
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sub-1.1 nm ultrathin porous CoP nanosheets with dominant reactive {200} facets: a high mass activity and efficient electrocatalyst for the hydrogen evolution reaction†
Chao
Zhang
,
Yi
Huang
,
Yifu
Yu
,
Jingfang
Zhang
,
Sifei
Zhuo
and
Bin
Zhang
 *
*
Department of Chemistry, School of Science, Tianjin Key Laboratory of Molecular Optoelectronic Science, Tianjin University and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, China. E-mail: bzhang@tju.edu.cn
First published on 25th January 2017
Abstract
The exploration of a facile strategy to synthesize porous ultrathin nanosheets of non-layered materials, especially with exposed reactive facets, as highly efficient electrocatalysts for the hydrogen evolution reaction (HER), remains challenging. Herein we demonstrate a chemical transformation strategy to synthesize porous CoP ultrathin nanosheets with sub-1.1 nm thickness and exposed {200} facets via phosphidation of Co3O4 precursors. The resultant samples exhibit outstanding electrochemical HER performance: a low overpotential (only 56 and 131 mV are required for current densities of 10 and 100 mA cm−2, respectively), a small Tafel slope of 44 mV per decade, a good stability of over 20 h, and a high mass activity of 151 A g−1 at an overpotential of 100 mV. The latter is about 80 times higher than that of CoP nanoparticles. Experimental data and density functional theory calculations reveal that a high proportion of reactive {200} facets, high utilization efficiency of active sites, metallic nature, appropriate structural disorder, facile electron/mass transfer and rich active sites benefiting from the unique ultrathin and porous structure are the key factors for the greatly improved activity. Additionally, this facile chemical conversion strategy can be developed to a generalized method for preparing porous ultrathin nanosheets of CoSe2 and CoS that cannot be obtained using other methods.
1. Introduction
Hydrogen produced from water electrolysis is considered to be a promising alternative energy source to fossil fuels by virtue of its environmental benignity and sustainable features.1–3 Pt is an excellent electrocatalyst for the hydrogen evolution reaction (HER),4 but its practical use is hindered by its high price and rarity. Since the pioneering report of MoS2 as an electrocatalyst for HER,5,6 low-cost promising candidates composed of earth-abundant elements including metal chalcogenides,7–10 carbides,11–15 phosphides,16–22 phosphosulphides,23 and oxides24,25 have attracted increasing attention. Although fascinating advances have been made in the search for novel alternatives and the improvement of their HER performances via various methods, the materials are mainly restricted to nanoparticles, polycrystalline one-dimensional nanomaterials and thick sheets. Thus, the relatively large sizes and low numbers of active sites of some current catalysts mean that improvement of HER activity, especially the mass activity, is needed. In addition, the catalytic performance of a material is mainly dependent on its exposed crystal facets.26 However, the development of a type of electrocatalyst with a large proportion of exposed active crystal planes with high mass activity for HER is still highly desirable.Two-dimensional (2D) ultrathin nanosheets with thicknesses of several nanometers have been extensively studied as ideal materials for both the fundamental understanding of structure–activity relationships and their promising applications in various fields because of their large specific surface areas, richness of active sites, short electron/carrier transfer distance, structural defects and predominantly exposed crystal facets.27–36 For example, greatly enhanced catalytic performances have been achieved by the pioneering 2D ultrathin nanosheets made by the Xie,27–30 Wei,27,28,31 Yang32 and Zhang33,34 groups. A huge mass activity for the oxygen evolution reaction has been achieved by well-designed ultrathin CoOOH solid nanosheets.31 Thus, some rationally-designed methods including liquid exfoliation,27–30 graphene oxide-assisted growth,34 topotactic reduction35 and conversion33 have been successfully developed to produce inorganic ultrathin nanosheets. However, the products are mainly solid, rather than in porous single-crystalline form. In addition, compared to solid materials, porous nanostructures can possess many more active sites and exhibit more facile mass transfer, therefore exhibiting improved chemical and physical properties.37,38 More importantly, making ultrathin 2D sheets porous can not only generate more coordinated-unsaturated active atoms, but also allows easy electrolyte infiltration into the inside of the catalysts,39 which contributes to providing more active sites to participate in the catalytic reactions and hence ensures energy conversion operating with high-efficiency. However, until now, the development of a facile chemical conversion route to prepare single-crystalline 2D non-layered nanosheets as highly active HER materials, especially endowing them with both ultrathin and porous characteristics, is still a big challenge.
