
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Manganese-catalyzed allylation via sequential C–H and C–C/C–Het bond activation†
Qingquan
Lu
 ,
Felix J. R.
Klauck
and
Frank
Glorius
*
,
Felix J. R.
Klauck
and
Frank
Glorius
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de
First published on 24th February 2017
Abstract
Manganese-catalyzed sequential C–H and C–C/C–Het bond activation to synthesize allylic alcohols, allylated arenes, functionalized cyclopentenes and skipped dienes is reported. This protocol can be readily scaled up and various coupling partners are applied in manganese catalysis for the first time. Moreover, manganese-catalyzed alkenyl C(sp2)–H activation is also shown. Complimentary to the standard solution-based protocols, these reactions also proceed efficiently under neat conditions, which is unprecedented for abundant metal catalyzed C–H activation reactions.
Complimentary to the noble fifth- and sixth-row metals, direct C–H activation1 using 3d-transition metal catalysis has fascinated chemists owing to their abundance, low price and low toxicity, as well as to their potential to promote novel reactivity.2 Over the past years, the goal of achieving sustainability in organic synthesis has propelled important research in this field and significant progress has been made. Base-metals with flexible redox ability, such as Fe,3 Co,4 Ni,5 and Cu6 are extensively used in organometallic C–H activation today. In contrast to being the third most abundant transition metal, manganese is comparatively underutilized.7
Manganese-mediated stoichiometric C–H activation has been explored since the 1970s, however, catalytic variants of these reactions have proved challenging.7 Recently, the groups of Kuninobu and Takai, Wang, Ackermann and others have significantly advanced this field of research.8 Manganese catalysts have been found to be versatile as they can display unique reactivity and enable C–H functionalization with a variety of coupling partners containing polar multiple bonds.8 Mechanistically, these reactions mainly involve the formal addition of a metallacycle to an unsaturated reaction partner or a substitution reaction.8 In recent years, considerable efforts have been made to develop processes that can merge C–H activation with challenging C–C/C–Het cleavage reactions, which could allow for the efficient introduction of two different functional groups into one molecule in a single step.9 However, most of the examples reported to date suffer from the requirement for precious transition metal catalysts and stoichiometric activators. Very recently, a manganese-catalyzed substitutive C–H allylation through highly selective C–H/C–O functionalization was achieved by Ackermann et al.8k Owing to our continuous interest in 3d-transition metal catalysis, we questioned whether manganese catalysis can serve as an alternative route to integrating C–H activation with β-carbon/-hetero atom elimination, which is largely unexplored in this field.
To date, cyclometalation has been the most straightforward and common method for the activation of C–H bonds. Such processes rely mainly on solvent-based techniques. From a sustainability perspective, solvent-free C–H activation processes are highly desirable. Recently, rhodium(III) and iridium(III) catalyzed C–H functionalizations under solvent-free conditions using a ball mill have been reported by Bolm and co-workers.10 However, to the best of our knowledge, first-row transition metal catalyzed C–H activation under neat conditions has not been developed thus far. Herein, our manganese catalyzed coupling offers an environmentally friendly, operationally simple alternative to the more traditional solvent-based protocols, featuring a cheap catalytic system. In this report, various coupling partners, including vinyl-1,3-dioxolan-2-one, 2-vinyloxirane, vinylcyclopropane and diazabicycle, are applied in manganese catalysis for the first time, leading to the convenient synthesis of allylic alcohols, allylated arenes, functionalized cyclopentenes and skipped dienes (Scheme 1).
