
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and stabilities of peptide-based [1]rotaxanes: molecular grafting onto lasso peptide scaffolds†
Fumito
Saito
and
Jeffrey W.
Bode
 *
*
Laboratorium füer Organische Chemie, Department of Chemistry and Applied Biosciences, ETH Zürich, Zürich, Switzerland 8093. E-mail: bode@org.chem.ethz.ch; Web: http://www.bode.ethz.ch
First published on 26th January 2017
Abstract
The chemical synthesis of peptide-based [1]rotaxanes (lasso peptides) was achieved by [2]rotaxane formation followed by two chemoselective ligation reactions. Our approach enabled incorporation of various peptide sequences into a common rotaxane structure. The synthetic lasso peptides were characterized by NMR, chromatography, and partial degradation by proteases. A linear peptide epitope grafted onto the rotaxane scaffold showed significantly improved proteolytic stability.
Introduction
Microcin J25 (MccJ25) and related structures are bacterially produced lasso peptides with a unique, interlocked structure that imparts remarkable thermal and proteolytic stabilities (Fig. 1A).1,2 These peptides, consisting of 15–24 amino acids, are characterized by a threaded structure where a C-terminal region of the peptide threads through an N-terminal macrolactam ring. Amazingly, they are ribosomally synthesized followed by post-translational, enzymatic cyclization and cleavage of a leader peptide. The steric bulk of amino acid side chains maintains the lasso topology. | ||
| Fig. 1 Lasso peptides. (A) Structure of microcin J25 and (right) 3D structure adopted from PDB 1Q71. (B) This work: synthesis of lasso peptides by grafting with chemoselective ligations. | ||
Lasso peptides represent an extreme example of the many strategies employed to stabilize α-peptide-based natural products. Unmodified linear peptides are generally unstable in vivo, and this intrinsic disadvantage hampers the use of peptides as drugs despite of their high selectivity and potency.3 A number of methods have been proposed to enhance the stability of peptides including cyclization, backbone N-methylation, and incorporation of unnatural amino acids. To take advantage of these strategies, scientists have recently adopted a molecular grafting approach where peptides are incorporated into a stable peptide/protein scaffold.4 Marahiel et al. have demonstrated a molecular grafting of the RGD-motif onto the MccJ25 scaffold using bacterial biosynthetic machinery.5 This approach made the otherwise unstable RGD peptide resistant to proteolytic degradation while showing integrin binding in the nanomolar range. Though the molecular grafting on MccJ25 was achieved by biosynthesis, there has been no report on chemical synthesis of natural lasso peptides simply because formation of the threaded structure is extremely difficult.
In this manuscript, we report chemical syntheses of peptide-based [1]rotaxanes (lasso peptides) reminiscent of natural lasso peptides (Fig. 1B). Chromatography, NMR, and analysis of partial enzymatic degradation products confirmed the successful formation of the threaded structure and evaluation of the physicochemical properties of the synthetic lasso peptides revealed excellent thermal and proteolytic stabilities.
Results and discussion
Synthetic strategy to prepare lasso peptides
Initially, we considered two strategies to construct lasso peptides (Fig. 2):6 (1) formation of unthreaded peptide Aun resulting from a covalent link between a macrocycle and a peptide-containing segment. Aun is in a thermodynamic equilibrium with pseudo[1]rotaxane A where the successful trapping of A with a capping agent B would lead to lasso peptide C; (2) formation of [2]rotaxane D followed by the attachment of a linear peptide E by two sequential chemical ligations to afford lasso peptide C. | ||
| Fig. 2 Strategies for synthesis of lasso peptides. un = unthreaded. | ||
Pioneering studies from Leigh, Aucagne and others on the preparation and properties of peptide rotaxanes provided promising precedent for the planned studies.7 Importantly, the synthesis of lasso peptides has been accomplished by Coutrot et al.,8 who incorporated triglycine residues into a [1]rotaxane scaffold by a self-entanglement strategy. However, the method for locking a molecule into the lasso structure was not practical, and the diversity of peptide sequences limited.
After careful consideration, we adopted the second approach, as with the first strategy the topology-determining step potentially produces Cun and C that are difficult to differentiate. In contrast, in the latter approach, the threaded structure of [2]rotaxane D – which can be easily characterized – is retained through the synthesis provided that the [2]rotaxane structure is thermally and chemically stable. More importantly, the latter approach enables modular synthesis of lasso peptides. Once an appropriate synthetic scheme is established, the “stitching” strategy enables incorporation of an almost unlimited number of peptide sequences into the stable lasso peptide scaffold.
