vanparijs, L. Albertazzi, J. Grooten, F. Caruso and B. G. De Geest, Angew. Chem., Int. Ed., 2016, 55, 1334–1339 CrossRef CAS PubMed.
- S. Kim, Y. T. Lim, E. G. Soltesz, A. M. De Grand, J. Lee, A. Nakayama, J. A. Parker, T. Mihaljevic, R. G. Laurence, D. M. Dor, L. H. Cohn, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2004, 22, 93–97 CrossRef CAS PubMed.
- B. Ballou, L. A. Ernst, S. Andreko, T. Harper, J. A. J. Fitzpatrick, A. S. Waggoner and M. P. Bruchez, Bioconjugate Chem., 2007, 18, 389–396 CrossRef CAS PubMed.
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, ACS Nano, 2008, 2, 2415–2423 CrossRef CAS PubMed.
- C. Wu, S. J. Hansen, Q. Hou, J. Yu, M. Zeigler, Y. Jin, D. R. Burnham, J. D. McNeill, J. M. Olson and D. T. Chiu, Angew. Chem., Int. Ed., 2011, 50, 3430–3434 CrossRef CAS PubMed.
- I. C. Wu, J. Yu, F. Ye, Y. Rong, M. E. Gallina, B. S. Fujimoto, Y. Zhang, Y. Chan, W. Sun, X. H. Zhou, C. Wu and D. T. Chiu, J. Am. Chem. Soc., 2014, 137, 173–178 CrossRef PubMed.
- Q. Pei, G. Yu, C. Zhang, Y. Yang and A. J. Heeger, Science, 1995, 269, 1086 CrossRef CAS PubMed.
- C. D. Muller, A. Falcou, N. Reckefuss, M. Rojahn, V. Wiederhirn, P. Rudati, H. Frohne, O. Nuyken, H. Becker and K. Meerholz, Nature, 2003, 421, 829–833 CrossRef PubMed.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- J. S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864–11873 CrossRef CAS.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS PubMed.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- J. Yu, Y. Rong, C. T. Kuo, X. H. Zhou and D. T. Chiu, Anal. Chem., 2016, 89, 42–56 CrossRef PubMed.
- C. Wu, T. Schneider, M. Zeigler, J. Yu, P. G. Schiro, D. R. Burnham, J. D. McNeill and D. T. Chiu, J. Am. Chem. Soc., 2010, 132, 15410–15417 CrossRef CAS PubMed.
- G. Jin, D. Mao, P. Cai, R. Liu, N. Tomczak, J. Liu, X. Chen, D. Kong, D. Ding, B. Liu and K. Li, Adv. Funct. Mater., 2015, 25, 4263–4273 CrossRef CAS.
- X. Feng, G. Yang, L. Liu, F. Lv, Q. Yang, S. Wang and D. Zhu, Adv. Mater., 2012, 24, 637–641 CrossRef CAS PubMed.
- L. Feng, L. Liu, F. Lv, G. C. Bazan and S. Wang, Adv. Mater., 2014, 26, 3926–3930 CrossRef CAS PubMed.
- D. Ding, D. Mao, K. Li, X. Wang, W. Qin, R. Liu, D. S. Chiam, N. Tomczak, Z. Yang, B. Tang, D. Kong and B. Liu, ACS Nano, 2014, 8, 12620–12631 CrossRef CAS PubMed.
- H. Shi, X. Ma, Q. Zhao, B. Liu, Q. Qu, Z. An, Y. Zhao and W. Huang, Adv. Funct. Mater., 2014, 24, 4823–4830 CrossRef CAS.
- H. Yuan, B. Wang, F. Lv, L. Liu and S. Wang, Adv. Mater., 2014, 26, 6978–6982 CrossRef CAS PubMed.
- J. Geng, C. Sun, J. Liu, L. D. Liao, Y. Yuan, N. Thakor, J. Wang and B. Liu, Small, 2015, 11, 1603–1610 CrossRef CAS PubMed.
