
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed oxidative C(sp3)–H/C(sp2)–H cross-coupling en route to carbocyclic rings†
Rui
Wang
 ,
Yan
Li
,
Ruo-Xing
Jin
and
Xi-Sheng
Wang
*
,
Yan
Li
,
Ruo-Xing
Jin
and
Xi-Sheng
Wang
*
Department of Chemistry, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui 230026, China. E-mail: xswang77@ustc.edu.cn
First published on 15th March 2017
Abstract
Copper-catalyzed selective coupling of C(sp3)–H bonds with C(sp2)–H bonds has been developed. An aniline module was used as a directing group to generate an aminyl radical, which selectively cleaves the secondary and tertiary C(sp3)–H bonds via a 1,5-HAT process to forge six-membered carbocyclic rings.
The selective construction of C–C bonds, the essential link in organic molecules, in more efficient ways is always the central topic in synthetic chemistry.1 In the past several decades, transition-metal-catalyzed C–C bond construction via the activation of ubiquitous C–H bonds has attracted increasing attention, due to the atom and step economy.2 While the recent fast developments in this area offered various transformations involving mainly activation of one C–H bond,3 C–C bond formation via direct C–H/C–H cross-coupling is no doubt the most efficient and ideal method. Inspired by this strategy, cross-dehydrogenative coupling (CDC), termed by Li, was developed and used for the construction of diverse functional molecules.4 However, at least one relatively active C–H bond, such as C–H bonds adjacent to heteroatoms and carbonyl groups, or at the benzylic and allylic positions, is typically required in these reactions, and the direct cross-coupling of two inert C–H bonds still faces many challenges from issues such as reactivity and regioselectivity.
Transition-metal-catalyzed oxidative coupling of inert C–H bonds has emerged as a powerful method to construct C–C bonds in an intra- and inter-molecular manner. While the coupling of both aryl C–H bonds has been well documented for biaryl bond formation,5 the cross-coupling of inert aromatic and aliphatic C–H bonds selectively still remains an unsolved problem. For instance, Li and co-workers reported an intermolecular CDC of arenes with simple unactivated alkanes in 2008.6 While the selective activation of C(sp2)–H bonds could be realized using pyridine as the ortho-directing group, the regioselective activation of inert C(sp3)–H bonds was still an unsurmountable problem in this transformation. Only two examples of intramolecular C(sp2)–C(sp3) couplings involving Pd(II)-catalyzed tandem activation of C(sp2)–H and C(sp3)–H bonds successively have been reported,7 in which only relatively active heteroaryl C(sp2)–H and primary C(sp3)–H bonds were compatible to produce five-membered-ring products accordingly (path A, Scheme 1). Different from the metal-catalyzed C(sp2)–H/C(sp3)–H oxidative couplings that were initiated from C(sp2)–H bond activation, we conceive that the C(sp3)–H bond could be cleaved firstly via radical hydrogen-atom abstraction,8,9 and the alkyl radical is then trapped by aryl rings to produce the final C(sp2)–C(sp3) coupling product after the following oxidation and deprotonation. While the inert C(sp3)–H bonds are almost impossible to distinguish from other aliphatic C–H bonds on the alkyl side chain, the 1,n-hydrogen-atom-transfer (1,n-HAT)9 strategy offers us a reliable solution, in which heteroatoms might be installed on the starting materials to generate the heteroatom radicals for directed selective cleavage of inert C(sp3)–H bonds (path B, Scheme 1).10
 | ||
| Scheme 1 Transition-metal-catalyzed intramolecular C(sp3)–H/C(sp2)–H cross-coupling. | ||