Synthesis of [2]rotaxane with a potassium acyltrifluoroborate (KAT) as capping agent
We initially focused on preparation of [2]rotaxanes by a simple protocol. We selected the molecular recognition between benzo-21-crown-7 (B21C7) and secondary dialkylammoniums, as they have been frequently used for rotaxane syntheses.9 A preliminary study revealed the coupling of a potassium acyltrifluoroborate (KAT) and an azide in the presence of HBF4·OEt2 (ref. 10) was suitable for forming a [2]rotaxane.11 This result prompted us to prepare 4-fluorophenyl KAT 1, crown ether 2 and azide 3 (Fig. 3). 2 and 3 are pre-functionalized with a protected hydroxylamine and thioester, respectively, which are the key functional groups for the subsequent ligation steps. These functional groups did not interfere with the formation of the [2]rotaxane, and 4 was obtained in 21% yield as a mixture of diastereomers 4a and 4b. The axle component 5 was concomitantly formed in the same reaction in 31% yield and used for synthesis of branched-cyclic peptides.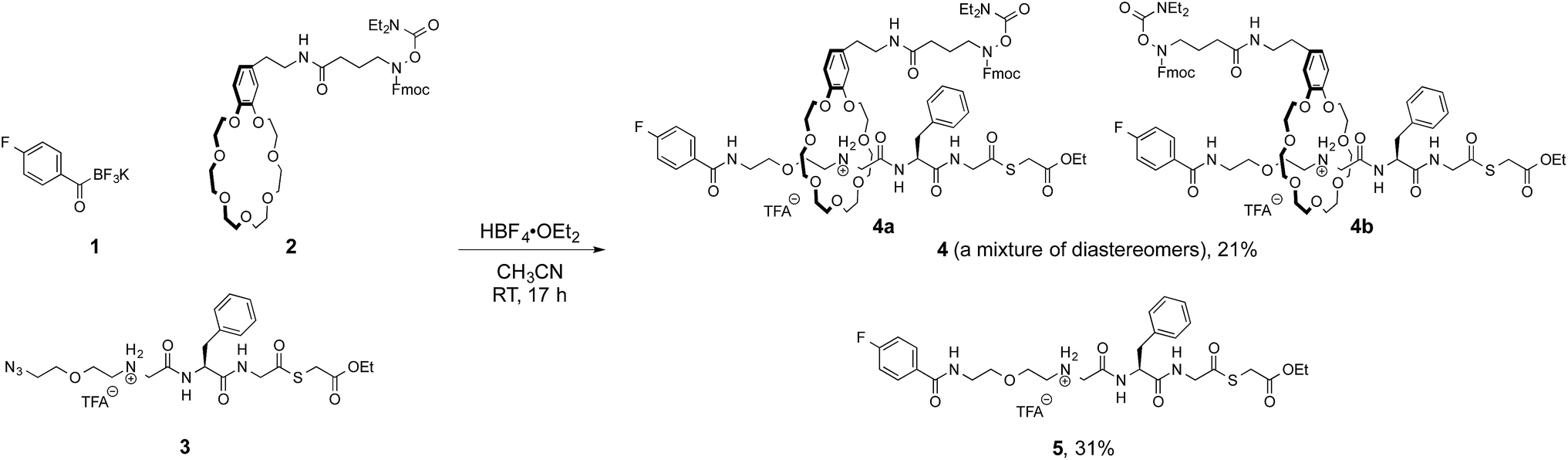 | ||
| Fig. 3 Formation of [2]rotaxane 4 by KAT–azide coupling. | ||
Synthesis of lasso peptides from [2]rotaxane 4
With a reliable protocol for the preparation of [2]rotaxanes in hand, we derivatized [2]rotaxane 4 to form lasso peptide by two amide-forming ligations, the α-ketoacid-hydroxylamine (KAHA) ligation12 and native chemical ligation (NCL)13 (Fig. 4). Fmoc cleavage by 5% Et2NH in DMSO revealed unprotected hydroxylamine 6 in 78% yield, poised for KAHA ligation. Importantly, the thioester remained intact under the Fmoc cleavage condition employed. Peptide α-ketoacid 7 was prepared by Fmoc SPPS following the protocol recently reported by our group.14 The peptide sequence of 7 was adapted from a commercially available peptide drug, octreotide, a somatostatin analog used for the treatment of diseases such as acromegaly.15 We found that a cysteine side chain protecting group was necessary; in this case the S-unprotected variant of 7 did not undergo clean KAHA ligation, presumably because of intramolecular cyclization between the N-terminal cysteine and the C-terminal leucine α-ketoacid. | ||
| Fig. 4 Synthesis of lasso peptide L1. For simplicity, only one of the diastereomers is shown. All products were obtained as TFA salts after HPLC purification. TFAs were omitted from the structures for clarity. NaPi = sodium phosphate buffer. | ||