Herein, by choosing CoP, one of the most efficient electrocatalysts for HER and other applications,40–42 as the model target, we present a convenient chemical transformation approach to synthesize ultrathin porous CoP nanosheets (CoP UPNSs) with a high proportion of exposed {200} facets, two unit cell-thin thickness and modest distorted atomic structures via low-temperature phosphidation of Co3O4 precursors. Our theoretical calculations and experimental results demonstrate that CoP UPNSs are highly efficient catalysts for HER with a huge mass activity of 151 A g−1 at an overpotential of 100 mV. The 2D ultrathin structure with abundant pores and active sites, high fraction of exposed active facets, modest structural disorder of CoP NSs and facile ion/electron transfer are the key factors for the superior catalytic performance. Furthermore, this facile chemical conversion method can be extended to prepare UPNSs of CoSe2 and CoS.
2. Experimental
2.1 Material synthesis
2.2 Material characterization
The structures of the samples were determined using a Hitachi S-4800 scanning electron microscope (SEM, 3 kV). Powder X-ray diffraction (XRD) patterns were collected using a Bruker D8 Focus Diffraction System using a Cu Kα source (λ = 0.154178 nm). Transmission electron microscopy (TEM), higher-magnification transmission electron microscopy (HRTEM) and elemental distribution mapping images were taken on a JEOL-2100F system equipped with EDAX Genesis XM2. The thickness of the nanosheets was determined using atomic force microscopy (AFM) (Bruker multimode 8). X-ray photoelectron spectroscopy (XPS) measurements were conducted with a PHI-1600 X-ray photoelectron spectrometer equipped with Al Kα radiation. All binding energies were referenced to the C 1s peak at 284.8 eV.2.3 Electrochemical measurements
Electrochemical measurements were performed with a CHI 660D electrochemical workstation (CH Instruments, Austin, TX) and a typical one-component three-electrode cell was used, including a working electrode, a saturated calomel electrode (SCE) as the reference electrode, and a glassy carbon counter electrode in the presence of 0.5 M H2SO4 as the electrolyte. The reference electrode was calibrated with respect to an in situ reverse hydrogen electrode (RHE), by using