- Y. Liu, K. Li, J. Pan, B. Liu and S. S. Feng, Biomaterials, 2010, 31, 330–338 CrossRef CAS PubMed.
- Y. Liu, K. Li, B. Liu and S. S. Feng, Biomaterials, 2010, 31, 9145–9155 CrossRef CAS PubMed.
- D. Ding, K. Li, Z. S. Zhu, K. Y. Pu, Y. Hu, X. Jiang and B. Liu, Nanoscale, 2011, 3, 1997–2002 RSC.
- K. Pu, A. J. Shuhendler, J. V. Jokerst, J. Mei, S. S. Gambhir, Z. Bao and J. Rao, Nat. Nanotechnol., 2014, 9, 233–239 CrossRef CAS PubMed.
- K. Pu, J. Mei, J. V. Jokerst, G. Hong, A. L. Antaris, N. Chattopadhyay, A. J. Shuhendler, T. Kurosawa, Y. Zhou, S. S. Gambhir, Z. Bao and J. Rao, Adv. Mater., 2015, 27, 5184–5190 CrossRef CAS PubMed.
- Q. Q. Miao, Y. Lyu, D. Ding and K. Y. Pu, Adv. Mater., 2016, 28, 3662–3668 CrossRef CAS PubMed.
- C. Xie, P. K. Upputuri, X. Zhen, M. Pramanik and K. Pu, Biomaterials, 2016, 119, 1–8 CrossRef PubMed.
- J. Zhang, X. Zhen, P. K. Upputuri, M. Pramanik, P. Chen and K. Pu, Adv. Mater., 2016 DOI:10.1002/adma.201604764.
- Y. Lyu, Y. Fang, Q. Miao, X. Zhen, D. Ding and K. Fu, ACS Nano, 2016, 10, 4472–4481 CrossRef CAS PubMed.
- K. Pu, A. J. Shuhendler and J. Rao, Angew. Chem., Int. Ed., 2013, 52, 10325–10329 CrossRef CAS PubMed.
- X. Zhen, C. Zhang, C. Xie, Q. Miao, K. L. Lim and K. Pu, ACS Nano, 2016, 10, 6400–6409 CrossRef CAS PubMed.
- Y. Wang, B. Liu, A. Mikhailovsky and G. C. Bazan, Adv. Mater., 2010, 22, 656–659 CrossRef CAS PubMed.
- B. Stender, S. F. Völker, C. Lambert and J. Pflaum, Adv. Mater., 2013, 25, 2943–2947 CrossRef CAS PubMed.
- P. J. Wu, S. Y. Kuo, Y. C. Huang, C. P. Chen and Y. H. Chan, Anal. Chem., 2014, 86, 4831–4839 CrossRef CAS PubMed.
- S. Y. Liou, C. S. Ke, J. H. Chen, Y. W. Luo, S. Y. Kuo, Y. H. Chen, C. C. Fang, C. Y. Wu, C. M. Chiang and Y. H. Chan, ACS Macro Lett., 2016, 5, 154–157 CrossRef CAS.
- X. Zhang, J. Yu, Y. Rong, F. Ye, D. T. Chiu and K. Uvdal, Chem. Sci., 2013, 4, 2143–2151 RSC.
- H. Y. Liu, P. J. Wu, S. Y. Kuo, C. P. Chen, E. H. Chang, C. Y. Wu and Y. H. Chan, J. Am. Chem. Soc., 2015, 137, 10420–10429 CrossRef CAS PubMed.
- H. Zhu, Y. Fang, X. Zhen, N. Wei, Y. Gao, K. Q. Luo, C. Xu, H. Duan, D. Ding, P. Chen and K. Pu, Chem. Sci., 2016, 7, 5118–5125 RSC.
- Y. Jin, F. Ye, M. Zeigler, C. Wu and D. T. Chiu, ACS Nano, 2011, 5, 1468–1475 CrossRef CAS PubMed.