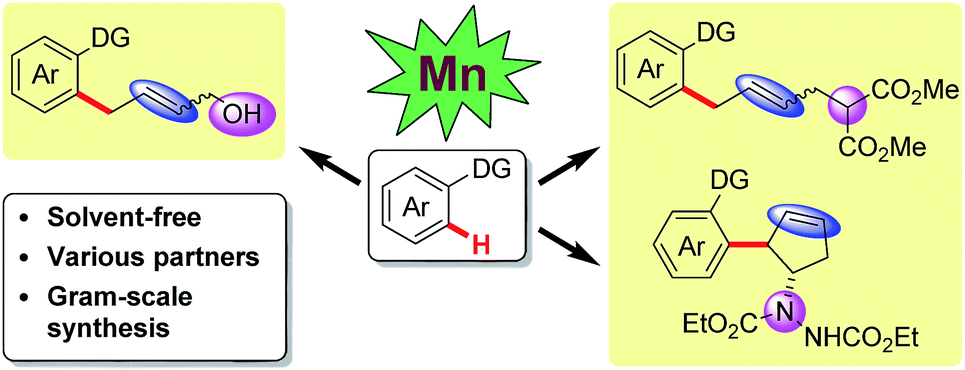 | ||
| Scheme 1 Mn-catalyzed sequential C–H and C–C/C–Het activation. | ||
We started our investigation by reacting N-(2-pyridyl)-indole (1a) with vinyl-1,3-dioxolan-2-one (2aa) under [Mn2(CO)10] catalysis in diethyl ether at 80 °C. This transformation involves the cleavage of a stable C–O bond to form an easily modifiable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and an alcohol moiety. To our delight, the desired product 3a could be isolated in 41% yield (for details, see Table S1 in the ESI†). Subsequently, employing [MnBr(CO)5] as the catalyst precursor afforded product 3a in 84% yield in the presence of sodium acetate. The yield of 3a could be further improved to 90% by increasing the temperature to 90 °C. Notably, the reaction occurred most efficiently in the absence of solvent and 3a could be isolated in 92% yield. Further experiments showed that Mn(OAc)2·4H2O or Cp*Mn(CO)3in lieu of [MnBr(CO)5] are ineffective. Additionally, manganese is essential for this transformation as in its absence no reaction occurred.
C bond and an alcohol moiety. To our delight, the desired product 3a could be isolated in 41% yield (for details, see Table S1 in the ESI†). Subsequently, employing [MnBr(CO)5] as the catalyst precursor afforded product 3a in 84% yield in the presence of sodium acetate. The yield of 3a could be further improved to 90% by increasing the temperature to 90 °C. Notably, the reaction occurred most efficiently in the absence of solvent and 3a could be isolated in 92% yield. Further experiments showed that Mn(OAc)2·4H2O or Cp*Mn(CO)3in lieu of [MnBr(CO)5] are ineffective. Additionally, manganese is essential for this transformation as in its absence no reaction occurred.
With the optimized reaction conditions in hand, the generality of this protocol was first tested by the reaction of indole heterocycles with 2aa and the results are given in Scheme 2. Compared to the neat conditions, our studies showed that the product 3a could be isolated in higher E/Z ratios when diethyl ether was used. Indoles substituted with reactive electrophilic functional groups which can undergo subsequent functionalization, such as the halides (–F, –Br, –I) and cyano substituents, were well tolerated. Introduction of a methyl or formyl group at the 3-position of the indole ring had no influence on the reaction efficiency (3f–3h), indicating that the reaction tolerates steric bulk in proximity of the reaction center of 1. Moreover, this protocol was not restricted to indole substrates but also amenable to benzene- and thiophene-containing substrates, furnishing the corresponding products 3i–3k in good yields. Furthermore, an N-pyrimidyl moiety could be employed as a directing group and the expected product 3l was isolated in 79% yield. Notably, this reaction also exhibited high efficiency on a large scale. The desired product 3a could be isolated in 91% yield (1.44 g) on a 6 mmol scale. In addition, this protocol was successfully applicable to 2-vinyloxiranes, as the coupling partner, and the products 3a and 3g were isolated in 54% and 69% yield, respectively.
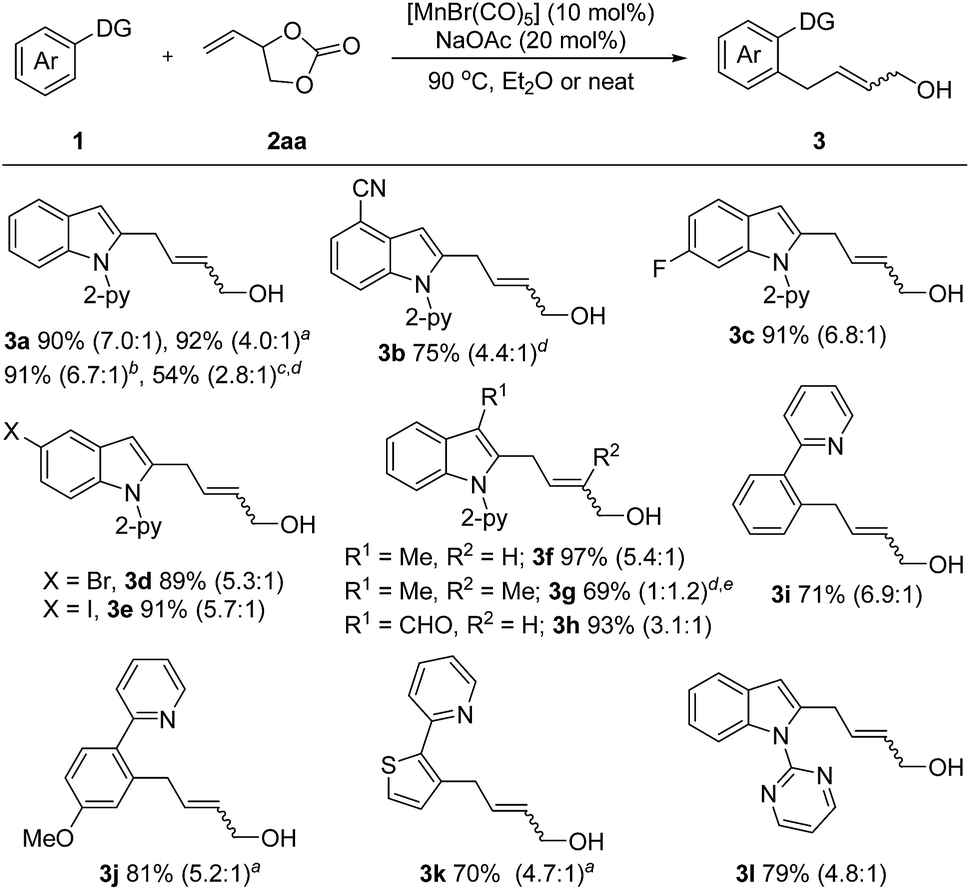 | ||
| Scheme 2 Unless otherwise specified, all reactions were carried out using 1 (0.2 mmol), 2 (0.3 mmol), [MnBr(CO)5] (10 mol%), and NaOAc (20 mol%) in Et2O (1.0 mL) at 90 °C under argon for 5 h, isolated yield, E/Z value given in parentheses. a Neat. b Reaction performed on a 6 mmol scale with a 47 h reaction time. c 2-Vinyloxirane (0.6 mmol) was used instead of 2aa. d 10 h. e 2-Methyl-2-vinyloxirane (0.6 mmol) was used instead of 2aa. | ||
Encouraged by these inspiring results, we then pursued a more challenging successive C–H/C–C activation. Gratifyingly, fine tuning of the reaction conditions allowed the coupling of 1a with dimethyl 2-vinylcyclopropane-1,1-dicarboxylate (2ba) to proceed smoothly under solvent free or concentrated DMF conditions (Scheme 3). Both electron-rich (–OMe) and electron deficient (–F, –Br, –I) N-(2-pyridyl)-indoles are amenable to this method, giving the corresponding products 4b–4e in 52–90% yields. A 3-methyl substituent did not diminish the reactivity, as demonstrated by the formation of the desired products 4f and 4g in excellent yields. Moreover, the scope of the arene substrate could further be extended to phenylpyridine and thiophene derivatives, affording the corresponding products 4h–4j in moderate yields.
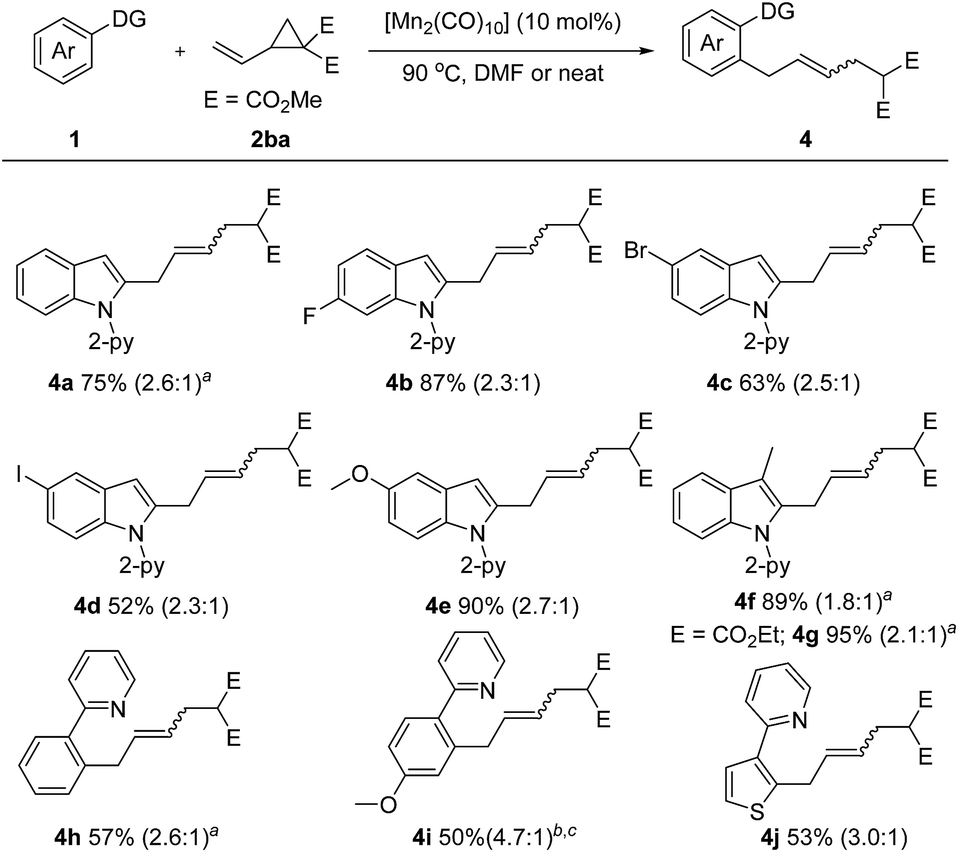 | ||
| Scheme 3 Unless otherwise specified, all reactions were carried out using 1 (0.2 mmol), 2 (0.3 mmol), and [Mn2(CO)10] (10 mol%) in DMF (0.2 mL) at 90 °C under argon for 24 h, isolated yield, E/Z value given in parentheses. a [MnBr(CO)5] (10 mol%) and NaOAc (20 mol%) were used under neat conditions. b Neat. c [Mn2(CO)10] (20 mol%). | ||
Considering the importance of nitrogen-containing compounds and the ease of further transformation on this moiety, we next sought to apply our developed protocol to introduce a vicinal 2-arylated cyclopentenylamine unit. These are known to be key structures found within biologically active small molecules and are key intermediates in the synthesis of pharmaceutically important cyclopentane derivatives.9e Pleasingly, when diazabicycle 2ca was utilized, the desired aryl- and amine-substituted cyclopentenes 5a–5g were obtained in 70–94% yields (Scheme 4). Not only N-(2-pyridyl)-indoles, but also phenylpyridines were suitable substrates. It is important to note that this reaction also proved to be efficient under neat conditions. Arguably, this is the first example of a first-row transition metal catalyzed C–H activation/six-membered ring scission sequence.
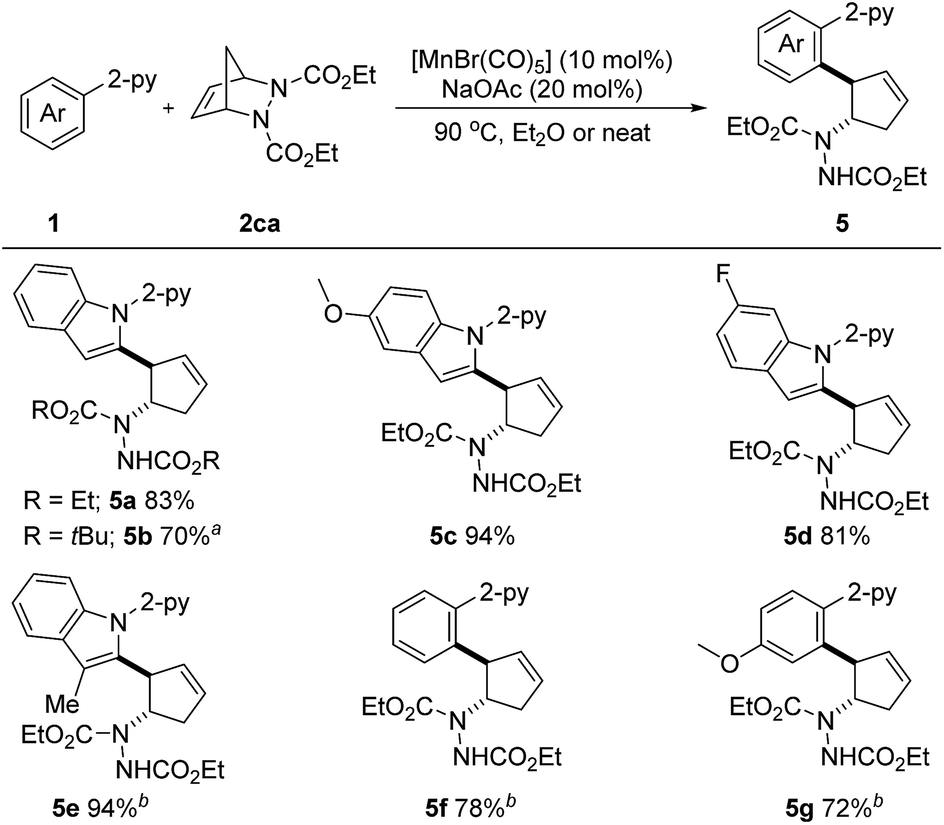 | ||
| Scheme 4 Unless otherwise specified, all reactions were carried out using 1 (0.2 mmol), 2 (0.3 mmol), [MnBr(CO)5] (10 mol%) and NaOAc (20 mol%) in Et2O (1.0 mL) at 90 °C under argon for 10 h, isolated yield. a 37 h. b [MnBr(CO)5] (20 mol%) and NaOAc (40 mol%) were used under neat conditions at 100 °C for 37 h. | ||
Olefinic C–H activation could also be achieved under these conditions. For example, 2-(prop-1-en-2-yl)pyridine (1m) performed well in this transformation and the desired products 3m and 5h, a skipped diene with a valuable handle for further derivatization, were obtained in 75% and 62% yield respectively (Scheme 5).
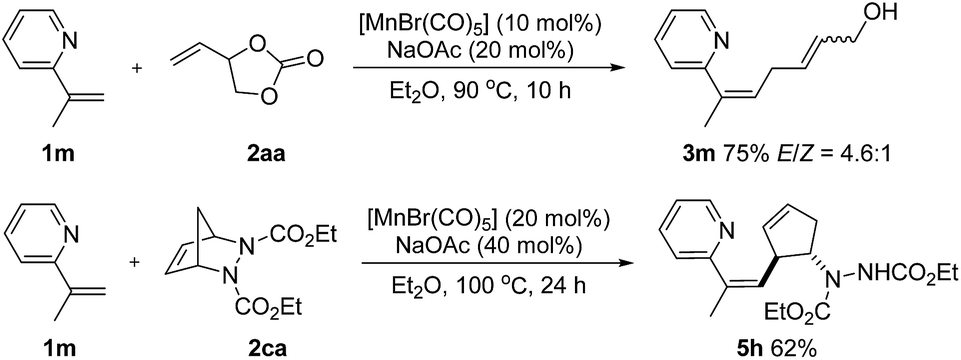 | ||
| Scheme 5 Manganese-catalyzed alkenyl C–H activation. | ||
This transformation is presumed to proceed through an organometallic C–H activation process, as was supported by radical trapping experiments (for details, see Scheme S1 in the ESI†). The reaction of 1a and 2aa under standard conditions was found to be compatible with radical scavengers 2,4-di-tert-butyl-4-methylphenol (BHT) and 1,1-diphenylethylene.
To gain more insight into the reaction mechanism, H/D scrambling experiments were next conducted (for details, see the ESI†). No H/D exchange at the 2-position of 1a was observed when 1a was simply mixed with CD3OD and sodium acetate. Approximately 71% deuterium was incorporated into the 2-position of recovered 1a when sodium acetate was replaced with [MnBr(CO)5]. Furthermore, a larger deuterium incorporation (85%) at the 2-position of 1a was observed when 1a was treated with CD3OD in the presence of both [MnBr(CO)5] and sodium acetate. These results suggest that the C–H activation step is reversible and might occur via a base-assisted cyclometalation process. In addition, a kH/kD = 1.0 was observed from parallel reactions of 1a and [D]-1a with 2aa, respectively, which suggests that the cleavage of the C–H bond is not involved in the rate-determining step. Furthermore, when (3-pyridyl)-thiophene was utilized, the reaction occurred exclusively at the more electron rich 2-position (Scheme 6). On the basis of the above-mentioned results, we may draw the conclusion that the olefin coordination and insertion step is the rate-determining step, and that β-elimination is a facile process.11
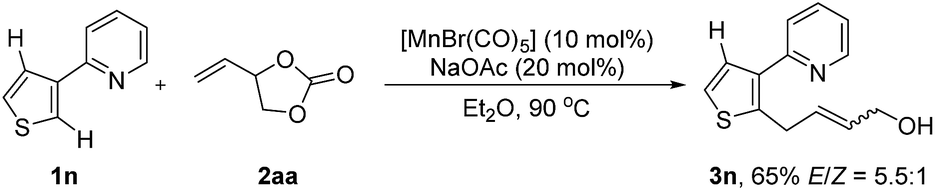 | ||
| Scheme 6 Intramolecular C–H competition experiment. | ||
To acquire a better understanding of the observed reaction selectivity, a series of experiments were performed. As shown in Table 1, no obvious isomerization of the CC bond was observed regardless of prolonged reaction time or decreased reaction temperature (entries 1–3, Table 1). On the contrary, solvent was found to have a dramatic effect on the E/Z selectivity (entries 3–5, Table 1). Compared to the neat reaction, DMF had a negative effect on the final E/Z selectivity, while diethyl ether had a positive effect. From these results, we inferred that the involvement of the π-allylmanganese intermediate in the reaction mechanism might be excluded and that the solvent imparts selectivity during this transformation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Temp./°C | Time/h | Solvent | Yield | Ratio E/Z |
| a All reactions were carried out using 1a (0.2 mmol), 2aa (0.3 mmol), [MnBr(CO)5] (10 mol%) and NaOAc (20 mol%) under different conditions, isolated yield, E/Z ratio is determined by 1H NMR. | |||||
| 1 | 90 | 5 | Et2O | 89 | 7.0 |
| 2 | 90 | 10 | Et2O | 89 | 6.9 |
| 3 | 60 | 10 | Et2O | 57 | 6.8 |
| 4 | 90 | 10 | DMF | 55 | 3.4 |
| 5 | 90 | 5 | — | 92 | 4.0 |

Conclusions