Herein we report a copper-catalyzed intramolecular cross-coupling of inert aliphatic and aryl C–H bonds with yields as high as 95%. An aniline module was used as a directing group to generate the aminyl radical, which selectively cleaves the secondary and tertiary C(sp3)–H bonds via a 1,5-HAT process to produce 6-membered-ring products. The initial nitrogen radical could be generated with the assistance of a catalytic amount of Cu(OAc)2 directly from an N–H bond in aniline,11 and pre-functionalized or in situ generated N–X bonds were not required.8
Our initial study commenced with 1a as the pilot substrate in the presence of a catalytic amount of Cu(OAc)2. After careful reaction optimization (Tables S1–S4, see ESI†), we found that the combination of 1a with Cu(OAc)2 (20 mol%) and Ag2CO3 (1.5 equiv.) in 1,2-dichloroethane (DCE, 2.0 mL) at 135 °C for 25 h afforded the desired product 2a with the best yield (92%, Table 1, entry 1). Control experiments and the effects of each parameter were then examined (Table 1). It is revealed that Cu(OAc)2 as the catalyst together with Ag2CO3 as the oxidant was crucial for this reaction. No product could be observed in the absence of Cu(OAc)2 (entry 2), and other copper salts failed to catalyze the reaction. If a stoichiometric amount of Cu(OAc)2 (3 equiv.) was used without the addition of Ag2CO3, only a trace amount of 2a was detected (entry 5). Variation of the reaction temperature decreased the yield slightly (entries 9–10). When the reaction was quenched at 12 h, the desired product 2a was isolated in 77% yield (entry 11). Upon reducing the catalyst loading of Cu(OAc)2 or the amount of oxidant Ag2CO3, the yields decreased only slightly, furnishing 2a in 83% yield even with only 5 mol% of Cu(OAc)2 (entry 13) and also in 83% yield with 1 equivalent of Ag2CO3 (entry 14).
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yield (%) |
| a Unless otherwise noted, the reaction conditions were as follows: 1a (0.1 mmol), Cu(OAc)2 (20 mol%) and Ag2CO3 (1.5 equiv.) in 1,2-DCE (2.0 mL) at 135 °C for 25 h. Isolated yields were reported. n.d., not detected. | ||
| 1 | None | 92 |
| 2 | Without Cu(OAc)2 | n.d. |
| 3 | Cu(OTf)2 instead of Cu(OAc)2 | n.d. |
| 4 | Cu(acac)2 instead of Cu(OAc)2 | Trace |
| 5 | Cu(OAc)2 instead of Ag2CO3 | Trace |
| 6 | AgOAc instead of Ag2CO3 | 38 |
| 7 | O2 instead of Ag2CO3 | 18 |
| 8 | TEMPO instead of Ag2CO3 | n.d. |
| 9 | 120 °C instead of 135 °C | 60 |
| 10 | 150 °C instead of 135 °C | 78 |
| 11 | 12 h instead of 25 h | 77 |
| 12 | 10 mol% instead of 20 mol% Cu(OAc)2 | 87 |
| 13 | 5 mol% instead of 20 mol% Cu(OAc)2 | 83 |
| 14 | 100 mol% instead of 150 mol% Ag2CO3 | 83 |

With the optimized conditions in hand, we next sought to analyze the scope of this protocol (Table 2). With regards to the substituent effect on a phenyl ring, various substituted groups were compatible with this transformation. As for the substituents on the para-site of the phenyl ring, both electron-donating and -withdrawing groups were well tolerated with moderate to excellent yields, including alkyl (2b–d), phenyl (2e), methylthio (2f), halogen (2g–i) and trifluoromethyl (2j). In particular, the presence of halogen atoms (Br, Cl, and F) in products 2 offers the potential for further synthetic elaboration via known transition-metal-catalyzed coupling methods. Generally, some anilines, 1, with different substituents, such as methyl, cyclopropyl, chloro and trifluoromethyl, at the meta-site on the phenyl ring could be cyclized completely at the less sterically hindered position with good yields (2k–n), which can be explained by the steric effect of such substituents.
|
|
|---|
| a Reaction conditions: for 2a–q, 2s–t, and 2v–y, reactions were performed on 1 (0.1 mmol), Cu(OAc)2 (20 mol%) and Ag2CO3 (1.5 equiv.) in 1,2-DCE (2.0 mL) at 135 °C for 25 h; for 2r, 2u, and 2z–ah, reactions were performed on 1 (0.1 mmol), Cu(OAc)2 (25 mol%) and Ag2CO3 (2 equiv.) in 1,2-DCE (2.0 mL) at 150 °C for 35 h. Isolated yields were reported. b 20 h. c 135 °C. d 125 °C. e Single diastereoisomer. |

|

Yet intriguingly, the meta-MeO-substituted aniline, 1o, afforded both 5- and 7-methoxyl cyclic products in an excellent combined yield (95%, 2o and 2o′), but with a compromised selectivity to give the cyclized product at the more sterically hindered position as the major isomer. Even though this selectivity was not significant (1.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), it indicated that the electronic effect also plays an important role in this transformation. The cyclization of naphthalen-2-yl-derived aniline, 1p, also proceeded smoothly to give a mixture of both tetrahydrophenanthren and tetrahydroanthracen-type products with a good total yield (60%, 2p and 2p′). Similar to the meta-methoxy substrates, aniline 2p that cyclized at the relatively more sterically hindered position was the major isomer. The selective activation of the C(sp3)–H bond in the cycloalkanes was also successful for cross-coupling of the phenyl ring, providing a synthetically difficult spiro skeleton in 62% yield (2q). A symmetric aniline with two of the same C(sp3)–H bonds suitable for 1,5-hydrogen-atom abstraction in both alkyl side chains was also tested in this catalytic system. It was interesting to find that both C(sp3)–H bonds coupled with the ortho-C(sp2)–H bonds on the phenyl ring smoothly, followed by in situ elimination of aniline to afford an attractive 2,3,5,6-tetrahydro-1H-phenalene structure (2r).
:1), it indicated that the electronic effect also plays an important role in this transformation. The cyclization of naphthalen-2-yl-derived aniline, 1p, also proceeded smoothly to give a mixture of both tetrahydrophenanthren and tetrahydroanthracen-type products with a good total yield (60%, 2p and 2p′). Similar to the meta-methoxy substrates, aniline 2p that cyclized at the relatively more sterically hindered position was the major isomer. The selective activation of the C(sp3)–H bond in the cycloalkanes was also successful for cross-coupling of the phenyl ring, providing a synthetically difficult spiro skeleton in 62% yield (2q). A symmetric aniline with two of the same C(sp3)–H bonds suitable for 1,5-hydrogen-atom abstraction in both alkyl side chains was also tested in this catalytic system. It was interesting to find that both C(sp3)–H bonds coupled with the ortho-C(sp2)–H bonds on the phenyl ring smoothly, followed by in situ elimination of aniline to afford an attractive 2,3,5,6-tetrahydro-1H-phenalene structure (2r).
The substituents at the α-position of the nitrogen atom had an obvious effect on the transformation. While the corresponding yields decreased along with the increase of steric hindrance of the alkyl groups (2s–u), the cyclization of N-α,α-biphenyl aniline proceeded quite smoothly with excellent yield (90%, 2v). It is important to point out that the hydrogen atom at the α-position of the nitrogen atom, well known to be activated via a radical path, could be well tolerated in this reaction (75%, 2w), which further indicated that the regioselectivity of this method was determined by a 1,5-HAT process. In order to demonstrate the synthetic potential of this method, we tried to synthesize 2x, an analogue of UCI-30002, which has been proven to play an inhibition role on several acetylcholine receptors.12 As expected, 2x could be obtained in 53% yield under our standard conditions. While aniline 1x could be prepared from readily available compounds like benzonitrile and 4-nitroaniline through a simple and easy-to-handle process (see ESI,† pp. 18), this result definitely showed the feasibility of variation on the directing aniline part. As a common removable protecting group on a nitrogen atom, para-MeOPh (PMP) was also well compatible with this method (69%, 2y).15
It is also important to note that the secondary C(sp3)–H bonds could also be selectively activated to furnish the corresponding cyclized products as single diastereoisomers in acceptable yields (40–62%, 2z–ag, for details, see ESI†). A number of substituents were readily compatible with this protocol. Remarkably, the subjection of substrate 1ah, which possesses a tertiary C(sp3)–H bond at the 4-position and a secondary C(sp3)–H center at the 5-position to the nitrogen atom, to the standard conditions only produced the 5-position-reacting product 2ah. This result also clearly showed that the site-selectivity of C(sp3)–H bond activation on the alkyl chain was determined by 1,5-HAT solely, which further illustrates the excellent site-selectivity of our methodology.
