
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rh-catalyzed regiodivergent hydrosilylation of acyl aminocyclopropanes controlled by monophosphine ligands†
Hiroki
Kondo
a,
Kenichiro
Itami
ab and
Junichiro
Yamaguchi
 *c
*c
aDepartment of Chemistry, Graduate School of Science and Institute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Chikusa, Nagoya 464-8602, Japan
bJST, ERATO, Itami Molecular Nanocarbon Project, Nagoya University, Chikusa, Nagoya 464-8602, Japan
cDepartment of Applied Chemistry, Waseda University, 3-4-1 Ohkubo, Shinjuku, Tokyo 169-8555, Japan. E-mail: junyamaguchi@waseda.jp
First published on 15th March 2017
Abstract
A Rh-catalyzed regiodivergent hydrosilylation of acyl aminocyclopropanes has been developed. Acyl aminocyclopropanes were reacted with hydrosilanes in the presence of Rh catalysts to afford ring-opened hydrosilylated adducts through carbon–carbon (C–C) bond cleavage of the cyclopropane ring. The regioselectivity of the addition of silanes (linear or branched) can be switched by changing the monophosphine ligand. This C–C bond cleavage/hydrosilylation methodology is applicable to the synthesis of silanediol precursors.
Introduction
Catalytic carbon–carbon (C–C) bond activation can be an efficient way to create valuable bonds from readily available starting materials.1 The development of methodologies for both the breaking of C–C bonds and the forming of new C–C and C–heteroatom bonds enables the synthesis of complex organic molecules in a novel and more efficient manner. In previous studies of transition-metal catalyzed C–C bond cleavage reactions, strained molecules have usually been employed.2,3 Particularly, cyclopropanes have been well investigated due to their high reactivity derived from a high strain energy. Various types of ring-opening reactions of “activated” cyclopropanes such as alkylidenecyclopropanes and donor–acceptor cyclopropanes have been reported, enabling the construction of diverse frameworks. However, mono-substituted cyclopropanes, which have inert C–C bonds compared to the “activated” cyclopropanes described above, are still difficult to functionalize and have limited application in organic synthesis.4 Since mono-substituted cyclopropanes such as cyclopropylamine are readily available ($5 per gram from Sigma-Aldrich), its synthetic utility might be enhanced if it can be coaxed into a new C–C bond activation mode. Recently, the Bower group reported excellent examples for a C–C activation/alkene formation sequence using acyl aminocyclopropanes.4j,kHydrosilylation is a classical and well-studied reaction, particularly for double bonds such as alkenes, alkynes, ketones, and imines under transition-metal catalysis.5 However, regarding the hydrosilylation of C–C single bonds, only a narrow scope of selected cyclopropanes has been studied. Catalytic hydrosilylation of “activated” cyclopropanes is known, and the site-selectivity of the C–C bond cleavage depends on the substituents on both the substrate and the catalyst (Scheme 1A).6,7 Specifically, the catalytic hydrosilylation of mono-substituted cyclopropanes has only been demonstrated in one report.7b In a related literature precedent, the Chirik group reported a two-step catalytic hydrosilylation of cyclopropyl alcohol in the presence of Wilkinson's catalyst (Scheme 1B).4c
 | ||
| Scheme 1 Transition-metal catalyzed hydrosilylation of cyclopropanes via C–C bond activation. | ||
They discovered that the proximal bond (C(α)–C(β) bond) of the cyclopropane can be cleaved selectively under the influence of the Rh catalyst by using PPh2 as a directing group, followed by hydrosilylation of the alkene to give the resulting alkylsilane.
During our efforts to develop C–H functionalization of aminocyclopropanes,8 we serendipitously discovered a Rh-catalyzed hydrosilylation of acyl aminocyclopropanes via C–C bond cleavage (Scheme 1C). We have found that the regioselectivity of the addition of silanes can be controlled by changing the monophosphine ligand to give linear or branched alkylsilanes. To the best of our knowledge, switching the regioselectivity by ligand control in a catalytic hydrosilylation of mono-substituted cyclopropanes has not been reported thus far. Additionally, since the functional motif of the resulting α-aminosilane is known to exhibit biological activity and is a prominent feature in silanediol protease inhibitors (Scheme 1D),9 many strategies for its preparation have been reported.10 This hydrosilylation methodology would introduce various silyl groups in an atom-economical fashion without the use of organometallic reagents.
Results and discussion
At the outset of our studies, we chose cyclopropylamine 1A and silane 2a as model substrates (Table 1). To our delight, hydrosilylated product 3Aa, which was formed through C(α)–C(β) bond cleavage and addition of the silyl group to the terminal carbon C(γ), was obtained in 40% yield with 2.5 mol% [Rh(cod)Cl]2 in THF at 120 °C for 6 h (Table 1, entry 1).11|
|
|||
|---|---|---|---|
| Entry | Ligand | 3Aa /% | 4Aa /% |
| a Conditions: 1A (0.35 mmol), 2a (2.0 equiv.), [Rh(cod)Cl]2 (2.5 mol%), ligand (bidentate: 5 mol%, monodentate: 10 mol%), THF (2.0 mL), 120 °C, 6 h. b Yields were determined by 1H NMR analysis of the crude product using CH2Br2 as an internal standard. c Isolated yield. d PCy3·HBF4 was used as the precursor. | |||
| 1 | None | 40c | <1 |
| 2 | Phen | <1 | <1 |
| 3 | dppb | 10 | 25 |
| 4 | PCy2Ph | 42 | 6 |
| 5d | PCy3 | 61 (56)c | <1 |
| 6 | PPh3 | 17 | 83 |
| 7 | P(1-nap)3 | 9 | 82 (81)c |

When examining ligand effects, we observed that 1,10-phenanthroline (phen), which is a bidentate nitrogen ligand, is ineffective (entry 2). When 1,4-bis(diphenylphosphino)butane (dppb) was employed, a mixture of linear product 3Aa and branched product 4Aa was obtained (entry 3). The monodentate phosphine ligand PCy2Ph provided 3Aa as the major product (entry 4). Moreover, when a more electron-rich and bulky ligand, PCy3, was used, regioselective hydrosilylation proceeded to yield the linear 3Aa in higher yield (56% isolated yield, entry 5).12 On the other hand, when PPh3 and tri(naphthalen-1-yl)phosphine (P(1-nap)3) were used as ligands, the regioselectivity dramatically changed: branched product 4Aa, which has a silyl group at the α position to the nitrogen, was obtained in high yields (entries 6 and 7). In this manner, we accomplished a switching of the regioselectivity when reacting cyclopropylamine 1A with hydrosilane 2avia C–C bond cleavage solely by changing the ligand on the Rh catalyst.
With optimized reaction conditions in hand, we evaluated the scope of cyclopropylamines and silanes under the [Rh(cod)Cl]2/P(1-nap)3 catalytic system (Scheme 2).
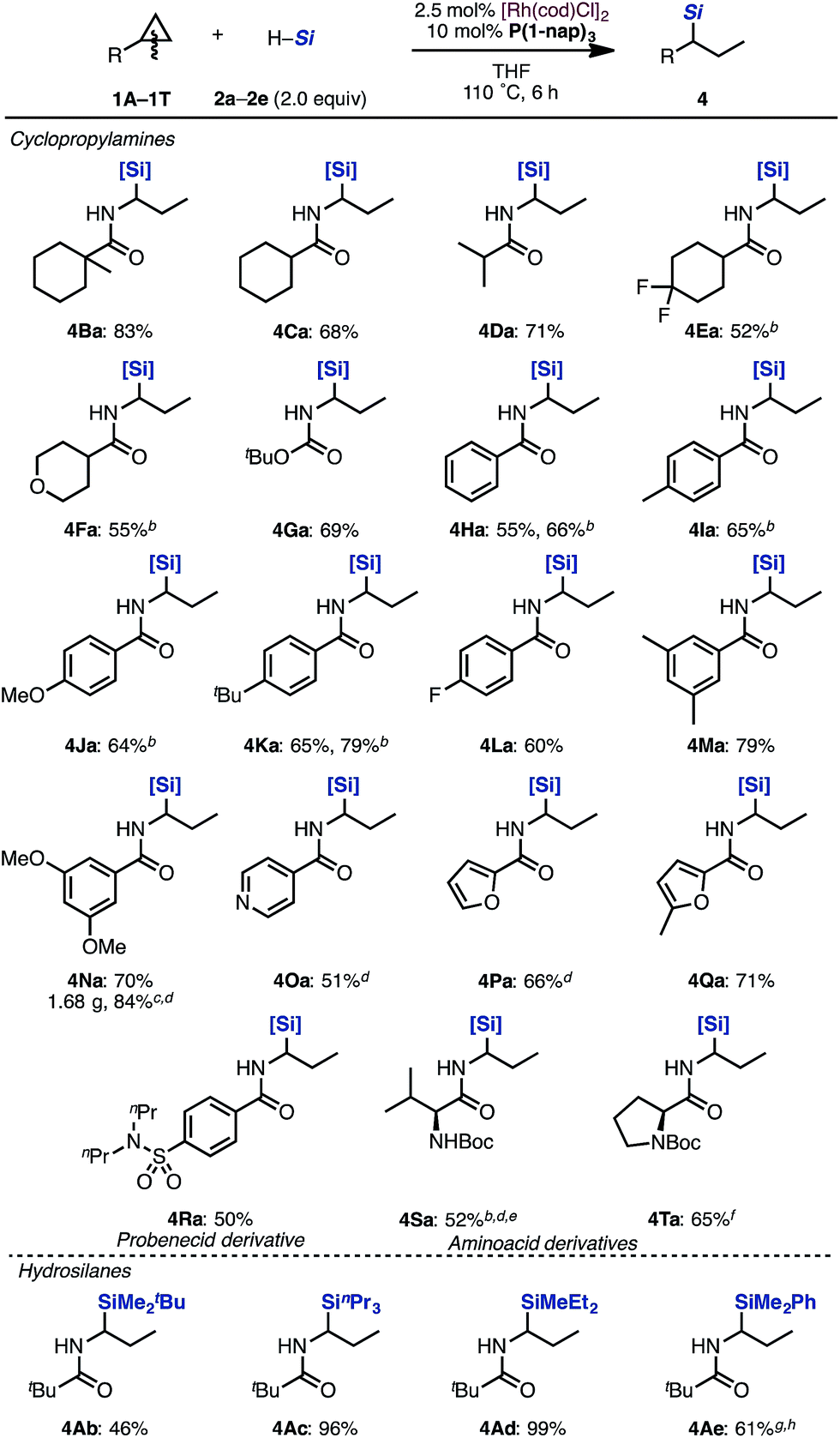 | ||
Scheme 2 Scope of cyclopropylamines and hydrosilanes in the hydrosilylation by a Rh/P(1-nap)3 catalysta. aIsolated yields. b[Rh(cod)OMe]2 (1.25 mol%), and P(1-nap)3 (5 mol%) were used. c[Rh(cod)Cl]2 (1.25 mol%), and P(1-nap)3 (5 mol%) were used. d18 h. eDiastereomer mixtures. fMixture of diastereomers (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40). g[Rh(cod)Cl]2 (5 mol%), and P(1-nap)3 (20 mol%) were used. h12 h. [Si] = SiMe(OSiMe3)2. :40). g[Rh(cod)Cl]2 (5 mol%), and P(1-nap)3 (20 mol%) were used. h12 h. [Si] = SiMe(OSiMe3)2. | ||
Cyclopropanes bearing cyclohexyl, isopropyl and tetrahydropyranyl groups were applicable in this reaction (4Ba–4Fa). Even carbamate 1G performed well to give the corresponding product 4Ga in 69% yield. Aminocyclopropanes protected by various benzoyl groups were tolerated to produce the desired products in good yields (4Ha–4Na). Cyclopropylamines with heteroaromatic substituents such as pyridine and furan rings were tolerated under the reaction conditions to afford hydrosilylated adducts in moderate yields (4Oa–4Qa). Probenecid derivative 1R could be transformed to alkylsilane 4Ra. Valine derivative 1S and proline derivative 1T can also be applied to this reaction, furnishing 4Sa and 4Ta in moderate yields. Various trialkylsilanes 1b–1e were also applicable and gave the corresponding hydrosilylated products in moderate to excellent yields (4Ab–4Ae). Typically, the regioselectivity of branched: linear products was in the range of 10:1 to 3:1.13
Next, the substrate scope of aminocyclopropanes in the [Rh(cod)Cl]2/PCy3·HBF4 catalytic system was examined (Scheme 3). The reaction of cyclopropylamines bearing various acyl protecting groups proceeded smoothly with virtually complete regioselectivities to provide the corresponding linear products (3Aa, 3Ba, 3Ma, 3Na, 3Ua, and 3Va) in moderate yields.14
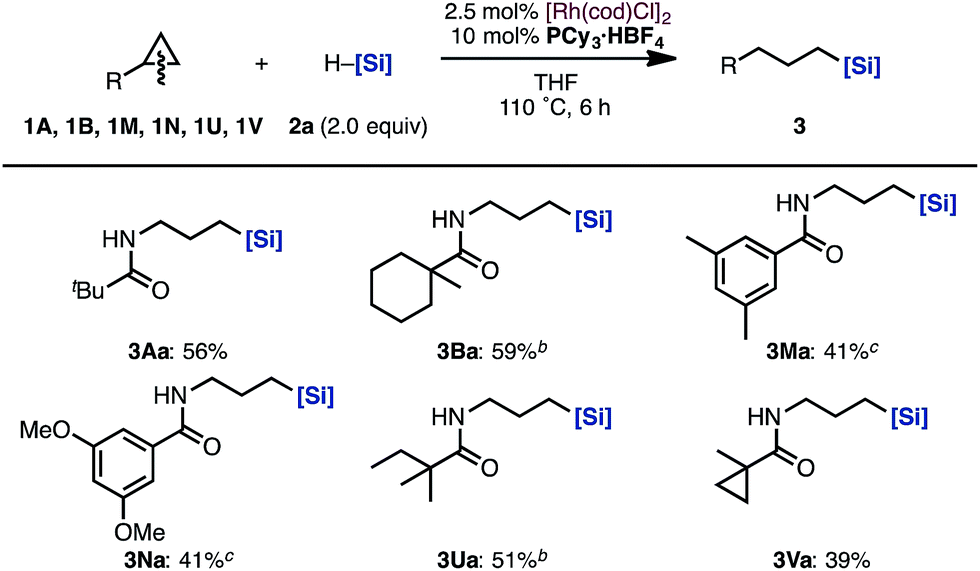 | ||
| Scheme 3 Scope of cyclopropylamines in the hydrosilylation by a Rh/PCy3·HBF4 catalysta. aIsolated yields. b12 h. c25 h. [Si] = SiMe(OSiMe3)2. | ||
To demonstrate the synthetic applicability of the linear product 3, alkylsilane 3Ma was converted to the corresponding alcohol 5Ma by treatment with Bu4NF/H2O2/KHCO3 in THF/MeOH (Scheme 4).15 In this manner, this hydrosilylation methodology can give access to a versatile building block for further transformation.
 | ||
| Scheme 4 Tamao oxidation of aminosilane 3Ma. | ||
To gain insight into the reaction mechanism, several experiments were conducted (Scheme 5). Firstly, we tried to isolate the reaction intermediates. Gratifyingly, hydrosilylated product 4Ab as well as (E)- and (Z)-enamide 6A were isolated when the reaction of 1A with tert-butyldimethylsilane (2b) in the presence of [Rh(cod)Cl]2/P(1-nap)3 was terminated at only 3 h of reaction time (Scheme 5A). This result indicated that the C–C bond cleavage generates the corresponding olefins in situ. Next, the reaction intermediates were subjected to our standard conditions (Scheme 5B). When (E)-6A and (Z)-6A were reacted with silane 2a by using P(1-nap)3 as the ligand, the branched 4Aa was obtained in 80% yield. This result is consistent with that of the hydrosilylation of 1A (Table 1, entry 7).16 On the other hand, when PCy3·HBF4 was employed, the hydrosilylated products did not form.12 Additionally, when we subjected allylamine 7A, which is another possible intermediate, under the same reaction conditions,173Aa was obtained in 21% yield along with 41% yield of 4Aa. This suggested that the isomerization of the olefin took place in situ. On the other hand, the hydrosilylation of 7A using PCy3·HBF4 afforded the linear product 3Aa with high regioselectivity.12
 | ||
| Scheme 5 Mechanistic exploration. aIsolated yields. bYields were determined by 1H NMR analysis of the crude product using CH2Br2 as an internal standard. | ||
According to our observations, a possible reaction mechanism is depicted (Scheme 5C). First, oxidative addition of the C(α)–C(β) bond to the Rh(I) catalyst occurs to generate rhodacycle B. Allylamine E and enamide F could be formed via β-H elimination, followed by reductive elimination from C and D. Both E and F would exist in equilibrium through intermediate G. Subsequently, Rh-catalyzed hydrosilylation of E or F would proceed by a modified Chalk–Harrod mechanism, giving H and I.18 Finally, a reductive elimination step yields 3 and 4 as the products.
Although the ligand effect remains unclear, we propose the following explanation: when PCy3 is used as the ligand, an electron-rich Rh complex with a high reduction ability is formed. Thus, enamides and imines, generated by isomerization, are hydrogenated immediately. This undesired pathway competes with the formation of the branched 4, leading to the high regioselectivity of the linear 3. On the other hand, enamide 6A was selectively formed in the reaction of allylamine 7A under [Rh(cod)Cl]2/P(1-nap)3 catalytic conditions.19 This result suggests that P(1-nap)3 is promoting the isomerization of allylamines to enamides. Furthermore, the regioselectivity might be correlated to the cone angle of the ligands (Fig. 1).20 Ligands with larger cone angles favor the linear product 3Aa, whereas ones with smaller cone angles tend to produce branched product 4Aa. Also, the IR stretching frequency of the Ni(CO)3(L) (L = ligand) catalysts did not show any relationship between the regioselectivity and the electronic cis effect of the phosphines.20 These factors suggest that the bulkiness of the ligand suppresses the hydrosilylation of the more sterically hindered enamide 6A compared to allylamine 7A.
 | ||
| Fig. 1 Relationship between the cone angle of ligands, the IR stretching frequency of the Ni(CO)3(L) catalysts, and the regioselectivity of hydrosilylation of acyl aminocyclopropanes. | ||
Conclusions
We have successfully developed a ligand-controlled, Rh-catalyzed regiodivergent hydrosilylation of acyl aminocyclopropanes with a wide range of silanes. Initial mechanistic investigations suggest that selective C–C bond cleavage takes place to generate allylamine and enamide, which are hydrosilylated by the Rh catalyst. Precise elucidation of ligand effects and application to asymmetric hydrosilylation are ongoing in our laboratory.Acknowledgements
This work was supported by JSPS KAKENHI Grant No. JP16H01011 and JP16H04148 (to J. Y.), the ERATO program from JST (K. I.), and a JSPS research fellowship for young scientists (to H. K.). ITbM is supported by the World Premier International Research Center (WPI) Initiative, Japan.Notes and references
- (a) W. D. Jones, Nature, 1993, 364, 676 CrossRef; (b) M. Murakami and Y. Ito, Top. Organomet. Chem., 1999, 3, 97 CrossRef CAS; (c) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870 CrossRef; (d) C. Perthuisot, B. L. Edelbach, D. L. Zubris, N. Simhai, C. N. Iverson, C. Müller, T. Satoh and W. D. Jones, J. Mol. Catal. A: Chem., 2002, 189, 157 CrossRef CAS; (e) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed; (f) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (g) T. Satoh and M. Miura, Top. Organomet. Chem., 2005, 14, 1 CAS; (h) T. Kondo and T. Mitsudo, Chem. Lett., 2005, 34, 1462 CrossRef CAS; (i) C.-H. Jun and J. W. Park, Top. Organomet. Chem., 2007, 24, 117 CrossRef CAS; (j) D. Necas and M. Kotora, Curr. Org. Chem., 2007, 11, 1566 CrossRef CAS; (k) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed; (l) H. Yorimitsu and K. Oshima, Bull. Chem. Soc. Jpn., 2009, 82, 778 CrossRef CAS; (m) M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100 RSC; (n) A. Dermenci, J. W. Coe and G. Dong, Org. Chem. Front., 2014, 1, 567 RSC; (o) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed; (p) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef CAS PubMed; (q) M. E. O'Reilly, S. Dutta and A. S. Veige, Chem. Rev., 2016, 116, 8105 CrossRef PubMed; (r) Y. Xia, G. Lu, P. Liu and G. Dong, Nature, 2016, 539, 546 CrossRef CAS PubMed; (s) C–C Bond Activation, ed. G. Dong, Springer, 2014 Search PubMed; (t) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.6b00599.
- For reviews for cyclopropanes, see: (a) I. Nakamura and Y. Yamamoto, Adv. Synth. Catal., 2002, 344, 111 CrossRef CAS; (b) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed; (c) A. Masarwa and I. Marek, Chem.–Eur. J., 2010, 16, 9712 CrossRef CAS PubMed; (d) D. J. Mack and J. T. Njardarson, ACS Catal., 2013, 3, 272 CrossRef CAS; (e) D.-H. Zhang, X.-Y. Tang and M. Shi, Acc. Chem. Res., 2014, 47, 913 CrossRef CAS PubMed; (f) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804 RSC; (g) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed; (h) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317 CrossRef CAS PubMed; (i) I. Marek, A. Masarwa, P.-O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414 CAS; (j) M. H. Shaw and J. F. Bower, Chem. Commun., 2016, 52, 10817 RSC.