- M. Palner, K. Pu, S. Shao and J. Rao, Angew. Chem., 2015, 127, 11639–11642 CrossRef.
- L. Xiong, F. Cao, X. Cao, Y. Guo, Y. Zhang and X. Cai, Bioconjugate Chem., 2015, 26, 817–821 CrossRef CAS PubMed.
- Y. Zhang, F. Ye, W. Sun, J. Yu, I. C. Wu, Y. Rong, Y. Zhang and D. T. Chiu, Chem. Sci., 2015, 6, 2102–2109 RSC.
- S. Li, X. Q. Shen, L. Li, P. Y. Yuan, Z. P. Guan, S. Q. Yao and Q. H. Xu, Langmuir, 2014, 30, 7623–7627 CrossRef CAS PubMed.
- M. K. Lowery, A. J. Starshak, J. N. Esposito, P. C. Krueger and M. E. Kenney, Inorg. Chem., 1965, 4, 128 CrossRef CAS.
- Y. Dong, W. Cai, X. Hu, C. Zhong, F. Huang and Y. Cao, Polymer, 2012, 53, 1465–1472 CrossRef CAS.
- J. Liu, J. Cao, S. Shao, Z. Xie, Y. Cheng, Y. Geng, L. Wang, C. Jing and F. Wang, J. Mater. Chem., 2008, 18, 1659–1666 RSC.
- C. Wu, C. Szymanski, Z. Cain and J. McNeill, J. Am. Chem. Soc., 2007, 129, 12904–12905 CrossRef CAS PubMed.
- C. Bouteiller, G. Clave, A. Bernardin, B. Chipon, M. Massonneau, P. Y. Renard and A. Romieu, Bioconjugate Chem., 2007, 18, 1303–1317 CrossRef CAS PubMed.
- C. Pan, K. Sugiyasu and M. Takeuchi, Chem. Commun., 2014, 50, 11814–11817 RSC.
- Q. Xu, L. M. Ensign, N. J. Boylan, A. Schön, X. Gong, J. C. Yang, N. W. Lamb, S. Cai, T. Yu, E. Freirel and J. Hanes, ACS Nano, 2015, 9, 9217–9227 CrossRef CAS PubMed.
- M. A. Miller, Y. R. Zheng, S. Gadde, C. Pfirschke, H. Zope, C. Engblom, R. H. Kohler, Y. Iwamoto, K. S. Yang, B. Askevold, N. Kolishetti, M. Pittet, S. J. Lippard, O. C. Farokhzad and R. Weissleder, Nat. Commun., 2015, 6, 8692 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- L. Q. Xiong, A. J. Shuhendler and J. H. Rao, Nat. Commun., 2012, 3, 1193 CrossRef PubMed.
- M. G. Harisinghani, J. Barentsz, P. F. Hahn, W. M. Deserno, S. Tabatabaei, C. H. van de Kaa and R. Weissleder, N. Engl. J. Med., 2003, 348, 2491–2499 CrossRef PubMed.
- D. L. Morton, J. F. Thompson, A. J. Cochran, N. Mozzillo, R. Elashoff, R. Essner, D. S. Reintgen, O. E. Nieweg, D. F. Roses, H. J. Hoekstra, C. P. Karakousis, D. S. Reintgen, B. J. Coventry, E. C. Glass and H. J. Wang, N. Engl. J. Med., 2006, 355, 1307–1317 CrossRef CAS PubMed.
- T. N. Erpelding, C. Kim, M. Pramanik, L. Jankovic, K. Maslov, Z. Guo, J. A. Margenthaler, M. D. Pashley and L. V. Wang, Radiology, 2010, 256, 102–110 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, 1H NMR and spectra of monomers, DLS, absorption and emission spectra of blended NIR800 Pdots, single particle fluorescence imaging. See DOI: 10.1039/c7sc00441a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |