
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn N-nitrosation reactivity-based two-photon fluorescent probe for the specific in situ detection of nitric oxide†
Zhiqiang
Mao
a,
Hong
Jiang
a,
Zhen
Li
a,
Cheng
Zhong
b,
Wei
Zhang
a and
Zhihong
Liu
 *a
*a
aKey Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China. E-mail: zhhliu@whu.edu.cn
bHubei Key Laboratory on Organic and Polymeric Optoelectronic Materials, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China
First published on 20th April 2017
Abstract
In situ fluorescence imaging of nitric oxide (NO) is a powerful tool for studying the critical roles of NO in biological events. However, the selective imaging of NO is still a challenge because most currently available fluorescent probes rely on the o-phenylenediamine (OPD) recognition site, which reacts with both NO and some abundant reactive carbonyl species (RCS) (such as dehydroascorbic acid and methylglyoxal) and some reactive oxygen/nitrogen species (ROS/RNS). To address this problem, a new fluorescent probe, NCNO, based on the N-nitrosation of aromatic secondary amine was designed to bypass the RCS, ROS, and RNS interference. As was expected, the probe NCNO could recognize NO with pronounced selectivity and sensitivity among ROS, RNS, and RCS. The probe was validated by detecting NO in live cells and deep tissues owing to its two-photon excitation and red-light emission. It was, hence, applied to monitor NO in ischemia reperfusion injury (IRI) in mice kidneys by two-photon microscopy for the first time, and the results vividly revealed the profile of NO generation in situ during the renal IRI process.
Introduction
In situ fluorescence imaging of reactive nitrogen species (RNS) produced in living systems is a useful strategy for studying the accurate roles of RNS in complex biosystems.1 Among the various RNS, nitric oxide (NO), which has not only been recognized as a highly reactive free radical but also as a celebrated signalling molecule, has revealed diverse functions in the cardiovascular, immune, and central nervous systems.2 However, further insights into the biological role of NO are still impeded due to the fact that the role may vary according to the location, time, and amount, especially in complex microenvironments such as tissues and live organisms. Therefore, it is meaningful to develop capable tools, such as optical probes with visualization, for the in situ detection of NO with high selectivity and sensitivity.To this end, a number of fluorescent probes have been exploited in the past few decades for the detection and imaging of NO owing to the advantages of fluorescence microscopy with the assistance of fluorescent probes, including simplicity and high sensitivity, as well as allowing the noninvasive visualization of biological molecules and processes with high temporal–spatial resolution.3 The current state-of-the-art fluorescent probes for NO are based on the specific reaction of NO with the o-phenylenediamine (OPD) moiety, metal–ligand complex and so on.4 Among these, the OPD moiety serving as an NO reaction site and fluorescence quencher has been harnessed in a variety of fluorescent probes for NO imaging both in cells and in tissues. However, concerns about the OPD-based probe still exist. First, the OPD moiety is also responsive to ascorbic acid (AA), dehydroascorbic acid (DHA) and MGO, which can unexpectedly light up the emission of probes.5 Actually, high levels of ascorbic acid (∼mM), DHA (∼μM) and MGO (∼μM) can cause serious false positive signals in comparison with the much lower level of NO (nM to μM) in biological systems.6 Furthermore, the OPD moiety is also easily oxidized by some ROS and RNS, such as H2O2 and ONOO−.7 This poor selectivity has provoked broad concern about the OPD-dependent design of the NO probe. Therefore, it is still highly demanded and challenging to develop a fluorescent probe for NO with the desired selectivity.
In the present work, bearing the aforementioned concerns in mind, we designed and prepared a new fluorescent probe NCNO specifically for NO based on the N-nitrosation of aromatic amine. Moreover, we also developed a two-photon (TP) fluorophore, which could be excited through the simultaneous absorption of two near-infrared (700–900 nm) photons, in consideration of the advantages of two-photon microscopy (TPM), such as a deeper imaging depth, higher temporal–spatial resolution with minimum background fluorescence and longer observation time.8 To gain a higher signal-to-noise (S/N) ratio in fluorescence imaging, long-wavelength (red to NIR) emission is also essential for a probe, since the red-emissive region (>600 nm) can avoid cellular absorption and autofluorescence which usually locates in the blue to green regions (<600 nm).9 So far, however, most TP probes for bioimaging emit at the blue to yellow range, and red-emissive TP probes are rarely reported.10,18 We used a benzo[g]coumarin derivate (8-(dimethylamino)-2-oxo-2H-benzo[g]chromene-3-carboxylic acid) as a red-emissive TP fluoro-phore, and a p-phenylenediamine derivate as an NO recognition group. The probe showed a highly specific response to NO over other interference species, including AA, DHA and MGO. It was thus able to sensitively detect exogenous and endogenous NO in live cells. The probe was successfully applied to image the renal ischemia and reperfusion injury (IRI) of a mice model in situ with TPM, which vividly revealed the NO generation during the IRI process for the first time.
Results and discussion
Design and fabrication of the NCNO probe
From the chemistry perspective, the facile cyclization reaction between the OPD moiety and 1,2-dicarbonyls promotes the response of the OPD-based probes to AA, DHA and MGO.6a It was previously reported that an aromatic secondary amine can readily react with NO under aerobic conditions through the N-nitrosation reaction.11 With this in mind, we anticipated that an N-nitrosation-based NO recognition site would show much higher selectivity for NO over AA, DHA and MGO than that of OPD-based probes (Scheme 1). Herein, an N-nitrosation reactivity-based two-photon fluorescent NCNO probe was designed for the specific detection of nitric oxide. In the NCNO probe, benzo[g]coumarin derivate (8-(dimethylamino)-2-oxo-2H-benzo[g] chromene-3-carboxylic acid) was selected as the two-photon fluorophore due to its favourable two-photon absorption properties and red-light emission (>600 nm), while mono alkyl substituted p-phenylenediamine was designed as the recognition group for NO. The benzo[g]coumarin moiety and p-phenylenediamine moiety were covalently linked by an amide bond, which could improve the photoinduced electron transfer (PeT)-induced quenching efficiency due to the short distance between the fluorophore and p-phenylenediamine.12 The N-nitrosation reaction under mild conditions (room temperature, neutral pH) is a specific reaction for monoalkyl aniline and NO, which endows the N-nitrosation-reaction-based fluorescent probe with high selectivity for NO. The lowered reactivity of this structure to other ROS and RNS can be ascribed to the controlled electron density by the introduction of a carbonyl group.13 Furthermore, compared to the ortho-diamine structure (with two coordinating atoms (N, N) for metal ions to form a stable complex), mono alkyl aniline has a much weaker coordinating ability with metal ions, which is another reason for the high selectivity. The compound NCNO was synthesized and characterized by 1H NMR, 13C NMR and HRMS spectra (Scheme S1 and Fig. S15–S22†).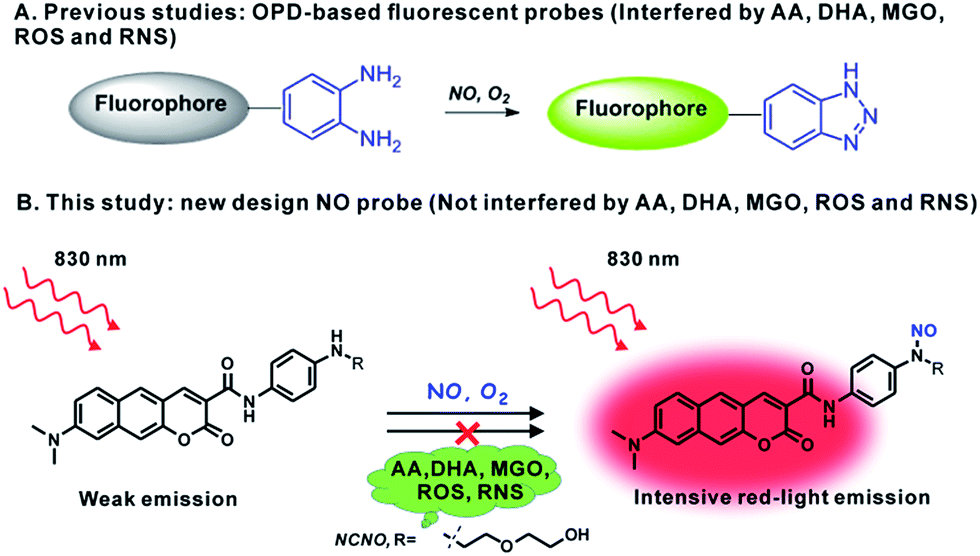 | ||
| Scheme 1 Schematic of the N-nitrosation-reactivity-based two-photon fluorescent probe NCNO for NO. | ||
Photophysical properties of NCNO
With NCNO in hand, its fluorescence response to NO was first evaluated in 10 mM PBS buffer (pH = 7.4, containing 10% CH3CN as the co-solvent). As expected, the NCNO probe showed extremely weak fluorescence (Φ = 0.008) at 608 nm and an absorption maxima at 473 nm (ε = 1.16 × 104 M−1 cm−1) (Fig. 1a). Upon the addition of excessive NO, the fluorescence quantum yield at 613 nm was increased by ca. 24-fold (Φ = 0.19), with the maxima absorption wavelength kept almost unchanged (475 nm, ε = 8.0 × 103 M cm−1) (Fig. 1b and Table S1†). The low background fluorescence and large enhancement was ascribed to PET, which was confirmed by the DFT calculations (Fig. S1†).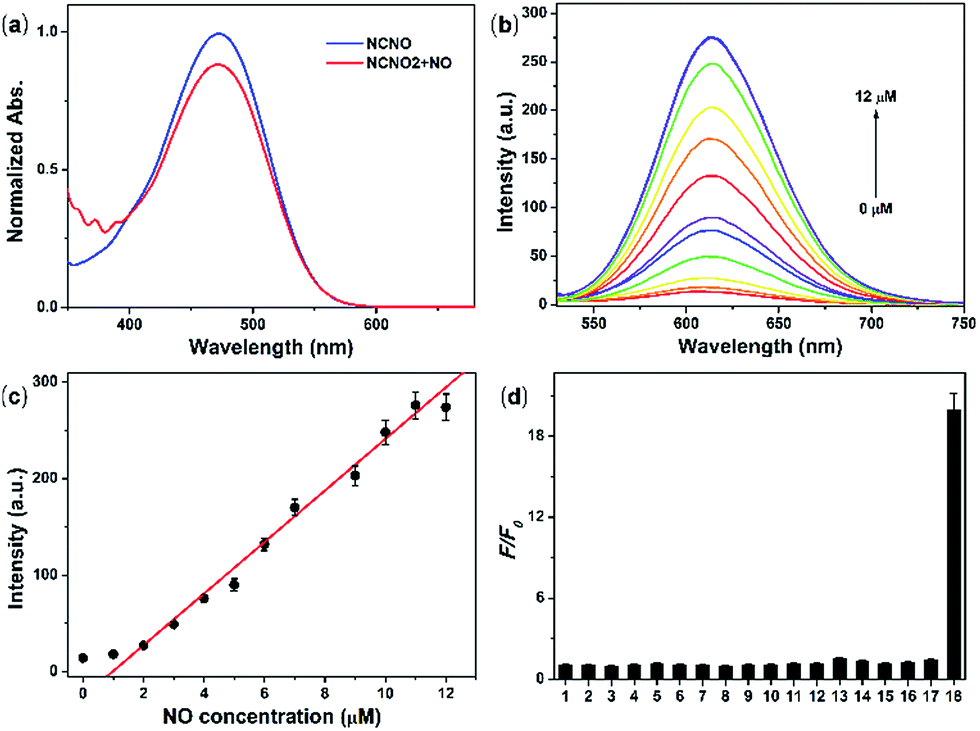 | ||