However, the selective cross-coupling of a primary C(sp3)–H bond (1ai, see ESI†) to an intramolecular phenyl ring provided only a trace amount of the corresponding product in this reaction. Taking into account all of the above results, the reactivity of aliphatic C–H bonds paralleled the thermodynamic stability of the carbon radicals produced and followed the order: tertiary > secondary > primary. Notably, as for the anilines bearing both tertiary C(sp3)–H and secondary or primary C(sp3)–H bonds (for primary C–H, see 1s; for secondary C–H, see 1t), the coupling reaction happened at the tertiary C–H position exclusively, which showed excellent chemoselectivity of this protocol for inert C(sp3)–H bond activation.
A series of control experiments were designed to probe the possible radical pathway. Firstly, some free radical inhibitors, including TEMPO, BHT and galvinoxyl, showed strong inhibition effects even with only a sub-stoichiometric amount (Table S5, see ESI†). It’s worth noting that the addition of 1.5 equivalents of TEMPO to the standard conditions afforded alkene 3a in 49% yield (Scheme 2A). As alkene 3a could be produced from the coupling adduct 3a′via elimination,13 this result was definitely consistent with our hypothesis about the radical path. To further verify the radical process, vinyl cyclopropane 1aj and cyclopropyl aniline 1ak were designed to trap the corresponding nitrogen and carbon radical one by one.14 Indeed, the reaction of 1aj gave the ring-opened products 2aj-1 and 2aj-2 in 14% and 18% yield, respectively (Scheme 2B). Moreover, while the subjection of 1ak to the standard conditions afforded none of the desired alkene, 2ak′, tricyclic compound 2ak was obtained successfully, albeit with low yield (10%, Scheme 2C). Given that 2ak could be transformed from 2ak′via 5-exo-trig cyclization14 followed by sequenced radical cyclization and oxidation (Fig. S2, see ESI†), 2ak′ was synthesized and subjected to the reaction system, furnishing 2ak in 51% yield accordingly, as shown in Fig. S2.† All these results clearly indicated that the corresponding nitrogen radical and carbon radical were involved in the catalytic cycle, thus supporting the selective radical C(sp3)–H activation path as in our design. Finally, no reaction occurred and all the starting material remained when tertiary amine 1al was subjected to the standard conditions (Scheme 2D), which showed the key role of a hydrogen atom on the nitrogen for radical generation.
 | ||
| Scheme 2 Control experiments designed to probe the radical path. | ||
Based on all of the results above and the previous reports, a plausible mechanism for the amine-directed selective activation of inert C(sp3)–H bonds for C(sp3)–H/C(sp2)–H cross-coupling is outlined in Scheme 3. The initial interaction of Cu(OAc)2 with amine 1 generates the nitrogen radical I,11 which selectively abstracts the hydrogen atom at the remote aliphatic C–H bond via a 1,5-hydrogen-atom transfer to afford the carbon center radical II. The carbon radical II is subsequently captured by a phenyl ring, and the final 6-membered product 2 is afforded after the following oxidation and deprotonation.