In conclusion, we have developed a general strategy to synthesize allylic alcohols, allylated arenes, functionalized cyclopentenes and skipped dienes, in which earth abundant manganese was utilized as the catalyst.12 This protocol represents a combination of C–H and C–C/C–Het bond activation. Both aromatic and olefinic substrates are functionalized in this reaction. This work broadens the scope of manganese catalysis to include a series of new coupling partners. Additionally, this reaction can occur efficiently under neat conditions yet does not require the use of a large excess of a coupling partner as the solvent, which is unprecedented in abundant metal catalysis. Finally, β-carbon and β-nitrogen elimination were shown to be feasible under low-valent manganese catalysis for the first time.Acknowledgements
This work was supported by the Alexander von Humboldt Foundation (Q. L.), the Deutsche Forschungsgemeinschaft (Leibniz Award for F. G.) and the Fonds der Chemischen Industrie (F. J. R. K.). We also thank Dr Kathryn M. Chepiga and Suhelen Vásquez-Céspedes for helpful discussions.Notes and references
- Selected reviews, see: (a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (d) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (e) L. McMurray, F. O’Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (f) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (g) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (h) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (i) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (j) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (k) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (l) C. Zhu, R. Wang and J. R. Falck, Chem.–Asian J., 2012, 7, 1502 CrossRef CAS PubMed; (m) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (n) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173 RSC; (o) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (p) G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169 RSC; (q) P. Gandeepan and C.-H. Cheng, Chem.–Asian J., 2016, 11, 448 CrossRef CAS PubMed; (r) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC.
- (a) A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087 CAS; (b) M. C. White, Adv. Synth. Catal., 2016, 358, 2364 CrossRef CAS.
- (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS PubMed; (b) E. Nakamura and N. Yoshikai, J. Org. Chem., 2010, 75, 6061 CrossRef CAS PubMed; (c) C. L. Sun, B. J. Li and Z. J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (d) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed.
- (a) N. Yoshikai, Synlett, 2011, 1047 CrossRef CAS; (b) L. Ackermann, J. Org. Chem., 2014, 79, 8948 CrossRef CAS PubMed; (c) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed; (d) P. Gandeepan and C.-H. Cheng, Acc. Chem. Res., 2015, 48, 1194 CrossRef CAS PubMed; (e) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2015, 498 Search PubMed; (f) N. Yoshikai, ChemCatChem, 2015, 7, 732 CrossRef CAS.
- C. L. C. Misal and C. Naoto, Chem. Lett., 2015, 44, 410 CrossRef.
- (a) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (b) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (c) S. Gaillard, C. S. J. Cazin and S. P. Nolan, Acc. Chem. Res., 2012, 45, 778 CrossRef CAS PubMed; (d) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed; (e) J. Liu, G. Chen and Z. Tan, Adv. Synth. Catal., 2016, 358, 1174 CrossRef CAS.
- (a) C. Wang, Synlett, 2013, 24, 1606 CrossRef CAS; (b) W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743 CrossRef CAS.