| Fig. 1 (a) UV-vis absorption spectra of 5.0 μM NCNO in the absence and presence of excess NO (15 μM). (b) Fluorescence spectra of 5.0 μM NCNO upon the addition of NO (0–12 μM). (c) Relative fluorescence intensity of 5.0 μM NCNO with varying amounts of NO (0–12 μM). (d) Relative fluorescence intensity of 5.0 μM NCNO incubated with various species (20 μM) for metal ions 1–4 (Ca2+, Mg2+, Zn2+, Fe2+), 50 μM reactive oxygen species 5–8 (ClO−, H2O2, ˙OH, O2−), 1.0 mM biological molecules 9–15 (GSH, Cys, Hcy, AA, DHA, MGO), 50 μM reactive nitrogen species 16–17 (NO2−, ONOO−) and 15 μM 18 (NO). Reaction time: 30 min. | ||
We further investigated the ability of NCNO to quantify the NO concentration in vitro. First, the reaction kinetics of the probe and NO were examined. Upon the addition of 3.0 eq. NO, the fluorescence intensity of the probe swiftly increased and reached a plateau in 120 s with an enhancement factor (F/F0) of ca. 20 (Fig. S2†), which was faster than that of most currently available OPD-based probes. As a control, we also performed the reaction between NCNO and NO under anaerobic conditions (Fig. S3†). Under this condition, the probe showed a much lower fluorescence enhancement (F/F0 = 1.7). These results suggested that the probe might react with N2O3 instead of NO directly, as discussed in the literature.14 The peak fluorescence intensity at 613 nm was linearly related to the NO concentration in the range of 1.0–11.0 μM (R2 = 0.98) (Fig. 1b and c). The calculated limit of detection (LOD, according to the 3σ criterion) for NO was 37 nM, which also represents a quite high sensitivity among all the fluorescent probes for NO. These results suggested that the newly developed N-nitrosation-based fluorescent NCNO probe might be suitable for tracking NO in biological systems at nM to μM concentrations.
The selectivity of NCNO towards NO is the most important performance for the accurate detection of NO in complicated cellular environments. The fluorescence response of NCNO to NO and other potentially biological interference species was inspected, including metal ions (Ca2+, Mg2+, Zn2+, Fe2+, Ag+, Hg2+, Cu2+, Fe3+, Cd2+), biomolecules (GSH, Cys, Hcy, AA, DHA, MGO), ROS (ClO−, H2O2, ˙OH, O2−) and RNS (NO2−, ONOO−). As depicted in Fig. 1d, S4 and S5,† only NO led to a significant fluorescence enhancement, while the other species caused negligible fluorescence restoration. Especially for those commonly known interfering substances, i.e. AA, DHA, MGO, ROS and RNS, to our delight, they did not cause any interference, even at a much higher concentration. This hence proved that the N-nitrosation-based NCNO probe possessed improved selectivity for NO over ROS, RNS and RCS than previously reported OPD-based NO probes. Subsequently, we checked the pH dependence of the emission profile of NCNO with or without NO. As shown in Fig. S6,† the fluorescence intensities of both NCNO and its reaction product with NO remained unchanged over a wide pH range of 5.0–8.2. The outstanding selectivity towards NO and the pH-independence of NCNO confirmed the rationality of our design and will be beneficial for NO tracking in living systems.