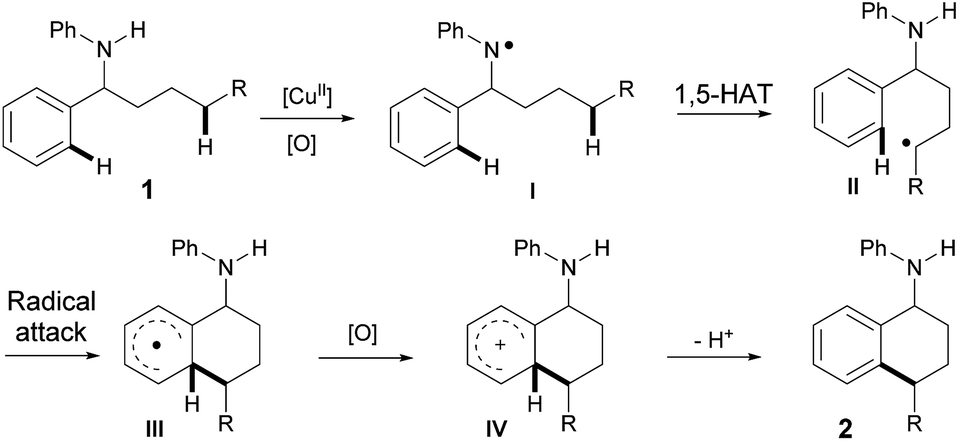 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In summary, we have developed a copper-catalyzed amine-directed selective activation of C(sp3)–H bonds for cross-coupling of inert C(sp3)–H and C(sp2)–H bonds. The catalytic cycle was initiated from the selective cleavage of inert secondary or tertiary C(sp3)–H bonds via a 1,5-HAT process, affording the 6-membered-ring products with high efficiency and broad scope. Further efforts to develop more novel transformations with this method are still ongoing in our lab.Acknowledgements
We gratefully acknowledge the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000), the National Basic Research Program of China (973 Program 2015CB856600), the National Science Foundation of China (21602213, 21522208, 21372209), and the Fundamental Research Funds for the Central Universities (WK2060190046) for financial support.Notes and references
- (a) R. L. Augustine, Carbon–Carbon Bond Formation, Dekker, New York, 1979, vol. 1 Search PubMed; (b) E. J. Corey and X. M. Cheng, The Logic of Chemical Synthesis, John Wiley & Sons, New York, 1989 Search PubMed; (c) B. M. Trost and I. Fleming, Comprehensive Organic Synthesis, Pergamon, Oxford, 1991, vol. 3 Search PubMed.
- For selected reviews on C–H activation: (a) A. E. Shilov and G. B. Shul’pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS PubMed; (b) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS PubMed; (c) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS PubMed; (d) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (e) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (f) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (g) L.-M. Xu, B.-J. Li, Z. Yang and Z.-J. Shi, Chem. Soc. Rev., 2010, 39, 712 RSC; (h) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC.
- For recent reviews: (a) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (b) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (c) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (d) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (e) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS PubMed.
- For selected reviews: (a) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (b) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; For selected examples: (c) Z. Li and C.-J. Li, J. Am. Chem. Soc., 2004, 126, 11810 CrossRef CAS PubMed; (d) L. Zhao, O. Baslé and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4106 CrossRef CAS PubMed; (e) G. Deng and C.-J. Li, Org. Lett., 2009, 11, 1171 CrossRef CAS PubMed; (f) Y.-Z. Li, B.-J. Li, X.-Y. Lu, S. Lin and Z.-J. Shi, Angew. Chem., Int. Ed., 2009, 48, 3817 CrossRef CAS PubMed; (g) Y.-X. Jia and E. P. Kündig, Angew. Chem., Int. Ed., 2009, 48, 1636 CrossRef CAS PubMed; (h) X. Guo and C.-J. Li, Org. Lett., 2011, 13, 4977 CrossRef CAS PubMed; (i) G. Zhang, J. Miao, Y. Zhao and H. Ge, Angew. Chem., Int. Ed., 2012, 51, 8318 CrossRef CAS PubMed; (j) A. P. Antonchick and L. Burgmann, Angew. Chem., Int. Ed., 2013, 52, 3267 CrossRef CAS PubMed; (k) Y. Aihara, M. Tobisu, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2014, 136, 15509 CrossRef CAS PubMed; (l) F. Mo and G. Dong, Science, 2014, 345, 68 CrossRef CAS PubMed.