- (a) Y. Kuninobu, Y. Nishina, T. Takeuchi and K. Takai, Angew. Chem., Int. Ed., 2007, 46, 6518 CrossRef CAS PubMed; (b) B. Zhou, H. Chen and C. Wang, J. Am. Chem. Soc., 2013, 135, 1264 CrossRef CAS PubMed; (c) R. He, Z. T. Huang, Q. Y. Zheng and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 4950 CrossRef CAS PubMed; (d) B. Zhou, P. Ma, H. Chen and C. Wang, Chem. Commun., 2014, 50, 14558 RSC; (e) W. Liu, J. Bang, Y. Zhang and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 14137 CrossRef CAS PubMed; (f) W. Liu, D. Zell, M. John and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 4092 CrossRef CAS PubMed; (g) L. Shi, X. Zhong, H. She, Z. Lei and F. Li, Chem. Commun., 2015, 51, 7136 RSC; (h) B. Zhou, Y. Hu and C. Wang, Angew. Chem., Int. Ed., 2015, 54, 13659 CrossRef CAS PubMed; (i) Y.-F. Liang, L. Massignan, W. Liu and L. Ackermann, Chem.–Eur. J., 2016, 22, 14856 CrossRef CAS PubMed; (j) W. Liu, S. C. Richter, R. Mei, M. Feldt and L. Ackermann, Chem.–Eur. J., 2016, 22, 17958 CrossRef CAS PubMed; (k) W. Liu, S. C. Richter, Y. Zhang and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7747 CrossRef CAS PubMed; (l) S. Sueki, Z. Wang and Y. Kuninobu, Org. Lett., 2016, 18, 304 CrossRef CAS PubMed; (m) N. P. Yahaya, K. M. Appleby, M. Teh, C. Wagner, E. Troschke, J. T. W. Bray, S. B. Duckett, L. A. Hammarback, J. S. Ward, J. Milani, N. E. Pridmore, A. C. Whitwood, J. M. Lynam and I. J. S. Fairlamb, Angew. Chem., Int. Ed., 2016, 128, 12643 CrossRef; (n) X. Yang, X. Jin and C. Wang, Adv. Synth. Catal., 2016, 358, 2436 CrossRef CAS; (o) Y. Hu and C. Wang, Sci. China: Chem., 2016, 59, 1301 CrossRef CAS.
- For examples, see: (a) C. Aïssa and A. Fürstner, J. Am. Chem. Soc., 2007, 129, 14836 CrossRef PubMed; (b) E. Jijy, P. Prakash, M. Shimi, P. M. Pihko, N. Joseph and K. V. Radhakrishnan, Chem. Commun., 2013, 49, 7349 RSC; (c) S. Yu and X. Li, Org. Lett., 2014, 16, 1200 CrossRef CAS PubMed; (d) S.-S. Zhang, J.-Q. Wu, Y.-X. Lao, X.-G. Liu, Y. Liu, W.-X. Lv, D.-H. Tan, Y.-F. Zeng and H. Wang, Org. Lett., 2014, 16, 6412 CrossRef CAS PubMed; (e) Y. Zhang, Q. Wu and S. Cui, Chem. Sci., 2014, 5, 297 RSC; (f) J.-Q. Wu, Z.-P. Qiu, S.-S. Zhang, J.-G. Liu, Y.-X. Lao, L.-Q. Gu, Z.-S. Huang, J. Li and H. Wang, Chem. Commun., 2015, 51, 77 RSC; (g) S.-S. Zhang, J.-Q. Wu, X. Liu and H. Wang, ACS Catal., 2015, 5, 210 CrossRef CAS; (h) L. Kong, S. Yu, G. Tang, H. Wang, X. Zhou and X. Li, Org. Lett., 2016, 18, 3802 CrossRef CAS PubMed; (i) S. Sharma, S. H. Han, Y. Oh, N. K. Mishra, S. Han, J. H. Kwak, S.-Y. Lee, Y. H. Jung and I. S. Kim, J. Org. Chem., 2016, 81, 2243 CrossRef CAS PubMed; (j) D. Zell, Q. Bu, M. Feldt and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7408 CrossRef CAS PubMed.
- (a) G. N. Hermann, P. Becker and C. Bolm, Angew. Chem., Int. Ed., 2015, 54, 7414 CrossRef CAS PubMed; (b) J. G. Hernandez and C. Bolm, Chem. Commun., 2015, 51, 12582 RSC; (c) G. N. Hermann, P. Becker and C. Bolm, Angew. Chem., Int. Ed., 2016, 55, 3781 CrossRef CAS PubMed.
- A mechanism was proposed in Scheme S3 in the ESI.†.
- Only a trace amount of the desired products can be detected when 4-(hex-1-en-2-yl)-1,3-dioxolan-2-one or dimethyl 2-(prop-1-en-2-yl)cyclopropane-1,1-dicarboxylate was utilized under the optimized conditions.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc00230k |
| This journal is © The Royal Society of Chemistry 2017 |