Rotaxane 6 and peptide 7 were ligated in DMSO/H2O (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) at 60 °C to form peptido[2]rotaxane 8. Unfortunately, the O-diethylcarbamoyl hydroxylamine was somewhat unstable under the KAHA ligation conditions (Fig. S4†). Nonetheless, the ligation product 8 was isolated in 23% yield as a major product. The deprotection-KAHA ligation sequence could be carried out in a one-pot manner, reducing the number of purification steps and avoiding possible decomposition of unprotected hydroxylamines upon lyophilization.
:4) at 60 °C to form peptido[2]rotaxane 8. Unfortunately, the O-diethylcarbamoyl hydroxylamine was somewhat unstable under the KAHA ligation conditions (Fig. S4†). Nonetheless, the ligation product 8 was isolated in 23% yield as a major product. The deprotection-KAHA ligation sequence could be carried out in a one-pot manner, reducing the number of purification steps and avoiding possible decomposition of unprotected hydroxylamines upon lyophilization.
The cyclization step was performed by NCL in the presence of an excess amount of 4-mercaptophenylacetic acid (MPAA) and TCEP. In situ deprotection of cysteine followed by ligation afforded the lasso peptide 9 in 71% yield. Alkylation of the sulfhydryl group present in 9 to suppress oxidation or dimerization of the cysteine residue gave L1 in 66% yield.
This reaction sequence was exploited to prepare branched-cyclic peptide B1; Fmoc-cleavage, KAHA ligation, NCL followed by cysteine alkylation afforded B1 (Scheme S4†). Both L1 and B1 were analyzed by NMR. Although we could not obtain compelling evidence for the threaded structure solely from NMR experiments, including 1H–15N HSQC and ROESY spectra, the 1H NMR spectrum of L1 was clearly different from that of B1. In addition, HRMS, HPLC retention time, and analysis of partial proteolytic degradation products further confirmed the formation of the threaded structure in L1.16
We investigated the possibility of incorporating various peptide sequences into our rotaxane scaffold. The peptide sequences were taken from an antibody Fc region binder,17 lassomycin,18 and somatostatin, which gave 9, 10 and 12-mer peptides, respectively, including the N-terminal cysteine and the C-terminal leucine α-ketoacid. The same reaction sequence was applicable and provided lasso peptides 10–12 (Fig. 5A).
 | ||
| Fig. 5 Peptide substrate scope. (A) Insertion of linear peptides with different length and side chain functional groups. (B) Trypsin digestion of 12. Degradation products were analyzed by HRMS (MALDI). The threaded structure of lasso peptides were retained in the form of [2]rotaxane. | ||
The threaded compounds were subjected to trypsin digestion and degraded into [2]rotaxanes (Fig. 5B, S76 and S90†). The results established the existence of the threaded structure in 10–12.
