
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bottom up approach towards artificial oxygenases by combining iron coordination complexes and peptides†
Olaf
Cussó
a,
Michael W.
Giuliano
b,
Xavi
Ribas
 a,
Scott J.
Miller
*c and
Miquel
Costas
*a
a,
Scott J.
Miller
*c and
Miquel
Costas
*a
aInstitut de Química Computacional i Catàlisi (IQCC), Departament de Química, Universitat de Girona, Campus Montilivi, Girona E-17071, Catalonia, Spain. E-mail: miquel.costas@udg.edu
bDepartment of Chemistry and Biochemistry, College of Charleston, South Carolina, USA
cDepartment of Chemistry, Yale University, P.O. Box 208107, New Haven, Connecticut 06520-8107, USA. E-mail: scott.miller@yale.edu
First published on 3rd March 2017
Abstract
Supramolecular systems resulting from the combination of peptides and a chiral iron coordination complex catalyze asymmetric epoxidation with aqueous hydrogen peroxide, providing good to excellent yields and high enantioselectivities in short reaction times. The peptide is shown to play a dual role; the terminal carboxylic acid assists the iron center in the efficient H2O2 activation step, while its β-turn structure is crucial to induce high enantioselectivity in the oxygen delivering step. The high level of stereoselection (84–92% ee) obtained by these supramolecular catalysts in the epoxidation of 1,1′-alkyl ortho-substituted styrenes, a notoriously challenging class of substrates for asymmetric catalysis, is not attainable with any other epoxidation methodology described so far. The current work, combining an iron center ligated to N and O based ligands, and a peptide scaffold that shapes the second coordination sphere, may be seen as a bottom up approach towards the design of artificial oxygenases.
Introduction
Metalloenzymes combine the rich reactivity of transition metal ions with highly structured polypeptide chains in order to perform selective transformations.1 The selectivity of some of these reactions, often unattainable with conventional reagents, makes them very attractive for organic synthesis, and has fueled interest in creating synthetic catalysts that harness the fundamental aspects of the enzymatic reactivity.2–7 A minimalistic approach towards this goal is the use of metallopeptides as catalysts.8–17 Peptides can be considered as low-molecular weight mimics of proteins. They can act as metal ligands and play a role in defining the reactivity of the metal ion, but they are also chemically versatile and can also shape the second coordination sphere of the metal with programmable functional groups capable of engaging in hydrogen bonding, electrostatic, hydrophobic and steric interactions with the substrates. The combination of these elements in turn may translate into distinctive chemo- and regioselective reactions. Furthermore, peptides are chiral and this confers metallopeptides with the potential ability of mediating stereoselective transformations.8–12 Traditional oxidants offer little selectivity in reactions of non-functionalized organic substrates, which remain, collectively, a great challenge in organic chemistry. Novel metallopeptide catalysts, by virtue of these discussed features, can offer effective solutions to this challenge.11Enzymatic oxidations catalyzed by iron oxygenases constitute paradigmatic examples of chemo-, regio- and stereoselective transformations that serve as inspiration for the design of iron oxidation catalysts (Scheme 1). Along this path, iron coordination complexes bearing nitrogen and oxygen-based ligands reproduce basic structural aspects of the first coordination sphere of the metal in non-heme oxygenases. Some of them have been shown to be capable of activating the O–O bond in peroxides to form metal-based oxidants that perform selective C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C oxidation reactions.2,18 Highly enantioselective epoxidations, including those of notoriously difficult substrates have been recently described, but the current substrate scope remains narrow.19–26 A current limitation of this approach is the high sensitivity of the H2O2 activation reaction to the catalyst structure. Unproductive consumption of H2O2, often accompanied by the generation of highly reactive radicals, is very commonly observed when modest changes are made in the first coordination sphere of the effective catalysts.27 The introduction of structural diversity in the second coordination sphere may represent a valuable alternative. In this regard, strategies that implement versatility while avoiding laborious ligand modifications will be particularly valuable. With these considerations in mind, herein we describe highly enantioselective supramolecular oxidation catalysts based on the combination of an iron coordination complex and a peptide. We illustrate the powerful reach of this approach in the highly enantioselective asymmetric epoxidation of 1,1′-alkyl substituted styrenes, a class of substrate that stands as a major problem for the asymmetric epoxidation systems described so far.28–31 Remarkably, high yield, and highly enantioselective epoxidation is accomplished in short reaction times (<30 min.) under mild experimental conditions using hydrogen peroxide as the oxidant with low catalyst loadings.