- For reviews for cyclobutanes, see: ref. 2c, i and (a) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS PubMed; (b) M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759 CrossRef CAS PubMed.
- For selected examples for selective proximal bond cleavage reactions, see: (a) T. Ichiyanagi, S. Kuniyama, M. Shimizu and T. Fujisawa, Chem. Lett., 1997, 26, 1149 CrossRef; (b) Y. Koga and K. Narasaka, Chem. Lett., 1999, 28, 705 CrossRef; (c) S. C. Bart and P. J. Chirik, J. Am. Chem. Soc., 2003, 125, 886 CrossRef CAS PubMed; (d) L. Liu and J. Montgomery, J. Am. Chem. Soc., 2006, 128, 5348 CrossRef CAS PubMed; (e) S. Ogoshi, M. Nagata and H. Kurosawa, J. Am. Chem. Soc., 2006, 128, 5350 CrossRef CAS PubMed; (f) L. Liu and J. Montgomery, Org. Lett., 2007, 9, 3885 CrossRef CAS PubMed; (g) T. Tamaki, M. Nagata, M. Ohashi and S. Ogoshi, Chem.–Eur. J., 2009, 15, 10083 CrossRef CAS PubMed; (h) A. B. Chaplin and A. S. Weller, Organometallics, 2010, 29, 2332 CrossRef CAS; (i) T. Tamaki, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2011, 50, 12067 CrossRef CAS PubMed; (j) M. H. Shaw, E. Y. Melikhova, D. P. Kloer, W. G. Whittingham and J. F. Bower, J. Am. Chem. Soc., 2013, 135, 4992 CrossRef CAS PubMed; (k) M. H. Shaw, N. G. McCreanor, W. G. Whittingham and J. F. Bower, J. Am. Chem. Soc., 2015, 137, 463 CrossRef CAS PubMed; (l) M. H. Shaw, R. A. Croft, W. G. Whittingham and J. F. Bower, J. Am. Chem. Soc., 2015, 137, 8054 CrossRef CAS PubMed; (m) G.-W. Wang, N. G. McCreanor, M. H. Shaw, W. G. Whittingham and J. F. Bower, J. Am. Chem. Soc., 2016, 138, 13501 CrossRef CAS PubMed; (n) M. H. Shaw, W. G. Whittingham and J. F. Bower, Tetrahedron, 2016, 72, 2731 CrossRef CAS; (o) R. Ye, B. Yuan, J. Zhao, W. T. Ralston, C.-Y. Wu, E. U. Barin, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2016, 138, 8533 CrossRef CAS PubMed; For selected examples for selective distal bond cleavage reactions: (p) M. Murakami, H. Amii, K. Shigeto and Y. Ito, J. Am. Chem. Soc., 1996, 118, 8285 CrossRef CAS; (q) Z. He and A. K. Yudin, Org. Lett., 2006, 8, 5829 CrossRef CAS PubMed.
- For reviews, see: (a) O. Riant, N. Mostefaï and J. Courmarcel, Synthesis, 2004, 2943 CrossRef CAS; (b) B. Marciniec, Coord. Chem. Rev., 2005, 249, 2374 CrossRef CAS; (c) S. Díez-González and S. P. Nolan, Acc. Chem. Res., 2008, 41, 349 CrossRef PubMed; (d) K. Riener, M. P. Högerl, P. Gigler and F. E. Kühn, ACS Catal., 2012, 2, 613 CrossRef CAS; (e) Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603 RSC.
- (a) A. G. Bessmertnykh, K. A. Blinov, Y. K. Grishin, N. A. Donskaya and I. P. Beletskaya, Tetrahedron Lett., 1995, 36, 7901 CrossRef CAS; (b) A. G. Bessmertnykh, K. A. Blinov, Y. K. Grishin, N. A. Donskaya, E. V. Tveritinova, N. M. Yur'eva and I. P. Beletskaya, J. Org. Chem., 1997, 62, 6069 CrossRef CAS; (c) Y. Nishihara, M. Itazaki and K. Osakada, Tetrahedron Lett., 2002, 43, 2059 CrossRef CAS; (d) M. Itazaki, Y. Nishihara and K. Osakada, J. Org. Chem., 2002, 67, 6889 CrossRef CAS PubMed; (e) S. Simaan, A. F. G. Goldberg, S. Rosset and I. Marek, Chem.–Eur. J., 2010, 16, 774 CrossRef CAS PubMed.