Structure modification of the crown ether
With the synthetic protocol in hand, we began to modify the core structure [2]rotaxane. We were especially concerned about the electron-rich nature of the catechol moiety in the crown ether. This moiety could be susceptible to enzymatic oxidation and might pose other stability issues. Based on this consideration, we prepared two crown ethers 13 and 14 without electron-rich moieties (Fig. 6A). Since the new crown ethers have larger ring sizes, a larger capping agent 15 was needed to form thermally stable [2]rotaxanes (Fig. S23†). The established protocol was applied to the synthesis of peptides L2 and L3 (Fig. 6B). Branched-cyclic peptides B2 and B3 (i.e. unthreaded variants of L2 and L3) were also prepared and used in the stability tests for comparison (vide infra). | ||
| Fig. 6 Modification of the core [2]rotaxane structure. (A) Structures of crown ethers 13 and 14 and KAT 15. (B) Structures of lasso peptides L2 and L3. Note that L2 was obtained as a mixture of diastereomers. | ||
Chromatographic characterization of lasso peptides
It has long been known that lasso peptides and their branched-cyclic counterparts are chromatographically distinct and show different retention times upon analysis by HPLC.19 This was true for our compounds; the retention times of the lasso peptides L1–L3 were found to be different from those of the branched-cyclic peptides B1–B3, respectively (Fig. S62–S64†).Thermal and enzymatic stabilities of lasso peptides
Many natural lasso peptides are also characterized by their thermal stability. For example, MccJ25 did not denature at 95 °C and maintained its threaded structure.20 As expected, the stopper groups used for the [2]rotaxane formation were large enough, and L1–L3 did not show dethreading even at 95 °C for 8 h (Fig. S117–S119†).We tested the proteolytic stability of lasso peptides L1–L3, linear peptides B1–B3, and the representative peptide macrocycle C1 against chymotrypsin, trypsin, and proteinase K.11 We were pleased to find that L1–L3 were significantly more stable than B1–B3. Lasso peptides L1–L3 and peptide macrocycle C1 were similarly stable against trypsin and proteinase K; but in case of chymotrypsin, L1–L3 were even more stable than macrocycle C1 (Fig. 7). In particular, L1 and L2 showed excellent resistance to degradation by chymotrypsin even after 24 h.
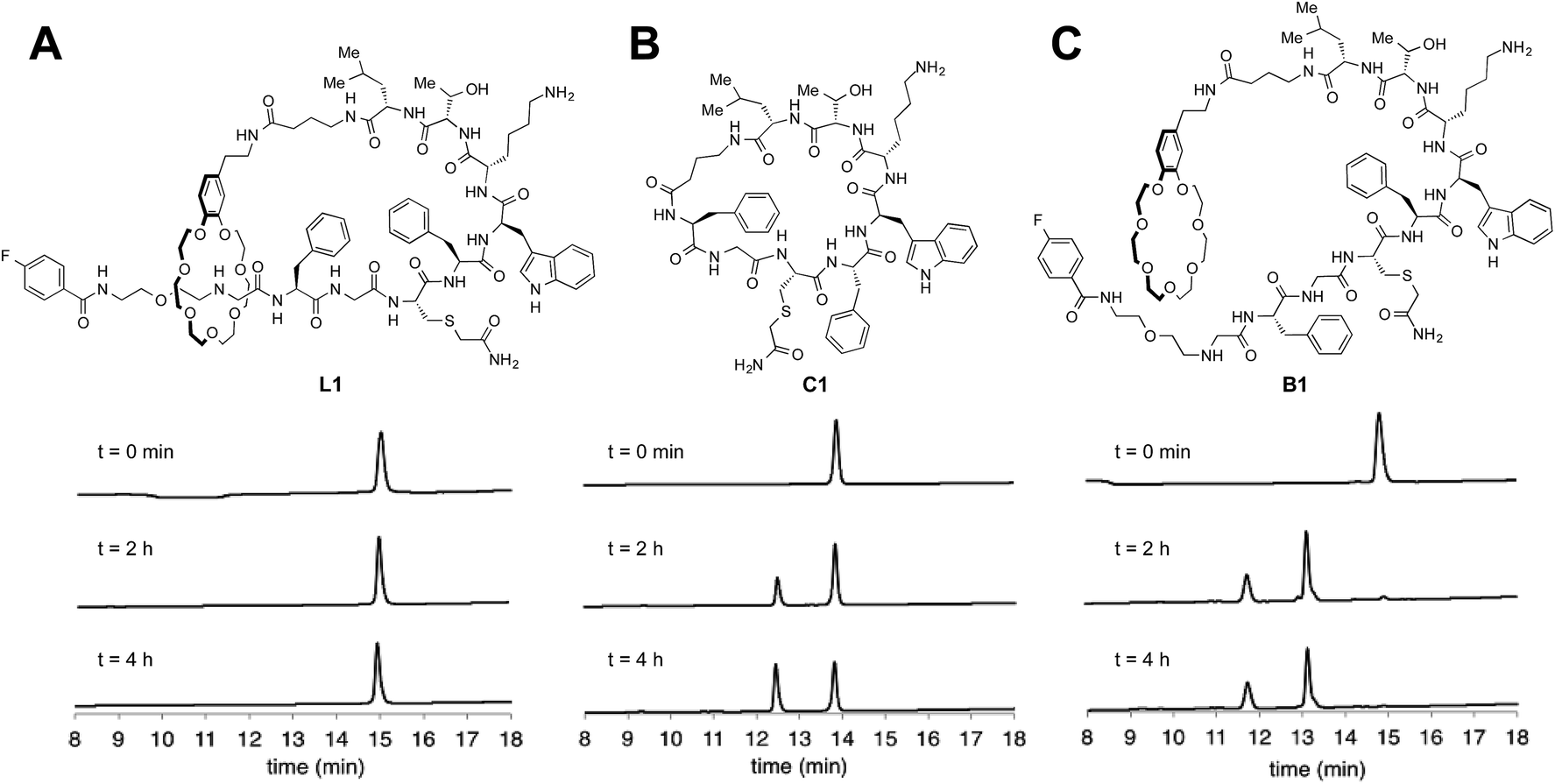 | ||
| Fig. 7 HPLC monitoring of degradation by chymotrypsin. (A) Lasso peptide L1. (B) Peptide macrocycle C1. (C) Branched-cyclic peptide B1. | ||