C oxidation reactions.2,18 Highly enantioselective epoxidations, including those of notoriously difficult substrates have been recently described, but the current substrate scope remains narrow.19–26 A current limitation of this approach is the high sensitivity of the H2O2 activation reaction to the catalyst structure. Unproductive consumption of H2O2, often accompanied by the generation of highly reactive radicals, is very commonly observed when modest changes are made in the first coordination sphere of the effective catalysts.27 The introduction of structural diversity in the second coordination sphere may represent a valuable alternative. In this regard, strategies that implement versatility while avoiding laborious ligand modifications will be particularly valuable. With these considerations in mind, herein we describe highly enantioselective supramolecular oxidation catalysts based on the combination of an iron coordination complex and a peptide. We illustrate the powerful reach of this approach in the highly enantioselective asymmetric epoxidation of 1,1′-alkyl substituted styrenes, a class of substrate that stands as a major problem for the asymmetric epoxidation systems described so far.28–31 Remarkably, high yield, and highly enantioselective epoxidation is accomplished in short reaction times (<30 min.) under mild experimental conditions using hydrogen peroxide as the oxidant with low catalyst loadings.
 | ||
| Scheme 1 | ||
Precedents of this approach for developing selective oxidation catalysts are scarce. The combination of iron salts and peptide libraries with the aim to produce stereoselective epoxidations has been previously investigated by following a combinatorial approach. The optimized catalyst produced trans-β-styrene epoxide in up to 78% yield and up to 20% ee.32 On the other hand, peptides are chemically robust structures that withstand oxidation conditions and indeed they are organocatalysts for the asymmetric epoxidation (AE) of enones via a mechanism that involves the generation of basic hydrogen peroxide (Scheme 2a).33–36 More recently, key to the present work, Miller and co-workers have developed a H2O2 activating relay in aspartate based peptide catalysts that enables the formation of peptide-based peracids37–40 and dioxiranes,41 which are mild electrophilic asymmetric epoxidation agents (Scheme 2b and c). It was envisioned that the incorporation of an iron catalyst in these structures may allow the generation of high-valent iron–oxo intermediates, which are powerful oxidizing species, in a structurally rich and chiral environment that will imbue them with enhanced selectivity properties. Key to this approach are recent reports describing the synergistic cooperation of carboxylic acids and chiral iron catalysts in activating H2O2 to perform selective C–H and CC oxidation reactions (Scheme 2d).26
 | ||
| Scheme 2 Peptide-based catalysts for asymmetric epoxidation. (a) Hydroperoxide oxidant through the Juliá-Colonna process.36 (b) Peracid peptide oxidant for AE.37 (c) Dioxirane-peptide oxidant for AE.41 (d) Peptide for AE with an iron catalyst. | ||
Results and discussion
Catalyst development
Our initial investigation of this approach entailed the epoxidation of α-methylstyrene S1, employing (S,S′)-[Fe(CF3SO3)2(Me2Npdp)] ((S,S′)Me2N1Fe) iron catalyst (Scheme 2d) and its analogous (R,R′) enantiomer (2 mol%), a peptide (4 mol%), (details on the preparation, and characterization of the peptides are collected in the ESI†), and hydrogen peroxide as oxidant (2.3 equiv. added via syringe pump during 30 min.) in acetonitrile solution at 0 °C. Iron complex (S,S′)Me2N1Fe was chosen because it has been recently shown to be particularly efficient in activating H2O2 requiring only catalytic amounts of carboxylic acid co-catalysts.42 The terminal carboxylic acid of the peptide was envisioned to play this role in the current catalytic system. On the other hand, peptides chosen in the initial screening (see ESI†) include peptides of different lengths, as well as peptides that may be biased toward secondary structures like β-turns.43,44 Reactions were analyzed using chiral HPLC to determine conversions and stereoselectivities. The results were collected in the ESI Table S1,† and results for representative examples of different peptides tested are shown in Scheme 3. | ||
| Scheme 3 (Top) Diagrams of five representative peptides studied. (Bottom) Enantioselectivities produced in the asymmetric epoxidation of α-methylstyrene (S1) with Me2N1Fe and the corresponding peptide. Conditions: 2 mol% of Me2N1Fe, 2.3 equiv. of H2O2, and 4 mol% of peptide in acetonitrile at 0 °C for 30 min. Dark blue bars show the results with enantiomer S,S′ of the catalyst and light blue with enantiomer R,R′ of the catalyst. ee’s determined using HPLC. | ||
Reactions finished immediately after peroxide addition and product conversions ranged from 57% to 87%, indicating that the system efficiently catalyzes the activation of H2O2 and epoxidizes the olefin with good chemoselectivity. Enantiomeric ratios range from a poor 52.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :47.5 (5% ee) to a moderate 72.5:27.5 (45% ee), indicating that the peptide plays a major role in dictating the enantioselectivity of the reactions (See ESI, Table S1†). In all cases, for a given peptide, the chirality of the major epoxide enantiomer is dependent on the chirality of the metal complex; the (S,S′)Me2N1Fe catalyst produces predominantly S-epoxide (S)-P1, while (R)-P1 is the main enantiomer of (R,R′)Me2N1Fe catalyzed reactions. In general, for a given peptide, the enantioselectivity of P1 obtained when using (S,S′)Me2N1Fe and (R,R′)Me2N1Fe differs modestly (<10%) but in specific cases differences in ee of > 25 percentage units are observed (see peptide 2 in Scheme 3). Interestingly, a comparative analysis of the results obtained with this series of peptides shows that relatively large and possibly more flexible peptides 1–4 provide worse enantioselectivities (6–38% ee) than the more rigid peptide 5, which provides up to 45% ee, suggesting that the β-turn structure is key for this improvement.