two platinum wire electrodes as the working and counter electrodes, which yields the relation E (V vs. RHE) = E (V vs. SCE) + 0.245 V. A glassy carbon electrode decorated with catalyst samples was used as the working electrode. In a typical procedure for the fabrication of the working electrode, 4 mg of CoP catalyst and 20 μL of 5% Nafion solution were dispersed in 1 mL of de-ionized water by sonication to generate a homogeneous ink. Then 5 μL of the dispersion (containing 20 μg catalyst) was transferred onto a glassy carbon electrode with a diameter of 3 mm (loading amount: 0.28 mg cm−2). The as-prepared catalyst film was dried at room temperature. Polarization data were collected at a sweep rate of 2 mV s−1. Electrochemical impedance spectroscopy (EIS) measurements were carried out in the same configuration at η = 56 mV or j = 10 mA cm−2 from 100 kHz to 0.1 Hz.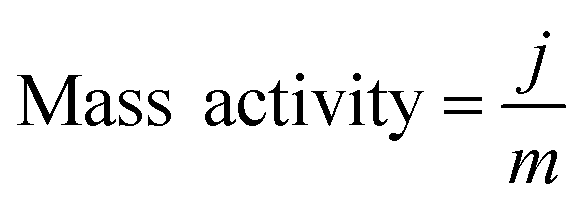
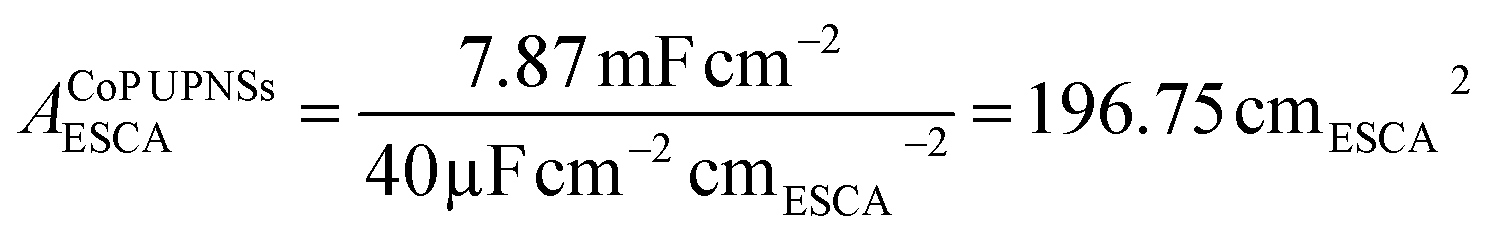
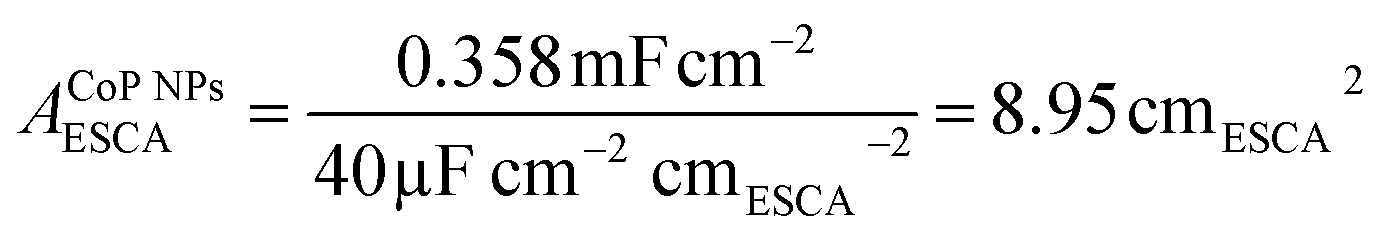
2.4 Theoretical calculations
Density functional theory (DFT) calculations were computed by the Vienna Ab initio Simulation Package (VASP). In the DFT calculations, the (100) surface was obtained by cutting bulk CoP along the [100] direction. The thickness of the surface slab was chosen to be that of a two-layer slab of the CoP unit. A vacuum layer as large as 12 Å was used along the c direction normal to the surface to avoid periodic interactions. A (2 × 2) supercell was used. The Gibbs free-energy change (ΔGads) of H on CoP (100) is defined as follows:| ΔGads = ΔEads + ΔEZPE − TΔS |
| ΔGads = ΔEads + 0.24 eV |
ΔEads is defined as follows:
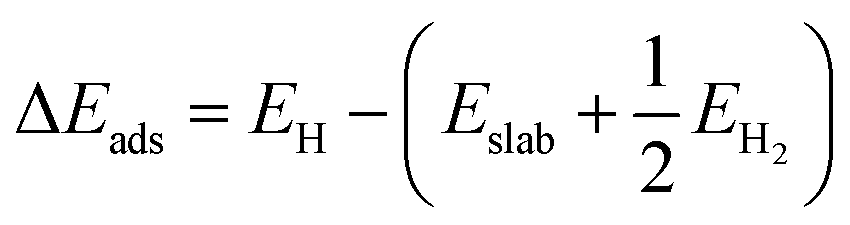
Since there are two surface structures of CoP (100), i.e. Co or P terminated, we have therefore calculated the surface energy of both surfaces by the following formula:46
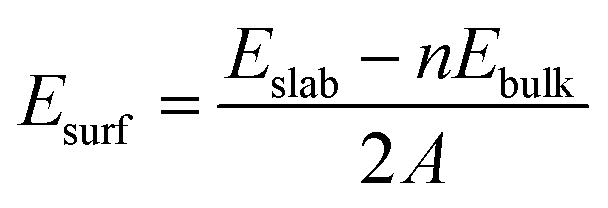
For the P terminated CoP (100) surface, only P atoms are exposed on the surface. Thus, H atoms will only locate at the top of the P atoms. In our model, there are eight surface P atoms available for H adsorption. The adsorption energy of one H atom (12.5% H coverage, Fig. S11b†) is −0.32 eV, and the adsorption energy will be further reduced to −0.11 eV as the coverage of H is above 75%.