- (a) D. R. Stuart and K. Fagnou, Science, 2007, 316, 1172 CrossRef CAS PubMed; (b) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540 CAS; (c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (d) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed.
- G. Deng, L. Zhao and C.-J. Li, Angew. Chem., Int. Ed., 2008, 47, 6278 CrossRef CAS PubMed.
- (a) B. Liégault and K. Fagnou, Organometallics, 2008, 27, 4841 CrossRef; (b) C. Pierre and O. Baudoin, Tetrahedron, 2013, 69, 4473 CrossRef CAS.
- For the Hofmann–Löffler–Freytag (HLF) reaction and its various examples: (a) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1883, 16, 558 CrossRef; (b) K. Löffler and C. Freytag, Ber. Dtsch. Chem. Ges., 1909, 42, 3427 CrossRef; (c) E. J. Corey and W. R. Hertler, J. Am. Chem. Soc., 1960, 82, 1657 CrossRef CAS; (d) M. E. Wolff, Chem. Rev., 1963, 63, 55 CrossRef CAS; (e) K. Chen, J. M. Richter and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 7247 CrossRef CAS PubMed; (f) K. Chen and P. S. Baran, Nature, 2009, 459, 824 CrossRef CAS PubMed; (g) H. F. Motiwala, B. Gülgeze and J. Aubé, J. Org. Chem., 2012, 77, 7005 CrossRef CAS PubMed; (h) C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2015, 54, 8287 CrossRef PubMed; (i) C. Yamamoto, K. Takamatsu, K. Hirano and M. Miura, J. Org. Chem., 2016, 81, 7675 CrossRef CAS PubMed; (j) W. Shu and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1881 CrossRef CAS PubMed.
- For selected reviews on hydrogen-atom-transfer: (a) J. Robertson, J. Pillai and R. K. Lush, Chem. Soc. Rev., 2001, 30, 94 RSC; (b) S. Chiba and H. Chen, Org. Biomol. Chem., 2014, 12, 4051 RSC; (c) M. Nechab, S. Mondal and M. P. Bertrand, Chem.–Eur. J., 2014, 20, 16034 CrossRef CAS PubMed.
- For amide-directed photoredox-catalyzed selective cleavage of sp3 C–H bonds, see: (a) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268 CrossRef CAS PubMed; (b) J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272 CrossRef PubMed.
- For copper-mediated generation of N-centered radicals from N–H bonds, see: (a) G. Speier and L. Párkányi, J. Org. Chem., 1986, 51, 218 CrossRef CAS; (b) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (c) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed.
- R. F. Yoshimura, D. J. Hogenkamp, W. Y. Li, M. B. Tran, J. D. Belluzzi, E. R. Whittemore, F. M. Leslie and K. W. Gee, J. Pharmacol. Exp. Ther., 2007, 323, 907 CrossRef CAS PubMed.
- (a) G. S. Ananchenko and H. Fischer, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3604 CrossRef CAS; (b) S. Maity, S. Manna, S. Rana, T. Naveen, A. Mallick and D. Maiti, J. Am. Chem. Soc., 2013, 135, 3355 CrossRef CAS PubMed.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef PubMed.
- The substrates containing other N-protecting groups different to arenes, including H, Et, Ac, (CO)CF3, and Ts, all failed to produce the desired products (Fig. S1, see ESI†).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc00250e |
| This journal is © The Royal Society of Chemistry 2017 |