:47.5 (5% ee) to a moderate 72.5:27.5 (45% ee), indicating that the peptide plays a major role in dictating the enantioselectivity of the reactions (See ESI, Table S1†). In all cases, for a given peptide, the chirality of the major epoxide enantiomer is dependent on the chirality of the metal complex; the (S,S′)Me2N1Fe catalyst produces predominantly S-epoxide (S)-P1, while (R)-P1 is the main enantiomer of (R,R′)Me2N1Fe catalyzed reactions. In general, for a given peptide, the enantioselectivity of P1 obtained when using (S,S′)Me2N1Fe and (R,R′)Me2N1Fe differs modestly (<10%) but in specific cases differences in ee of > 25 percentage units are observed (see peptide 2 in Scheme 3). Interestingly, a comparative analysis of the results obtained with this series of peptides shows that relatively large and possibly more flexible peptides 1–4 provide worse enantioselectivities (6–38% ee) than the more rigid peptide 5, which provides up to 45% ee, suggesting that the β-turn structure is key for this improvement.
The data also suggests that the relative position of the carboxylic acid moiety in the peptide also plays an important role in determining the stereoselectivity of the reactions. This is best exemplified by analyzing catalytic epoxidations under standard conditions using peptides 1–5. Pairs of peptides 1/2 and 3/4 contain analogous peptide structures but differ in the position of the carboxylic acid moiety. Within each of the two pairs, we observed substantial differences in terms of enantioselectivity (Scheme 3). These are complex peptides and structure–activity correlations could not be easily deduced. Furthermore, some caution should be taken when making this comparison because this change may also impact on the overall structure and rigidity of the peptide. However, the comparison among the series of peptides suggests that the enantioselectivity of the oxygen atom transfer event depends on the chemical architecture of the region of the peptide in close proximity to the carboxylic acid moiety. The simplest interpretation of this data is that the carboxylic acid moiety acts as a ligand of the iron catalyst, thus resembling the role of simple alkyl carboxylic acids with this kind of iron catalyst.20a,42,45–49
Reasoning that the β-turn is a key structural element for stereoselectivity in the epoxidation reaction with Me2N1Fe, a set of peptides sharing this basic secondary structure but bearing diverse modifications were studied (Scheme 4). Interestingly, peptide 7, which contains a 4-hydroxyproline and is conformationally different than the peptides with the Aib residue (5, 6, 8, 9 and 10), showed the best enantioselectivity, which was up to 58% ee when combined with (S,S′)Me2N1Fe. The combination of (R,R′)Me2N1Fe with 7 resulted in a substantially lower ee (32% ee), indicating an effective translation of the matching/mismatching of the chirality of 7 and Me2N1Fe in the enantioselectivity of the epoxidation. Also illustrative, we observed low enantioselectivities for peptide (10), which lacks the capacity to form a second hydrogen bond. Moreover the Dproline group is also important because when it is replaced by Dhomoproline an erosion of enantioselectivity was observed (peptide 8). Also the modification of the first or last residue (peptides 6 and 9) showed worse enantioselectivities in comparison with peptide 5. In conclusion, this set of reactions led to the identification of 7 as the optimal peptide within the series. Furthermore, enantioselectivity may correlate with the presence of a peptide with a rigid, well-defined secondary structure.
 | ||
| Scheme 4 (Top) Schematic diagrams of six different peptides derived from 5 (blue colored residues indicated sequence changes relative to peptide 5). (Bottom) Enantioselectivities for the asymmetric epoxidation of α-methylstyrene with these peptides and Me2N1Fe. Conditions: 2 mol% of Me2N1Fe, 2.3 equiv. of H2O2, and 4 mol% of peptide in acetonitrile at 0 °C for 30 min. Dark blue bars show the results with enantiomer S,S′ of the catalyst and light blue with enantiomer R,R′ of the catalyst. ee’s determined using HPLC. | ||
Characterization of the peptide–iron catalyst synergy
Control experiments were performed in order to provide understanding into the role of the peptide and the iron complex in the H2O2 activation step (Table 1). Standard reaction conditions employed for comparison involved the use of catalyst Me2N1Fe (2 mol%), peptide 7 (4 mol%) and H2O2 (2.3 equiv.) in acetonitrile at 0 °C. Under these conditions, epoxide (S)-P1 is obtained in 81% yield and 58% ee (Table 1, entry 1). Instead, in the absence of Me2N1Fe no epoxide product was formed (Table 1, entry 2). This rules out the possibility that H2O2 is activated solely by the peptide, for example by formation of basic peroxide, or via generation of an organic peracid. Also the replacement of Me2N1Fe by Fe(OTf)2(CH3CN)2 under standard reaction conditions did not produce epoxide product (Table 1, entry 3). Most interestingly, the reaction in the absence of peptide 7 only showed epoxide traces (Table 1, entry 4). Therefore, both the peptide and the iron complex Me2N1Fe are necessary for efficient activation of H2O2. Enantioselectivity is not affected when equimolar ratios of Me2N1Fe and peptide 7 are employed, but the use of a 2:1 ratio resulted in slightly decreased enantioselectivity (53%). This set of observations strongly suggests that the active catalysts result from the equimolar combination of iron complex and peptide. As expected, the importance of the terminal carboxylic acid moiety of the peptide in the H2O2 activation was demonstrated, because low yield (12%) and enantioselectivity (21% ee) of epoxide P1 was observed when a terminal ester group was used in its place (Table 1, entry 5). Furthermore, the observation of matching/mismatching between the stereoisomer of the iron catalyst and peptide 7 with respect to reaction enantioselectivity strongly suggests an inner sphere interaction via the binding of the iron center by the peptide carboxylate. Characterization of these complexes is difficult because of their labile nature and because of the paramagnetic nature of the iron species. However, evidence in favor of their formation could be gathered using high resolution mass spectrometry (HRMS) under catalytic conditions. HRMS spectra collected during a catalytic epoxidation under standard conditions using a combination of (S,S′)Me2N1Fe and 7 as catalyst show a dominant peak at m/z = 1200.4962, that could be assigned to {(S,S′)-[LFeIII(7′)](CF3SO3)}+ species (where 7′ = 7-H+ and L = MeNpdp) on the basis of their mass and isotopic pattern distribution. This species can be recognized as analogous to the carboxylate bound {(S,S′)-[LFeIII(OAc)](CF3SO3)}+ species, previously observed as the resting state in catalytic reactions with acetic acid.50 These species react with H2O2 producing the putative FeVO species finally responsible for the oxygen atom transfer to the olefin.42 Therefore, despite the fact that conclusive experimental evidence for carboxylate binding could not be obtained, the sum of the experimental observations led us to conclude that the catalytic activity should be attributed to {(S,S′)-[LFeIII(7′)](CF3SO3)}2+, in which the carboxylate moiety of the peptide presumably acts as a ligand. Therefore, we propose that this catalytic system reproduces key structural features of a number of non-heme iron dependent oxygenases; it contains a first coordination sphere that combines heterocyclic amines, carboxylate anions and labile sites, and the carboxylate group connects the metal center to an amino acid chain that participates in defining its second coordination sphere.51 The synergistic interplay of these elements is necessary to enable the efficient activation of H2O2 and the highly chemo- and enantioselective oxygen atom transfer.
| Entry | Iron source (2 mol%) | Pept. 7 (mol%) | H2O2 (eq.) | Conv. (yield) (%) | ee (%) |
|---|---|---|---|---|---|
| a Conditions: 2 mol% of iron source, 2.3 equiv. of H2O2, and 4% mol of peptide in acetonitrile at 0 °C for 30 min. b Not available. c The peptide contains a terminal ester instead of carboxylic acid (peptide 7-MeO, see ESI for peptide structure), also 5% yield of acetophenone was observed. Substrate conversion, yields and ee’s determined using HPLC. | |||||
| 1 | (S,S′)Me2N1Fe | 4 | 2.3 | 92(81) | 58 |
| 2 | — | 4 | 2.3 | —(—) | — |
| 3 | Fe(OTf)2(CH3CN)2 | 4 | 2.3 | —(—) | — |
| 4 | (S,S′)Me2N1Fe | — | 2.3 | Traces | NAb |
| 5c | (S,S′)Me2N1Fe | 4 | 2.3 | 23(12) | 21 |
The combination of (S,S′)Me2N1Fe or (R,R′)Me2N1Fe with the peptide will form different diastereoisomers, which a priori may exhibit different reactivities with H2O2, which in the current reaction is the rate determining step. Should this be the case, one of the diastereomers will dominate the outcome, resulting in non-linear effects on the overall enantioselectivity. In order to investigate this possibility, a study of the enantiomeric purity of the iron catalyst Me2N1Fe as a function of enantioselectivity in the epoxidation of S4 was performed. When the reaction was performed with a racemic mixture of Me2N1Fe and peptide 7, the epoxide was obtained with only 10% ee (see ESI† for details), and a good linear correlation between the ee of the iron catalysts and the epoxide was observed (see ESI Fig. SI. 2†) discarding the presence of non-linear effects. This analysis indicates that the activation of H2O2 by (S,S′)Me2N1Fe and 7, or (R,R′)Me2N1Fe and 7 occurs at indistinguishable rates, but the stereoselectivity of the oxygen atom transfer event is different.
Substrate scope
With the optimal peptide in hand, the asymmetric epoxidation of different α-methylstyrene derivatives was studied. Reactions were performed at −30 °C, in order to obtain improved enantioselectivities (Table 2). For meta-, para- and non-substituted derivatives (S1–S3), the enantioselectivities obtained were moderate, from 64% to 68% (Table 2, entry 1–3). But in the case of the ortho-methyl substituted styrene derivative (S4) the enantioselectivity increased up to 92% (Table 2, entry 4). The ability of peptide 7 is exclusive in order to provide high enantioselectivity in the epoxidation of ortho-susbtituted α-methylstyrene substrates. This is best illustrated when reactions were performed using carboxylic acid co-ligands previously described in asymmetric iron catalyzed oxidations; these included acetic acid, 2-ethylhexanoic acid, S-ibuprofen and Npha-Ileu-OH (see Fig. 1). The last three acids were identified in previous works as the best partners for Me2N1Fe in the asymmetric epoxidation of a wide range of substrates, including α-methylstyrene substrates.42,45 In all cases, the ee’s were substantially lower than those obtained with peptide 7 in the AE of S4. For example the best result at 0 °C was obtained with S-ibuprofen and (S,S′)Me2N1Fe, providing the corresponding epoxide with 68% ee that improved up to 78% ee at −30 °C. Under analogous conditions (0 °C), peptide 7 combined with (S,S′)Me2N1Fe provided a remarkable 81% ee, which improved up to 92% ee at −30 °C. The electron rich catalyst (S,S′)Me2N1Fe appears to be crucial in terms of yield and enantioselectivity; when (S,S′) and (R,R′)-[Fe(CF3SO3)2(pdp)] were employed instead of (S,S′)Me2N1Fe, the epoxidation of S4 proceeds with low yields and enantioselectivities (7% yield (31% ee) and 16% yield (9% ee), respectively).




 | ||
| Fig. 1 Structures of different carboxylic acids and the enantioselectivities obtained with both enantiomers of Me2N1Fe in the catalytic epoxidation of S4. Conditions: 2 mol% of catalyst, 2.3 equiv. of H2O2, and 4% mol of peptide in acetonitrile at 0 °C and −30 °C for 30 min. | ||
A series of ortho-substituted α-methylstyrene derivatives were tested in the asymmetric epoxidation reaction (Table 3). We observed good tolerance of the system to the introduction of different halogen groups (Cl, F and Br) in the ortho position, showing in all cases good to excellent yields and enantioselectivities, up to 82% and 90%, respectively (Table 3, entries 1–3). In the case of the nitro derivative S10, with a NO2 group in the meta-position, excellent yield and very good enantioselectivity, 99% yield and 86% ee (entry 6) were obtained. Finally, this methodology showed excellent enantioselectivities for cis-aromatic olefins (S11 and S12), which to the best of our knowledge are the best reported so far with iron catalysts, up to 91% ee (entry 7 and 8).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Isol. yield (%) | (ee)% |
| a Unless stated, reaction conditions are 2 mol%, (S,S′)Me2N1Fe, peptide (4 mol%), and H2O2 (2.3 equiv.) in CH3CN at −30 °C for 30 min. b Substrate conversion and epoxide yields (in parenthesis) determined using GC. ee’s determined using chiral GC and HPLC. | |||
| 1 |
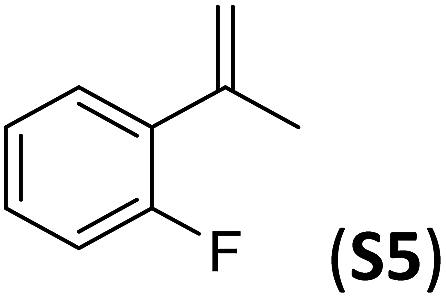
|
79(61)b | 86 |
| 2 |

|
82 | 84 |
| 3 |

|
67 | 90 |
| 4 |

|
88 | 90 |
| 5 |

|
66 | 90 |
| 6 |

|
99 | 86 |
| 7 |

|
100(84)b | 90 |
| 8 |

|
70 | 91 |

These results must be placed into context. α-Alkyl substituted styrenes are recognized as particularly challenging substrates for asymmetric catalysis.52 Furthermore, for the specific case of ortho-substituted α-methylstyrene derivatives there are no epoxidation systems in the literature describing high stereoselectivities, and enantioenriched products could only be obtained via the hydrolytic kinetic resolution of ortho-substituted styrene epoxide derivatives.53 Therefore, the current catalysts can be regarded as a promising platform for the identification of peptides to improve the enantioselectivity of Fe catalysts.