3. Results and discussion
Co3O4 was selected as the initial material because of its good thermal stability. Firstly, atomically-thick porous Co3O4 precursor sheets (Fig. S1, ESI†) were synthesized according to a rationally-designed scalable fast-heating strategy developed by the Xie group.43 Then the Co3O4 nanosheets were transformed into CoP through low temperature gas-phase phosphodation using NaH2PO2 as the phosphorus source. Transmission electron microscopy (TEM) images (Fig. 1a and b) demonstrate that the ultrathin 2D porous structure can be successfully prepared on a large scale, which is of great importance for potential catalytic applications. Moreover, a typical high-resolution TEM (HRTEM) image (Fig. 1c) shows that the nanosheets possess mesopores with diameters of several nanometers. Lattice spacings of 0.280 and 0.283 nm can be attributed to the (002) and (011) crystallographic planes of orthorhombic CoP, respectively (Fig. 1c). A close-up view (Fig. 1d and S2a†) reveals that a mixture of some atomic disorder structures and amorphous areas can be observed clearly. The appearance of structural distortion and the amorphous phase may be associated with strain release due to lattice mismatches of Co3O4 and CoP.47,48 Such structural defects should be conducive to decreasing the surface energy in order to improve the stability of ultrathin 2D sheets.29 The associated Fast Fourier Transform (FFT) pattern of the HRTEM image (inset in Fig. 1c) discloses that the porous nanosheet is in single crystalline form with a preferential [100] orientation. The atomic force microscopy (AFM) image and the corresponding height configuration (Fig. 1e and f) indicate that CoP UPNSs possess uniform thickness of about 1.01 nm. This value corresponds to the thickness of two unit cells along the [100] direction of orthorhombic CoP (a = 5.076 Å, b = 3.279 Å, c = 5.599 Å, in JCPDS no. 29-0497), further illustrating that the as-transformed nanosheets possess a preferentially exposed {200} crystal facet with a two unit cell-thin thickness. An obvious Tyndall light-scattering effect is observed by side-illuminating lighting (inset in Fig. 1e), suggesting the formation of a well-dispersed ultrathin 2D sheet colloid. The diffraction peaks in the X-ray diffraction (XRD) pattern (Fig. 1g) can be indexed as orthorhombic CoP (JCPDS no. 29-0497), thus demonstrating the successful conversion from Co3O4 into CoP. Point-scan energy dispersive X-ray spectroscopy (EDS) analysis (Fig. S3†) and the scanning transmission electron microscopy EDS (STEM-EDS) mapping images (Fig. 1h) indicate the existence and uniform distribution of Co and P. In addition, the specific surface area of UPNSs is 92.23 m2 g−1 (Fig. S4†). All these results imply that CoP UPNSs with a high proportion of exposed {200} facets and with sub-1.1 nm thickness have been successfully fabricated through the convenient chemical transformation route.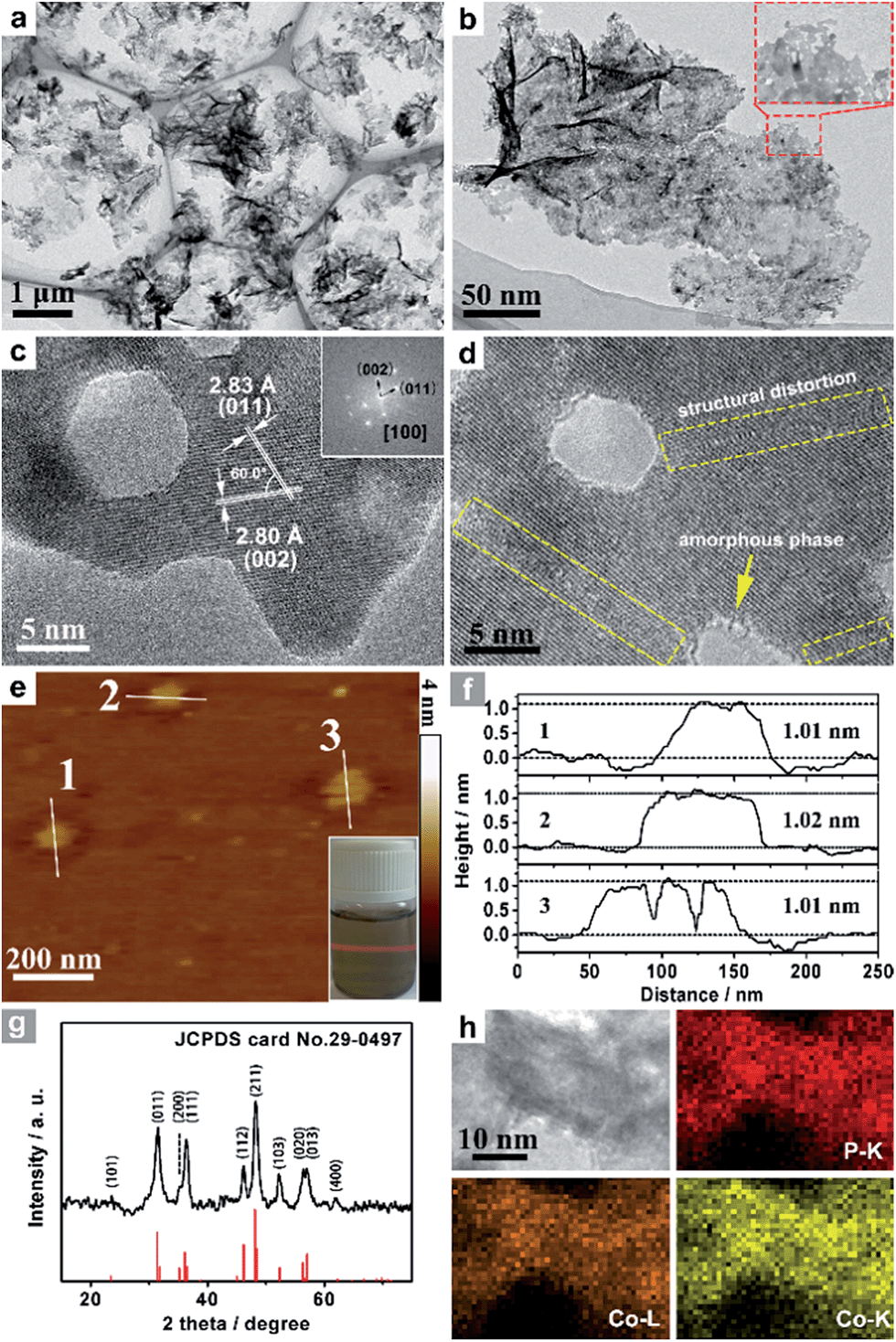 | ||
| Fig. 1 (a, b) TEM images, (c, d) HRTEM images and the associated FFT pattern (inset c) of CoP UPNSs. (e) AFM image and the side-illuminating lighting photo (inset e). (f) The corresponding height profiles of the nanosheets. (g) XRD pattern and (h) STEM-EDS elemental mapping images of CoP UPNSs. | ||
The electrocatalytic HER activity of CoP UPNSs was firstly examined by linear scan voltammetry (LSV) in 0.5 M H2-saturated H2SO4 solution. For comparison, CoP nanoparticles (NPs) (Fig. S5†) and commercial 20 wt% Pt/C deposited on glassy carbon (GC) electrodes with the same amount were also tested under the same conditions. As shown in the I–R corrected LSV polarization curves (Fig. 2a and S6†), Pt/C unquestionably exhibits the highest performance with negligible overpotential and bare GC is totally inactive towards HER. Surprisingly, for the as-obtained CoP UPNSs, the current densities of 10, and 100 mA cm−2 only require overpotentials of 56 mV and 131 mV, respectively, which are much lower than those required for CoP NPs and most reported TMPs under similar conditions (Table S1†). This performance is far superior to most other non-noble metal HER catalysts (Table S2†), indicating the high activity of CoP UPNSs. To probe the HER kinetics, Tafel slopes were calculated. As depicted in Fig. 2b, the Tafel slope for Pt/C is ∼32 mV per decade, which is consistent with the literature values.1–3,16–21 The Tafel slope for CoP UPNSs is calculated to be 44 mV per decade, indicating a first-class electrocatalytic activity towards HER with the Volmer–Heyrovsky mechanism.21 This value is smaller than that observed for CoP NPs (81 mV per decade) and those of other TMPs (Table S1†). Meanwhile, an extrapolation method applied to the Tafel plot reveals that the exchange current density (j0) is 0.61 mA cm−2, which is the best value for TMP electrocatalysts (Table S1†). Very surprisingly, CoP UPNSs possess a huge mass activity towards HER. For instance, a low overpotential of 100 mV can deliver a mass activity of 151 A g−1, which is over 80 times higher than that of CoP NPs (Fig. 2c). All these results demonstrate that CoP UPNSs are highly active for HER with an extremely large mass activity. Electrochemical impedance spectroscopy (EIS) results (Fig. 2d and S7†) demonstrate a smaller interfacial charge-transfer resistance of CoP UPNSs than that of CoP NPs. This greatly accelerating interfacial charge transfer can be ascribed to the improved conductivity29 and efficient interfacial contact of 2D porous materials with electrolyte. To evaluate the stability in a strong acid environment, a long-term cycling test was adopted by comparing the polarization curves before and after 2000 CV cycles. The final polarization curve of CoP UPNSs still overlaps with the original one (Fig. 2e). Fig. 2f shows that the catalytic performance remains unchanged for at least 24 h. Additional characterizations clearly show that the original ultrathin porous 2D architecture and composition can be maintained after long-term measurements (Fig. S8 and S9†), revealing that CoP UPNSs are highly stable for HER. Importantly, the CoP UPNSs exhibit almost 100% faradic efficiency for HER (Fig. S10†).
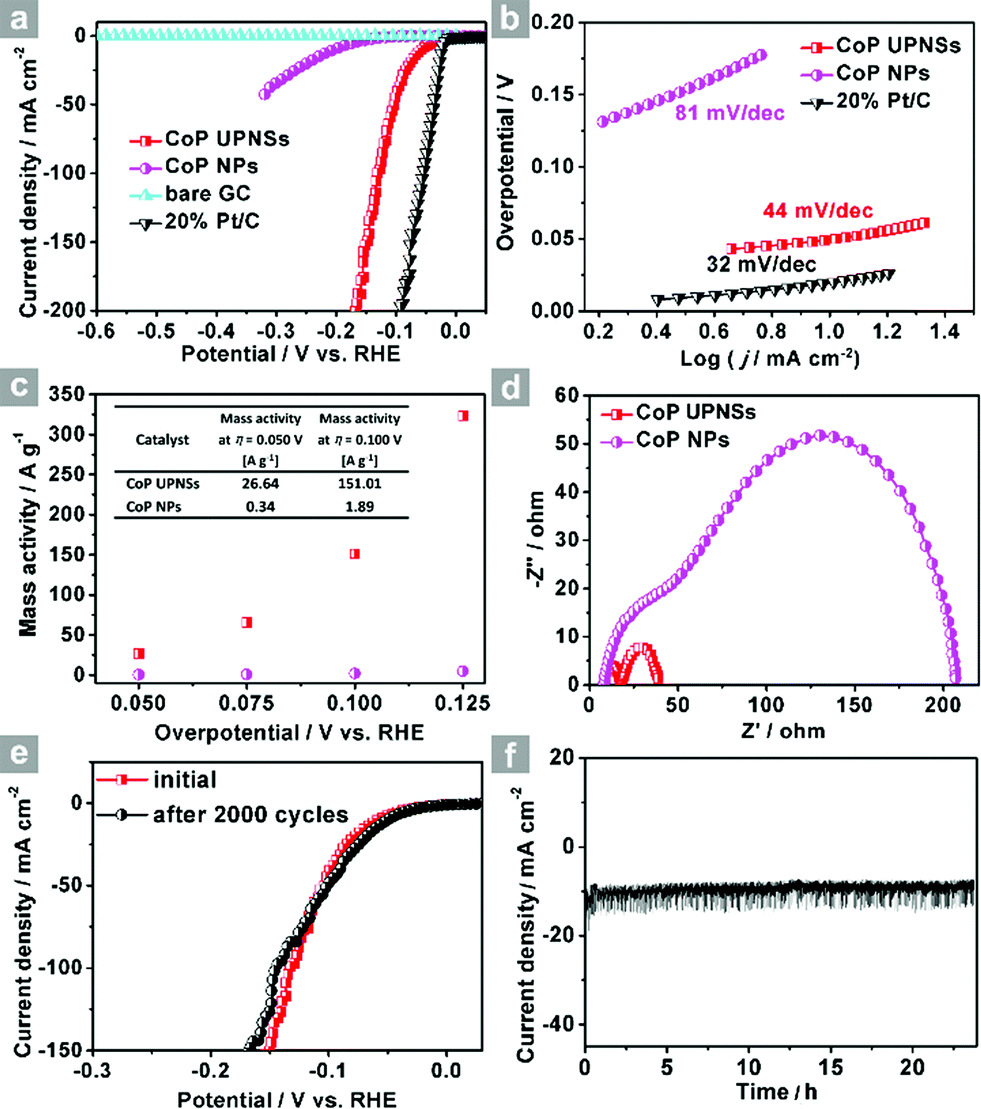 | ||
| Fig. 2 (a) I–R corrected polarization curves of CoP UPNSs, CoP NPs, bare GC and 20% Pt/C and (b) corresponding Tafel plots of CoP UPNSs, CoP NPs and 20% Pt/C in 0.5 M H2SO4 at a scan rate of 2 mV s−1. (c) Mass activity as a function of the overpotential for CoP UPNSs and NPs. (d) Electrochemical impedance spectra of CoP UPNSs and NPs. (e) Polarization curves of CoP UPNSs initially and after 2000 CV scans. (f) Time-dependent current density curve. | ||
To elucidate the origins of the outstanding performance of CoP UPNSs, we performed a series of experimental characterizations. The electrochemical active surface areas (ECSA) are usually evaluated by their electrochemical double layer capacitances (Cdl) because of their positive proportion relationship.49 As shown in Fig. 3c, CoP UPNSs display a Cdl value of 7.87 mF cm−2, which is 22 times higher than that of CoP NPs (0.358 mF cm−2), suggesting a much larger ECSA of CoP UPNSs over the corresponding NPs. Thus, the large ECSA that originates from both the ultrathin and porous characteristics of CoP UPNSs can play an important role on the high activity of the as-converted UPNSs. In addition, the structural distortions and amorphous areas observed in Fig. 1d should make significant contributions to the high activity.29 To determine the facet effect and exclude the influence of ECSA on the electrochemical activity of CoP towards HER, the currents were normalized to the relative ECSA. The normalization curves (Fig. 3d) reveal that CoP UPNSs still exhibit a slightly lower onset potential and smaller Tafel slope than CoP NPs. We speculate that the improvement of the normalized activity may be associated with a high proportion of exposed {200} facets in CoP UPNSs.