Serum stability tests were also conducted (Fig. 8). For the peptide sequence used in this study, N-, and C-terminal masking was effective enough to stabilize the peptides; N-, and C-terminal unmasked linear peptide R1 was degraded within 2 h in human serum. In contrast to lability against specific proteases mentioned above, branched-cyclic peptides B1–B3 were stable in human serum. All cyclic compounds except for L1 were stable under the condition for 24 h. L1, though reasonably stable, was gradually degraded possibly due to the catechol moiety.
 | ||
| Fig. 8 Serum stability tests. Stability is evaluated based on remaining peptide substrate monitored by HPLC. Data is averaged from two independent experiments and error bars show standard deviations. R1 is an N- and C-terminal unmasked linear peptide. | ||
Conclusions
In summary, we achieved the molecular grafting of linear peptide epitopes onto a lasso peptide scaffold. The combination of KAHA ligation and NCL enabled attachment of various peptide sequences from a common, readily prepared rotaxane. The synthetic lasso peptides were thermally stable, and the peptide epitope gained significant proteolytic stability upon grafting. A merger of our molecular grafting approach and other stability enhancement techniques would provide further improvements of physicochemical properties of peptides.Notes
A patent application covering portions of this work has been filed.Acknowledgements
This work was supported by ETH Zürich and the Commission for Technology and Innovation (CTI). We thank the LOC MS Service and the LOC NMR Service for analyses. X-ray services were provided by SMoCC – The Small Molecule Crystallography Center of ETH Zurich (http://www.smocc.ethz.ch). We are grateful to Alberto Osuna Galvez for providing the reagent 1 at the initial stage of the project and Dr Satomi Shimura for technical assistance.Notes and references
- Thermally unstable lasso peptides have been recently discovered: J. D. Hagemann, M. Zimmerman, X. Xie and M. A. Marahiel, J. Am. Chem. Soc., 2013, 135, 210–222 CrossRef PubMed.
- (a) M. O. Maksimov, S. J. Pan and A. J. Link, Nat. Prod. Rep., 2012, 29, 996–1006 RSC; (b) J. D. Hagemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef PubMed; (c) Y. Li, S. Zirah and S. Rebuffat, Lasso Peptides: Bacterial Strategies to make and maintain Bioactive Entangled Scaffolds, Springer, New York, 2015 Search PubMed.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CAS.
- (a) S. Gunasekera, F. M. Foley, R. J. Clark, L. Sando, L. J. Fabri, D. J. Craik and N. L. Daly, J. Med. Chem., 2008, 51, 7697–7704 CrossRef CAS PubMed; (b) C. T. T. Wong, D. K. Rowlands, C.-H. Wong, T. W. C. Lo, G. K. T. Nguyen, H.-Y. Li and J. P. Tam, Angew. Chem., Int. Ed., 2012, 51, 5620–5624 CrossRef CAS PubMed; (c) C. K. Wang, C. W. Gruber, M. Cemazar, C. Siatskas, P. Tagore, N. Payne, G. Sun, S. Wang, C. C. Bernard and D. J. Craik, ACS Chem. Biol., 2014, 9, 156–163 CrossRef CAS PubMed; (d) A. C. Conibear, S. Chaousis, T. Durek, K. J. Rosengren, D. J. Craik and C. I. Schroeder, Biopolymers, 2016, 106, 89–100 CrossRef CAS PubMed.