Conclusions
This work describes a supramolecular system based on the combination of a chiral iron coordination complex and a peptide with a β-turn design that activates H2O2 and performs high enantioselective epoxidation of α-alkyl-substituted and cis-substituted styrene derivatives. Reactions are performed in short reaction times, employing aqueous hydrogen peroxide as oxidant, and ee’s are remarkably high for substrates that are recognized as particularly difficult, and which lack satisfactory alternatives. The carboxylic acid moiety of the peptide is shown to help the iron center in the activation of the H2O2 while the rigid well-defined 3D architecture of the peptide appears to be crucial for eliciting high enantioselectivities. This combination of an iron center ligated to N and O based ligands, and a peptide scaffold that contributes to shaping the second coordination sphere may be seen as a bottom up approach towards the design of artificial peroxigenases. Furthermore, this approach is envisioned to have a powerful reach. Multiple catalytic systems could be envisioned, based on the chemical versatility of peptides, without the need to manipulate the iron catalyst. The current system appears to rely on the structural architecture of the peptide and operates on non-functionalized olefins. The rich functional group versatility of the peptides may serve to introduce residues with additional properties such as charge, H-bonding, and hydrophobic interactions, among others. These interactions are presently employed in highly enantioselective organocatalysis, and may boost the substrate scope and overall performance of the current catalytic system.Acknowledgements
MC acknowledge financial support from MINECO of Spain and Fondo Europeo de Desarrollo Regional (CTQ2015-70795-P/BQU), and the Catalan Department for Innovation, Universities and Enterprise (DIUE) of the Generalitat de Catalunya (2009SGR637). SJM is grateful to the NIGMS of the NIH for support (GM-096403). M. C. and X. R. thank ICREA-Acadèmia awards. We thank STR’s from UdG for technical support.Notes and references
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239 CrossRef CAS PubMed.
- L. Que Jr and W. B. Tolman, Nature, 2008, 455, 8 CrossRef PubMed.
- S. Sahu and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 11410 CrossRef CAS PubMed.
- X. Engelmann, I. Monte-Pérez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632 CrossRef CAS PubMed.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942 CrossRef CAS PubMed.
- Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855 CrossRef CAS PubMed.
- T. Heinisch and T. R. Ward, Curr. Opin. Chem. Biol., 2010, 14, 184 CrossRef CAS PubMed.
- R. Sambasivan and Z. T. Ball, J. Am. Chem. Soc., 2010, 132, 9289 CrossRef CAS PubMed.
- R. Sambasivan and Z. T. Ball, Angew. Chem., Int. Ed., 2012, 51, 8568 CrossRef CAS PubMed.
- N. S. Josephsohn, K. W. Kuntz, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2001, 123, 11594 CrossRef CAS PubMed.
- S. R. Gilbertson, S. E. Collibee and A. Agarkov, J. Am. Chem. Soc., 2000, 122, 6522 CrossRef CAS.
- B. Kim, A. J. Chinn, D. R. Fandrick, C. H. Senanayake, R. A. Singer and S. J. Miller, J. Am. Chem. Soc., 2016, 138, 7939 CrossRef CAS PubMed.
- J. C. Lewis, ACS Catal., 2013, 3, 2954 CrossRef CAS.
- J. Bos and G. Roelfes, Curr. Opin. Chem. Biol., 2014, 19, 135 CrossRef CAS PubMed.
- J. C. Lewis, Curr. Opin. Chem. Biol., 2015, 25, 27 CrossRef CAS PubMed.
- W. J. Shaw, Catal. Rev.: Sci. Eng., 2012, 54, 489 CAS.
- P. J. Deuss, R. den Heeten, W. Laan and P. C. Kamer, Chem.–Eur. J., 2011, 17, 4680 CrossRef CAS PubMed.
- W. N. Oloo and L. Que, Acc. Chem. Res., 2015, 48, 2612 CrossRef CAS PubMed.
- A. Fingerhut, O. V. Serdyuk and S. B. Tsogoeva, Green Chem., 2015, 17, 2042 RSC.