To verify the mechanism underlying the reaction of NCNO and NO, we first used the NO reaction site N-(4-(methylamino) phenyl)acetamide to react with NO in a mixture solution of PBS/CH3CN (pH = 7.4, v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). The results clearly suggested that N-(4-(methylamino)phenyl)acetamide converted to its N-nitrosation product with a high yield of 92% (Scheme S2 and Fig. S23–S28†). Then, we performed HPLC-MS analysis of the reaction of NCNO and NO, and the results supported that NCNO was quantitatively converted to its N-nitrosation product with an expected mass peak of m/z = 491 (Fig. S7†).
:2). The results clearly suggested that N-(4-(methylamino)phenyl)acetamide converted to its N-nitrosation product with a high yield of 92% (Scheme S2 and Fig. S23–S28†). Then, we performed HPLC-MS analysis of the reaction of NCNO and NO, and the results supported that NCNO was quantitatively converted to its N-nitrosation product with an expected mass peak of m/z = 491 (Fig. S7†).
Next, the two-photon absorption properties of the NCNO probe and its N-nitrosation product were investigated. The probe itself and the product of the probe and NO showed a maximal TP action cross-section (δΦ) of 2.4 GM and 54 GM at 830 nm, respectively, under the excitation at 760–900 nm, which proved that the probe could be employed in the detection of NO by TPM (Fig. S8†).
Exogenous and endogenous NO detection in live cells
Prior to cellular detection, the cytotoxicity of NCNO was investigated by MTT assay with HepG2 cells (Fig. S9†). The results supported that NCNO has a low cytotoxicity (0–20 μM, cell viability >90%) and is suitable for use in living cells.To monitor NO in a cellular environment under TPM, HepG2 cells were incubated with 5.0 μM NCNO, followed by further incubation with various amounts of NOC-9 (NO donor). As shown in Fig. 2a, NCNO-labelled HepG2 cells exhibited scant red fluorescence in cytoplasm, indicating the probe can readily enter into cells and then predominately localizes in cytoplasm. Notably, the fluorescence intensity of the NCNO-loaded cells increased gradually along with the increasing dosage of NOC-9 (Fig. 2a–g). It was noteworthy that the relative TP fluorescence intensity showed a positive correlation with the NOC-9 concentration in the range of 0–30 μM (Fig. 2h). These results indicated the capability of NCNO to sensitively detect exogenous NO concentration in living cells.
 | ||
| Fig. 2 (a–g) TP images of 5.0 μM NCNO-loaded HepG2 cells incubated with various amounts of NOC-9 (0, 5, 10, 20, 30, 40, 50 μM) for 30 min. (h) Relative TP fluorescence intensity in (a)–(g). The two-photon excitation wavelength was 830 nm, and the emission was collected at 600–700 nm. Scale bars, 10 μm. | ||
Following the success in detecting exogenous NO, we further attempted to harness the NCNO probe for the detection of endogenous NO in living cells. It has been documented that RAW 264.7 macrophages can produce NO upon stimulation with L-arginine (L-Arg), interferon-γ (IFN-γ) and lipopolysaccharide (LPS).15 As depicted in Fig. 3a, the NCNO-loaded RAW 264.7 cells also exhibited weak fluorescence. When the cells were pretreated with NO stimulants (0.5 mg mL−1L-Arg, 200 U mL−1 IFN-γ and 20 μg mL−1 LPS), a remarkably enhanced fluorescence signal was observed (Fig. 3b). This phenomenon may be ascribed to the promoted endogenous NO generation by the stimulants, which was instantly captured by NCNO, resulting in fluorescence enhancement. To verify this, we conducted further experiments by incubating the cells with NO stimulants as well as 10 μM L-NG-nitroarginine (L-NNA), which is a NO synthase (iNOS) inhibitor.16 In this case, the fluorescence intensity of the cells significantly decreased down to the initial level (Fig. 3c and e). We also tested another group where the cells were pretreated by NO stimulants together with a NO scavenger, 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide potassium salt (carboxyl-PTIO),17 before the probe loading. As expected, the fluorescence signal of this group was attenuated to the basal level (Fig. 3d and e). Altogether, these results have confirmed that NCNO was capable of monitoring the fluctuation of endogenous NO concentrations in living cells with high sensitivity.