- (a) T. A. Knappe, F. Manzenrieder, C. Mas-Moruno, U. Linne, F. Sasse, H. Kessler, X. Xie and M. A. Marahiel, Angew. Chem., Int. Ed., 2011, 50, 8714–8717 CrossRef CAS PubMed; (b) J. D. Hegemann, M. D. Simone, M. Zimmermann, T. A. Knappe, X. Xie, F. S. D. Leva, L. Marinelli, E. Novellino, S. Zahler, H. Kessler and M. A. Marahiel, J. Med. Chem., 2014, 57, 5829–5834 CrossRef CAS PubMed.
- For discussion about synthesis of [1]rotaxanes, see: P. Waelès, C. Clavel, K. Fournel-Marotte and F. Coutrot, Chem. Sci., 2015, 6, 4828–4836 RSC.
- (a) A. Fernandes, A. Viterisi, F. Coutrot, S. Potok, D. A. Leigh, V. Aucagne and S. Papot, Angew. Chem., Int. Ed., 2009, 48, 6443–6447 CrossRef CAS PubMed; (b) A. Fernandes, A. Viterisi, V. Aucagne, D. A. Leigh and S. Papot, Chem. Commun., 2012, 48, 2083–2085 RSC; (c) B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed; (d) G. De Bo, S. Kuschel, D. A. Leigh, B. Lewandowski, M. Papmeyer and J. W. Ward, J. Am. Chem. Soc., 2014, 136, 5811–5814 CrossRef CAS PubMed.
- C. Clavel, K. Fournel-Marotte and F. Coutrot, Molecules, 2013, 18, 11553–11575 CrossRef CAS PubMed.
- (a) C. Zhang, S. Li, J. Zhang, K. Zhu, N. Li and F. Huang, Org. Lett., 2007, 9, 5553–5556 CrossRef CAS PubMed; (b) Q. Zhou, P. Wei, Y. Zhang, Y. Yu and X. Yan, Org. Lett., 2013, 15, 5350–5353 CrossRef CAS PubMed.
- G. A. Molander, J. Raushel and N. M. Ellis, J. Org. Chem., 2010, 75, 4304–4306 CrossRef CAS PubMed.
- See ESI† for detailed procedures.
- J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CAS.
- T. G. Wucherpfennig, V. R. Pattabiraman, F. R. P. Limberg, J. Ruiz-Rodríguez and J. W. Bode, Angew. Chem., Int. Ed., 2014, 53, 12248–12252 CrossRef CAS PubMed.
- G. Weckbecker, I. Lewis, R. Albert, H. A. Schmid, D. Hoyer and C. Bruns, Nat. Rev. Drug Discovery, 2003, 2, 999–1017 CrossRef CAS PubMed.
- See ESI† for detailed characterization of L1.
- S. Menegatti, M. Hussain, A. D. Naik, R. G. Carbonell and B. M. Rao, Biotechnol. Bioeng., 2013, 110, 857–870 CrossRef CAS PubMed.
- E. Gavrish, C. S. Sit, S. Cao, O. Kandror, A. Spoering, A. Peoples, L. Ling, A. Fetterman, D. Hughes, A. Bissell, H. Torrey, T. Akopian, A. Mueller, S. Epstein, A. Goldberg, J. Clardy and K. Lewis, Chem. Biol., 2014, 21, 509–518 CrossRef CAS PubMed.
- K. J. Rosengren, R. C. Clark, N. L. Daly, U. Göransson, A. Jones and D. J. Craik, J. Am. Chem. Soc., 2003, 125, 12464–12474 CrossRef CAS PubMed.
- A. Blond, M. Cheminant, D. Destoumieux-Garzon, I. Segalas-Milazzo, J. Peduzzi, C. Goulard and S. Rebuffat, Eur. J. Biochem., 2002, 269, 6212–6222 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data of all new compounds. CCDC 1520783. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00021a |
| This journal is © The Royal Society of Chemistry 2017 |