- Non-transition metal mediated hydrosilylation of cyclopropanes: (a) J. R. Hwu, J. Chem. Soc., Chem. Commun., 1985, 452 RSC; (b) S. Nagahara, T. Yamakawa and H. Yamamoto, Tetrahedron Lett., 2001, 42, 5057 CrossRef CAS; (c) J. Clayden, D. W. Watson and M. Chambers, Tetrahedron, 2005, 61, 3195 CrossRef CAS; (d) C. Gómez, V. J. Lillo and M. Yus, Tetrahedron, 2007, 63, 4655 CrossRef.
- (a) S. Miyamura, M. Araki, T. Suzuki, J. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 846 CrossRef CAS PubMed; (b) S. Miyamura, M. Araki, Y. Ota, Y. Itoh, S. Yasuda, M. Masuda, T. Taniguchi, Y. Sowa, T. Sakai, T. Suzuki, K. Itami and J. Yamaguchi, Org. Biomol. Chem., 2016, 14, 8576 RSC.
- (a) M. W. Mutahi, T. Nittoli, L. Guo and S. M. Sieburth, J. Am. Chem. Soc., 2002, 124, 7363 CrossRef CAS PubMed; (b) S. M. Sieburth and C.-A. Chen, Eur. J. Org. Chem., 2006, 311 CrossRef CAS; (c) L. Nielsen and T. Skrydstrup, J. Am. Chem. Soc., 2008, 130, 13145 CrossRef CAS PubMed; (d) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (e) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388 CrossRef CAS PubMed.
- Recent examples for the synthesis of α-aminosilanes: (a) N. Niljianskul, S. Zhu and S. L. Buchwald, Angew. Chem., Int. Ed., 2015, 54, 1638 CrossRef CAS PubMed; (b) K. Kato, K. Hirano and M. Miura, Angew. Chem., Int. Ed., 2016, 55, 14400 CrossRef CAS PubMed and references therein.
- Another ring-opened product was obtained, see the ESI.†.
- N-Propylpivalamide, which was generated via C–C bond cleavage and hydrogenation, was formed, see the ESI.†.
- For details regarding the regioselectivities of each product, see the ESI.†.
- The main byproducts were desilylated products in 10–30% yield. When tert-butyl acrylate was added as a hydrogen scavenger, these byproducts were suppressed to less than 10% yield. However, in such cases, the product yields did not increase, as the amount of recovered starting material 1 increased.
- K. Tamao, N. Ishida, T. Tanaka and M. Kumada, Organometallics, 1983, 2, 1694 CrossRef CAS.
- Examples of Rh-catalyzed hydrosilylation of enamides: (a) T. Murai, T. Oda, F. Kimura, H. Onishi, T. Kanda and S. Kato, J. Chem. Soc., Chem. Commun., 1994, 2143 RSC; (b) G. W. Hewitt, J. J. Somers and S. M. Sieburth, Tetrahedron Lett., 2000, 41, 10175 CrossRef CAS; (c) G. K. Min and T. Skrydstrup, J. Org. Chem., 2012, 77, 5894 CrossRef CAS PubMed.
- An example of Rh-catalyzed hydrosilylation of allylamine: A. J. Huckaba, T. K. Hollis and S. W. Reilly, Organometallics, 2013, 32, 6248 CrossRef CAS.
- S. Sakaki, M. Sumimoto, M. Fukuhara, M. Sugimoto, H. Fujimoto and S. Matsuzaki, Organometallics, 2002, 21, 3788 CrossRef CAS.
- See the ESI.†.
- (a) C. A. Tolman, Chem. Rev., 1977, 77, 313 CAS. For the cone angle of P(1-nap)3; (b) H. Bahrmann and B. Fell, J. Mol. Catal., 1980, 8, 329 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, and spectral data for all compounds, including scanned images of 1H and 13C NMR spectra. See DOI: 10.1039/c7sc00071e |
| This journal is © The Royal Society of Chemistry 2017 |