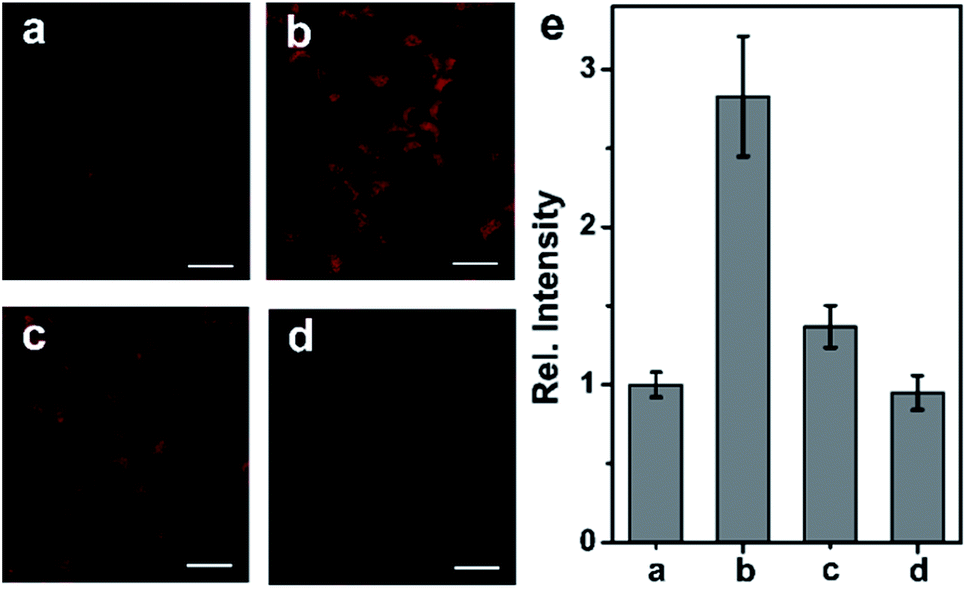 | ||
| Fig. 3 (a) TP image of RAW 264.7 cells incubated with 5.0 μM NCNO for 30 min. (b) TP image of RAW 264.7 cells pretreated with NO stimulants for 16 h, and then incubated with 5.0 μM NCNO for 30 min. (c) TP image of RAW 264.7 cells pretreated with NO stimulants and 10 μM L-NNA for 16 h, and then incubated with 5.0 μM NCNO for 30 min. (d) TP image of RAW 264.7 cells pretreated with NO stimulants followed by incubation with 0.5 mM carboxyl-PTIO for 30 min, and then incubated with 5.0 μM NCNO for 30 min. (e) Relative fluorescence intensity in (a–c). The images were obtained at 600–700 nm upon the excitation at 830 nm with a femtosecond laser. Scale bars, 20 μm. | ||
Tissue imaging study
The excellent performances of NCNO in detection of endogenous and exogenous cellular NO motivated us to use the probe for NO detection in live tissues under TPM. It's been established that two-photon microscopy features with less background fluorescence in contrast with one-photon microscopy, but researches have revealed that some intrinsic molecules (such as NADH, folic acid, and riboflavin, etc.) still can be two-photon excitable and result in blue to green background fluorescence in tissue imaging.18 We hereby firstly studied the tissue background fluorescence under two-photon excitation. A mouse brain tissue slice was chosen as a model. The tissue background fluorescence was collected at three different detection windows (blue: 400–500 nm; green: 500–600 nm; red: 600–700 nm) by changing the TP excitation wavelength from 700 to 850 nm (700, 750, 800, 850 nm) (Fig. S10†). Interestingly, all the images collected at green channel showed obvious fluorescence signal, while there was almost none fluorescence signal detected in the blue and red windows (Fig. S11†). It can be explained that most TP excitable intrinsic molecules emit at green region, whereas two-photon excitable biomolecules rarely emit at blue or red regions.9a,19 In connection with another obvious fact that red light has larger penetration depth in tissues than other shorter-wavelength lights, it is safe to highlight the necessity and advantages of two-photon excited red-emissive (>600 nm) fluorescent probes in tissue imaging.We then tried using NCNO to detect NO in tissue samples. A fresh mouse liver tissue slice was incubated with 20 μM NCNO for 60 min, and then treated with 50 μM NO for another 30 min. As shown in Fig. S12,† the TPM images reveal that the probe can well stain the tissue. Furthermore, the accumulated TP images obtained at z-direction revealed that the probe can image the target at a depth of 160 μm. Meanwhile, the NCNO-stained tissues treated with external NO showed much brighter fluorescence (Fig. S13a and b†). In addition to liver tissues, other mice tissues including brain, spleen, lung and kidney also can be stained by NCNO with high resolution (Fig. S14†). These results implied that NCNO could be a competent probe for NO in deep tissues.