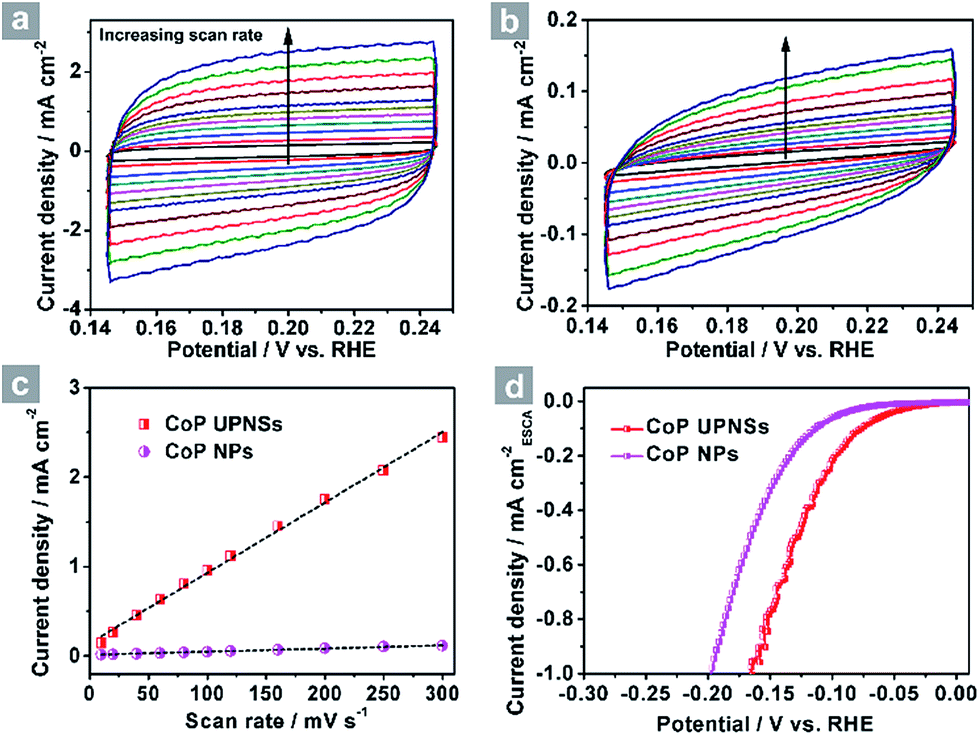 | ||
| Fig. 3 CV curves of CoP UPNSs (a) and CoP NPs (b) with various scan rates, (c) charging current density differences plotted against scan rates. The capacitive currents were measured at 0.10 V vs. RHE. (d) The LSV curves from Fig. 2a normalized to the electrochemical active surface area (ECSA). | ||
Next, density functional theory (DFT) calculations were adopted to fundamentally understand the role of the exposed {200} facets. For most electrocatalysts, ΔGH* and its coverage dependence are the key descriptors for HER activity.1–3,45,50–52 It is believed that the optimal value of |ΔGH*| is zero.45,50–52 For example, the best catalyst, Pt, possesses a ΔGH* value of about −0.09 eV.45,50–52 Surface formation energy calculations reveal that the stable plane for the (100) facet, one typical plane of {200} facets, is the P terminated CoP (100) surface (Fig. 4a). Fig. 4b shows the dependence of ΔGH* on the hydrogen coverage (θH*) of the CoP (100) facet. When one hydrogen atom is absorbed on the P terminated surface (12.5% hydrogen coverage), the calculated ΔGH* is −0.32 eV (Fig. S11b† and 4b). Although the ΔGH* value is comparable with that of most non-precious metal electrocatalysts (Table S3†), the relatively large negative value could not account for such outstanding HER performance.