- For selected examples of Fe-catalyzed epoxidation reactions see: (a) M. C. White, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 7194 CrossRef CAS PubMed; (b) G. Dubois, A. Murphy and T. D. P. Stack, Org. Lett., 2003, 5, 2469 CrossRef CAS PubMed; (c) G. Anilkumar, B. Bitterlich, F. G. Gelalcha, M. K. Tse and M. Beller, Chem. Commun., 2007, 3, 289 RSC; (d) P. Liu, E. L. Wong, A. W. Yuen and C. Che, Org. Lett., 2008, 10, 3275 CrossRef CAS PubMed; (e) K. Schröder, S. Enthaler, B. Join, K. Junge and M. Beller, Adv. Synth. Catal., 2010, 352, 1771 CrossRef; (f) K. Schroeder, S. Enthaler, B. Bitterlich, T. Schulz, A. Spannenberg, M. K. Tse, K. Junge and M. Beller, Chem.–Eur. J., 2009, 15, 5471 CrossRef CAS PubMed; (g) K. Schröder, B. Join, A. J. Amali, K. Junge, X. Ribas, M. Costas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 1425 CrossRef PubMed; (h) E. A. Mikhalyova, O. V. Makhlynets, T. D. Palluccio, A. S. Filatov and E. V. Rybak-Akimova, Chem. Commun., 2012, 48, 687 RSC; (i) B. F. Perandones, E. D. Nieto, C. Godard, S. Castillon, P. De Frutos and C. Claver, ChemCatChem, 2013, 5, 1092 CrossRef CAS; (j) I. Y. Skobelev, E. V. Kudrik, O. V. Zalomaeva, F. Albrieux, P. Afanasiev, O. A. Kholdeeva and A. B. Sorokin, Chem. Commun., 2013, 49, 5577 RSC; (k) D. Clemente-Tejeda, A. López-Moreno and F. A. Bermejo, Tetrahedron, 2013, 69, 2977 CrossRef CAS; (l) J. W. Kuck, A. Raba, I. I. E. Markovits, M. Cokoja and F. E. Kuhn, ChemCatChem, 2014, 6, 1882–1886 CrossRef.
- Fe systems showing moderate stereoselectivity: (a) C. Marchi-Delapierre, A. Jorge-Robin, A. Thibon and S. Ménage, Chem. Commun., 2007, 11, 1166 RSC; (b) H.-L. Yeung, K.-C. Sham, C.-S. Tsang, T.-C. Lau and H.-L. Kwong, Chem. Commun., 2008, 3801 RSC; (c) F. Oddon, E. Girgenti, C. Lebrun, C. Marchi-Delapierre, J. Pecaut and S. Menage, Eur. J. Inorg. Chem., 2012, 85 CrossRef CAS.
- (a) B. Wang, S. Wang, C. Xia and W. Sun, Chem.–Eur. J., 2012, 18, 7332 CrossRef CAS PubMed; (b) M. Wu, C.-X. Miao, S. Wang, X. Hu, C. Xia, F. E. Kühn and W. Sun, Adv. Synth. Catal., 2011, 353, 3014 CrossRef CAS; (c) O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2012, 2, 1196 CrossRef CAS; (d) Q. F. Cheng, X. Y. Xu, W. X. Ma, S. J. Yang and T. P. You, Chin. Chem. Lett., 2005, 16, 1467 CAS; (e) W. Dai, G. Li, B. Chen, L. Wang and S. Gao, Org. Lett., 2015, 17, 904 CrossRef CAS PubMed; (f) F. G. Gelalcha, G. Anilkumar, M. K. Tse, A. Brückner and M. Beller, Chem.–Eur. J., 2008, 14, 7687 CrossRef CAS PubMed; (g) F. G. Gelalcha, B. Bitterlich, G. Anilkumar, M. K. Tse and M. Beller, Angew. Chem., Int. Ed., 2007, 46, 7293 CrossRef CAS PubMed.
- Fe systems that employ hypervalent iodine oxidants: (a) T. Niwa and M. Nakada, J. Am. Chem. Soc., 2012, 134, 13538 CrossRef CAS PubMed; (b) B. Wang, Y.-M. Lee, M. S. Seo and W. Nam, Angew. Chem., Int. Ed., 2015, 54, 11740 CrossRef CAS PubMed.
- L. Luo and H. Yamamoto, Eur. J. Org. Chem., 2014, 35, 7803 CrossRef PubMed.
- Y. Nishikawa and H. Yamamoto, J. Am. Chem. Soc., 2011, 133, 8432 CrossRef CAS PubMed.
- O. Cusso, X. Ribas and M. Costas, Chem. Commun., 2015, 51, 14285 RSC.
- K. Chen, M. Costas, J. Kim, A. K. Tipton and L. Que Jr, J. Am. Chem. Soc., 2002, 124, 3026 CrossRef CAS PubMed.
- G. De Faveri, G. Ilyashenko and M. Watkinson, Chem. Soc. Rev., 2011, 40, 1722 RSC.
- Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199 CrossRef CAS PubMed.
- R. L. Davis, J. Stiller, T. Naicker, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2014, 53, 7406 CrossRef CAS PubMed.
- C. Wang and H. Yamamoto, Chem.–Asian J., 2015, 10, 2056 CrossRef CAS PubMed.
- M. B. Francis and E. N. Jacobsen, Angew. Chem., Int. Ed., 1999, 38, 937 CrossRef CAS.
- G. Carrea, S. Colonna, A. D. Meek, G. Ottolina and S. M. Roberts, Chem. Commun., 2004, 1412 RSC.
- E. Weitz and A. Scheffer, Ber. Dtsch. Chem. Ges., 1921, 54, 2327 CrossRef.
- Y. Apeloig, M. Karni and Z. Rapoport, J. Am. Chem. Soc., 1983, 105, 2784 CrossRef CAS.
- S. Julia, J. Masana and J. C. Vega, Angew. Chem., Int. Ed., 1980, 19, 929 CrossRef.
- G. Peris, C. E. Jakobsche and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 8710 CrossRef CAS PubMed.
- P. A. Lichtor and S. J. Miller, Nat. Chem., 2012, 4, 990 CrossRef CAS PubMed.
- P. A. Lichtor and S. J. Miller, J. Am. Chem. Soc., 2014, 136, 5301 CrossRef CAS PubMed.
- N. C. Abascal, P. A. Lichtor, M. W. Giuliano and S. J. Miller, Chem. Sci., 2014, 5, 4504 RSC.
- D. K. Romney and S. J. Miller, Org. Lett., 2012, 14, 1138 CrossRef CAS PubMed.
- O. Cussó, I. Garcia-Bosch, X. Ribas, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2013, 135, 14871 CrossRef PubMed.
- T. S. Haque, J. C. Little and S. H. Gellman, J. Am. Chem. Soc., 1996, 118, 6975 CrossRef CAS.
- (a) C. M. Wilmot and J. M. Thornton, J. Mol. Biol., 1988, 203, 221 CrossRef CAS PubMed; (b) A. J. Metrano, N. C. Abascal, B. Q. Mercado, E. K. Paulson, A. E. Hurtley and S. J. Miller, J. Am. Chem. Soc., 2017, 139, 492 CrossRef CAS PubMed.
- O. Cussó, X. Ribas, J. Lloret-Fillol and M. Costas, Angew. Chem., Int. Ed., 2015, 54, 2729 CrossRef PubMed.
- O. Cussó, M. Cianfanelli, X. Ribas, K. R. J. M. Gebbink and M. Costas, J. Am. Chem. Soc., 2016, 138, 2732 CrossRef PubMed.
- X. Wang, C. Miao, S. Wang, C. Xia and W. Sun, ChemCatChem, 2013, 5, 2489 CrossRef CAS.
- R. Mas-Balleste and L. Que Jr, J. Am. Chem. Soc., 2007, 129, 15964 CrossRef CAS PubMed.
- A. M. Zima, O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2017, 7, 60 CrossRef CAS.
- L. Gomez, I. Garcia-Bosch, A. Company, J. Benet-Buchholz, A. Polo, X. Sala, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2009, 48, 5720 CrossRef CAS PubMed.
- (a) E. L. Hegg and L. Que Jr, Eur. J. Biochem., 1997, 250, 625 CAS; (b) E. G. Kovaleva and J. D. Lipscomb, Nat. Chem. Biol., 2008, 4, 186 CrossRef CAS PubMed; (c) S. M. Barry and G. L. Challis, ACS Catal., 2013, 3, 2362 CrossRef CAS PubMed.
- Good ee’s in the asymmetric epoxidation of α-alkystyrenes with no substitutions at the o-position are limited to chloroperoxidase (up to 89% ee), A. F. Dexter, F. J. Lakner, R. A. Campbell and L. P. Hager, J. Am. Chem. Soc., 1995, 117, 6412 CrossRef CAS , Shi’s organocatalysts (up to 88% ee), B. Wang, O. A. Wong, M.-X. Zhao and Y. Shi, J. Org. Chem, 2008, 73, 9539 CrossRef PubMed , and O. A. Wong, B. Wang, M.-X. Zhao and Y. Shi, J. Org. Chem, 2009, 74, 6335 CrossRef PubMed , and the combination of (S,S′)Me2N1Fe with Npha-Ileu-OH (ref. 45).
- V. Capriati, S. Florio, F. M. Perna and A. Salomone, Chem.–Eur. J., 2010, 16, 9778 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of synthesis, characterization and NMR, HPLC and GC spectra. See DOI: 10.1039/c7sc00099e |
| This journal is © The Royal Society of Chemistry 2017 |