In situ detection of NO generation in the mouse model
Ischemia and reperfusion (IRI) is a pathological condition characterized by temporary blood cessation with energy depletion to an organ, followed by the subsequent restoration of perfusion and reoxygenation. The restoration of blood flow and reoxygenation often causes serious tissue injury and an acute inflammatory response, which is termed as “reperfusion injury”. It was proposed that the accumulation of reactive species, including ROS and RNS, exacerbate the reperfusion injury.20 However, there is still a lack of practical tools for the selective monitoring of NO generation in situ during the ischemia reperfusion injury (IRI) process. With this aim in mind, we continued to use our probe to detect the NO generation in situ during the IRI process. A midline incision was performed on the test mice, which were then subjected to renal ischemia for 60 min, during which both the renal arteries and veins were occluded by the renal clamps.21 Next, the kidney underwent reperfusion for varying time courses (0, 0.5, 1, 1.5, 2 h) by removal of the clamps. Then, the mice were injected with 0.2 mL of 1.0 mM NCNOvia the tail vein. After 10 min, the organ was isolated and cut into slices for TP imaging immediately. The control mice renal tissue slices showed weak fluorescence (Fig. 4a), whereas the IRI slices exhibited brighter fluorescence. As is vividly illustrated in Fig. 4, the fluorescence signal of NCNO-stained renal tissues showed an obvious increase (1.2-fold NO production compared to the control) after reperfusion for 0.5 h, indicating that NO was generated rapidly during the reperfusion process. Subsequently, compared with the control, the NCNO-stained tissues showed a continued fluorescence increase even after the restoration of reperfusion for 2 h, suggesting that the reperfusion injury caused a long-time NO generation and high NO level in reperfusion injury tissues (Fig. 4a–e and g), which was consistent with the findings of previous biology studies.22 Furthermore, as a negative control the renal tissues of the mice injected with 0.2 mL of 1.0 mM NCNO together with 1.0 mM carboxyl-PTIO showed a reduced fluorescence signal, which further confirmed that the fluorescence enhancement was induced by the generated NO (Fig. 4f). The above results can be evidence of the ability of the NCNO probe to detect NO generation in biological/pathological processes in situ. | ||
| Fig. 4 Mouse kidneys were subjected to ischemia for 1 h, and reperfusion for various times ((a–e) 0, 0.5, 1, 1.5, 2 h), then the mice were injected with 0.2 mL of 1.0 mM NCNO and incubated for 10 min. (f) The mice kidneys were subjected to ischemia for 1 h, and then reperfusion for 2 h and injected with 0.2 mL of 1.0 mM NCNO together with 1.0 mM carboxyl-PTIO. (g) The relative fluorescence intensity for TP images (a–f). The two-photon excitation wavelength was 830 nm, and the fluorescence emission was collected at 600–700 nm. Scale bar, 50 μm. | ||
Conclusion
In summary, we developed a red-emissive two-photon fluorescent NCNO probe for NO based on the N-nitrosation of an aromatic secondary amine. The newly developed NO probe overcomes the drawbacks of the OPD-based NO probe: it is free of interference from AA, RCS (DHA, MGO) and ROS, as well as RNS. The pronounced selectivity of NCNO towards NO among other ROS, RNS and RCS guarantees the accurate detection of NO in complicated biosystems. It also features quick reaction with NO and shows a linear response to NO in the concentration range of 1–11 μM with a detect limit of 37 nM. Furthermore, the probe was able to detect both exogenous and endogenous NO in living cells. It was also capable of imaging NO in deep tissues, taking the advantages of two-photon excitation and red-light emission. We demonstrated its applicability in monitoring the NO generation in an ischemia reperfusion injury (IRI) mouse model for the first time. Therefore, the red-emissive NCNO probe can be a practical tool for studying NO-related biological processes in situ.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21625503, 21535005, 51203121). We thank Fang Zhou of the Institute of Hydrobiology, CAS, for help with confocal microscopy observation.References
- (a) J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed; (b) L. E. McQuade, J. Ma, G. Lowe, A. Ghatpande, A. Gelperin and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8525–8530 CrossRef CAS PubMed; (c) A. J. Shuhendler, K. Pu, L. Cui, J. P. Uetrecht and J. Rao, Nat. Biotechnol., 2014, 32, 373–380 CrossRef CAS PubMed.
- (a) V. Calabrese, C. Mancuso, M. Calvani, E. Rizzarelli, D. A. Butter-field and A. M. Stella, Nat. Rev. Neurosci., 2007, 8, 766–775 CrossRef CAS PubMed; (b) C. Szabo, Nat. Rev. Drug Discovery, 2016, 15, 185–203 CrossRef CAS PubMed.
- (a) M. H. Lim, B. A. Wong, W. H. Pitcock Jr, D. Mokshagundam, M. H. Baik and S. J. Lippard, J. Am. Chem. Soc., 2006, 128, 14364–14373 CrossRef CAS PubMed; (b) C. Sun, W. Shi, Y. Song, W. Chen and H. Ma, Chem. Commun., 2011, 47, 8638–8640 RSC; (c) E. Sasaki, H. Kojima, H. Nishimatsu, Y. Urano, K. Kikuchi, Y. Hirata and T. Nagano, J. Am. Chem. Soc., 2005, 127, 3684–3685 CrossRef CAS PubMed; (d) W. Xu, Z. Zeng, J. H. Jiang, Y. T. Chang and L. Yuan, Angew. Chem., Int. Ed., 2016, 55, 13658–13699 CrossRef CAS PubMed; (e) S. Ma, D. C. Fang, B. Ning, M. Li, L. He and B. Gong, Chem. Commun., 2014, 50, 6475–6478 RSC; (f) H. W. Yao, X. Y. Zhu, X. F. Guo and H. Wang, Anal. Chem., 2016, 88, 9014–9021 CrossRef CAS PubMed; (g) H. Yu, Y. Xiao and L. Jin, J. Am. Chem. Soc., 2012, 134, 17486–17489 CrossRef CAS PubMed; (h) E. W. Seo, J. H. Han, C. H. Heo, J. H. Shin, H. M. Kim and B. R. Cho, Chem.–Eur. J., 2012, 18, 12388–12394 CrossRef CAS PubMed.
- (a) X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590–659 CrossRef CAS PubMed; (b) Z. Xu and L. Xu, Chem. Commun., 2016, 52, 1094–1119 RSC; (c) H. Li and L. Wan, Analyst, 2015, 140, 7129–7141 RSC; (d) X. Chen, F. Wang, J. Y. Hyun, T. Wei, J. Qiang, X. Ren, I. Shin and J. Yoon, Chem. Soc. Rev., 2016, 45, 2976–3016 RSC; (e) Z. Dai, L. Tian, B. Song, X. Liu and J. Yuan, Chem. Sci., 2017, 8, 1969–1976 RSC.
- (a) X. Ye, S. S. Rubakhin and J. V. Sweedler, J. Neurosci. Methods, 2008, 168, 373–382 CrossRef CAS PubMed; (b) X. Ye, S. S. Rubakhin and J. V. Sweedler, Analyst, 2008, 133, 423–433 RSC.
- (a) Y. Wang and C. T. Ho, Chem. Soc. Rev., 2012, 41, 4140–4149 RSC; (b) Y. Yang, S. K. Seidlits, M. M. Adams, V. M. Lynch, C. E. Schmidt, E. V. Anslyn and J. B. Shear, J. Am. Chem. Soc., 2010, 132, 13114–13116 CrossRef CAS PubMedY. Q. Sun, J. Liu, H. Zhang, Y. Huo, X. Lv, Y. Shi and W. Guo, J. Am. Chem. Soc., 2014, 136, 12520–12523 CrossRef CAS PubMed; (c) T. Wang, E. F. Douglass Jr, K. J. Fitzgerald and D. A. Spiegel, J. Am. Chem. Soc., 2013, 135, 12429–12433 CrossRef CAS PubMed.
- (a) S. Fornera and P. Walde, Anal. Biochem., 2010, 407, 293–295 CrossRef CAS PubMed; (b) H. Li, D. Zhang, M. Gao, L. Huang, L. Tang, Z. Li, X. Chen and X. Zhang, Chem. Sci., 2017, 8, 2199–2203 RSC.
- (a) H. M. Kim and B. R. Cho, Chem. Rev., 2015, 115, 5014–5055 CrossRef CAS PubMed; (b) M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed; (c) L. Qian, L. Li and S. Q. Yao, Acc. Chem. Res., 2016, 49, 626–634 CrossRef CAS PubMed; (d) Y.-Y. Ren, N.-W. Wu, J. Huang, Z. Xu, D.-D. Sun, C.-H. Wang and L. Xu, Chem. Commun., 2015, 51, 15153–15156 RSC.
- (a) W. R. Zipfel, R. M. Williams, R. Christie, A. Y. Nikitin, B. T. Hyman and W. W. Webb, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7075–7080 CrossRef CAS PubMed; (b) D. Kim, H. Moon, S. H. Baik, S. Singha, Y. W. Jun, T. Wang, K. H. Kim, B. S. Park, J. Jung, I. Mook-Jung and K. H. Ahn, J. Am. Chem. Soc., 2015, 137, 6781–67789 CrossRef CAS PubMed.
- (a) A. R. Sarkar, C. H. Heo, H. W. Lee, K. H. Park, Y. H. Suh and H. M. Kim, Anal. Chem., 2014, 86, 5638–5641 CrossRef CAS PubMed; (b) D. Kim, Q. P. Xuan, H. Moon, Y. W. Jun and K. H. Ahn, Asian J. Org. Chem., 2014, 3, 1089–1096 CrossRef CAS; (c) B. Sui, X. Yue, B. Kim and K. D. Belfield, ACS Appl. Mater. Interfaces, 2015, 7, 17565–17568 CrossRef CAS PubMed.
- (a) T. Nagano, H. Tnkizawa and M. Hirobe, Tetrahedron Lett., 1995, 36, 8239–8242 CrossRef CAS; (b) M. D. Pluth, L. E. McQuade and S. J. Lippard, Org. Lett., 2010, 12, 2318–2321 CrossRef CAS PubMed; (c) J. Miao, Y. Huo, X. Lv, Z. Li, H. Cao, H. Shi, Y. Shi and W. Guo, Biomaterials, 2016, 78, 11–19 CrossRef CAS PubMed.
- (a) A. R. Sarkar, D. E. Kang, H. M. Kim and B. R. Cho, Inorg. Chem., 2014, 53, 1794–1803 CrossRef CAS PubMed; (b) W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Chem. Rev., 2016, 116, 7768–7817 CrossRef CAS PubMed; (c) Z. Mao, L. Hu, X. Dong, C. Zhong, B.-F. Liu and Z. Liu, Anal. Chem., 2014, 86, 6548–6554 CrossRef CAS PubMed; (d) J. Zhang, H.-W. Liu, X.-X. Hu, J. Li, L.-H. Liang, X.-B. Zhang and W. Tan, Anal. Chem., 2015, 87, 11832–11839 CrossRef CAS PubMed; (e) H.-L. Un, S. Wu, C.-B. Huang, Z. Xu and L. Xu, Chem. Commun., 2015, 51, 3143–3146 RSC.
- (a) T. Peng, X. Chen, L. Gao, T. Zhang, W. Wang, J. Shen and D. Yang, Chem. Sci., 2016, 7, 5407–5413 RSC; (b) X. Wu, L. Li, W. Shi, Q. Gong and H. Ma, Angew. Chem., Int. Ed., 2016, 55, 14728–14732 CrossRef CAS PubMed; (c) K. Xu, M. Qiang, W. Gao, R. Su, N. Li, Y. Gao, Y. Xie, F. Kong and B. Tang, Chem. Sci., 2013, 4, 1079–1086 RSC.
- T. Itoh, Y. Matsuya, H. Maeta, M. Miyazaki, K. Nagata and A. Ohsawa, Chem. Pharm. Bull., 1999, 47, 819–823 CrossRef CAS.
- (a) G. K. Vegesna, S. R. Sripathi, J. Zhang, S. Zhu, W. He, F. T. Luo, W. J. Jahng, M. Frost and H. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 4107–4112 Search PubMed; (b) Y. Li, W. Wu, J. Yang, L. Yuan, C. Liu, J. Zheng and R. Yang, Chem. Sci., 2016, 7, 1920–1925 RSC; (c) X. Dong, C. H. Heo, S. Chen, H. M. Kim and Z. Liu, Anal. Chem., 2014, 86, 308–311 CrossRef CAS PubMed.
- U. Grohmann and V. Bronte, Immunol. Rev., 2010, 236, 243–264 CrossRef CAS PubMed.
- L. Yuan, W. Lin, Y. Xie, B. Chen and S. Zhu, J. Am. Chem. Soc., 2012, 134, 1305–1315 CrossRef CAS PubMed.
- Z. Mao, W. Feng, Z. Li, L. Zeng, W. Lv and Z. Liu, Chem. Sci., 2016, 7, 5230–5235 RSC.
- W. R. Zipfel, R. M. Williams and W. W. Webb, Nat. Biotechnol., 2003, 21, 1369–1377 CrossRef CAS PubMed.
- (a) H. K. Eltzschig and T. Eckle, Nat. Med., 2011, 17, 1391–1401 CrossRef CAS PubMed; (b) T. Kalogeris, C. P. Baines, M. Krenz and R. J. Korthuis, Int. Rev. Cell Mol. Biol., 2012, 298, 229–317 CAS.
- (a) T. Yano, Y. Nozaki, K. Kinoshita, S. Hino, Y. Hirooka, K. Niki, H. Shimazu, K. Kishimoto, M. Funauch and I. Matsumura, Lab. Invest., 2015, 95, 78–91 CrossRef CAS PubMed; (b) X. Xie, X. Yang, T. Wu, Y. Li, M. Li, Q. Tan, X. Wang and B. Tang, Anal. Chem., 2016, 88, 8019–8025 CrossRef CAS PubMed.
- (a) K. Fassbender, M. Fatar, A. Ragoschke, M. Picard, T. Bertsch, S. Kuehl and M. Hennerici, Stroke, 2000, 31, 2208–2211 CrossRef CAS PubMed; (b) M. Grandati, C. Verrecchia, M. L. Revaud, M. Allix, R. G. Boulu and M. Plotkine, Br. J. Pharmacol., 1997, 122, 625–630 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc00416h |
| This journal is © The Royal Society of Chemistry 2017 |