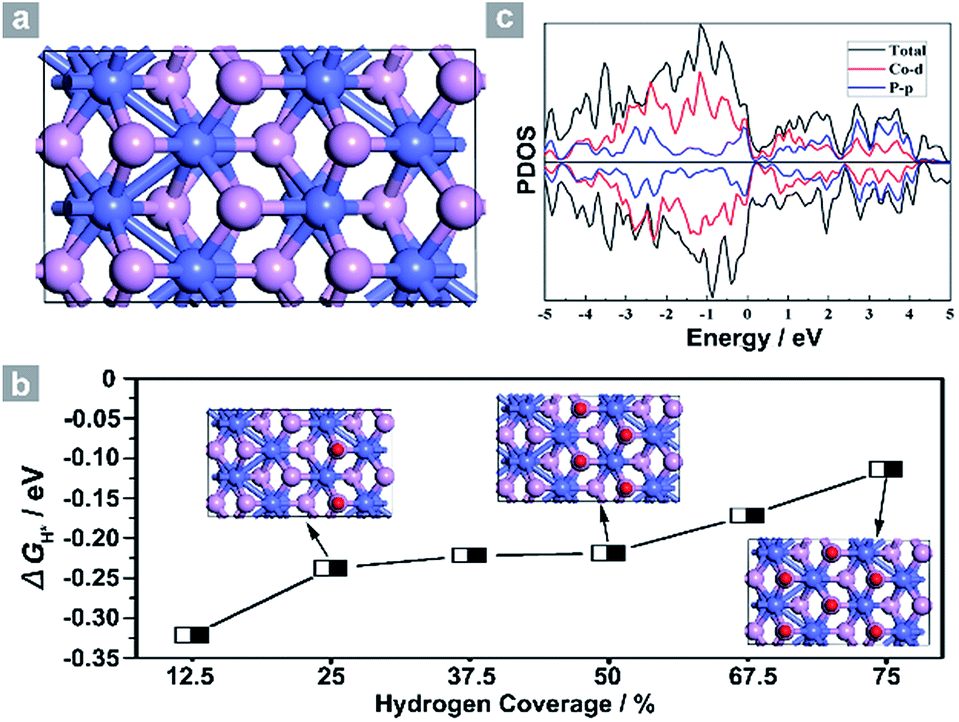 | ||
| Fig. 4 (a) Simulated structure and (b) the dependence of ΔGH* on hydrogen coverage θH*. (c) Projected density of states of the P terminated CoP (100) facet. Co atoms: blue, P atoms: purple and hydrogen atoms: red. | ||
Interestingly, the calculated ΔGH* values shift positively to −0.238 eV and −0.219 eV for the 25% and 50% hydrogen coverages, respectively. The obvious change in the positive direction has been theoretically predicted in some metal phosphides.45 Significantly, when θH* is increased to 75%, ΔGH* moves positively to −0.114 eV, which is one of the best values for non-precious metal electrocatalysts (Table S3†), suggesting that hydrogen atoms still adsorb strongly on the CoP (100) surface at high θH*. For other materials,45,51 the high coverage will make ΔGH* cross over 0 eV and become a positive value, thus causing the difficulty of hydrogen adsorption. However, for the CoP (100) surface, the near-zero ΔGH* at high θH* will lead to the high utilization efficiency of active sites, and thus makes CoP UPNSs with preferentially exposed {200} facets highly active electrocatalysts. In addition, the simulated band structure (Fig. S12†) and the projected density of states (Fig. 4c) reveal the metallic nature of the CoP (100) plane. These metallic characteristics can accelerate electron transfer and thus improve the electrocatalytic performance.
This easy chemical transformation strategy can also be utilized to synthesize other UPNSs. For example, the low-temperature selenylation or sulfuration of Co3O4 precursors can lead to the formation of single-crystalline-like porous ultrathin nanosheets of non-layered CoSe2 (Fig. 5 and S13†) and CoS (Fig. S14†), two of the most efficient electrocatalysts for energy conversion,53–55 suggesting the generality of our methodology.
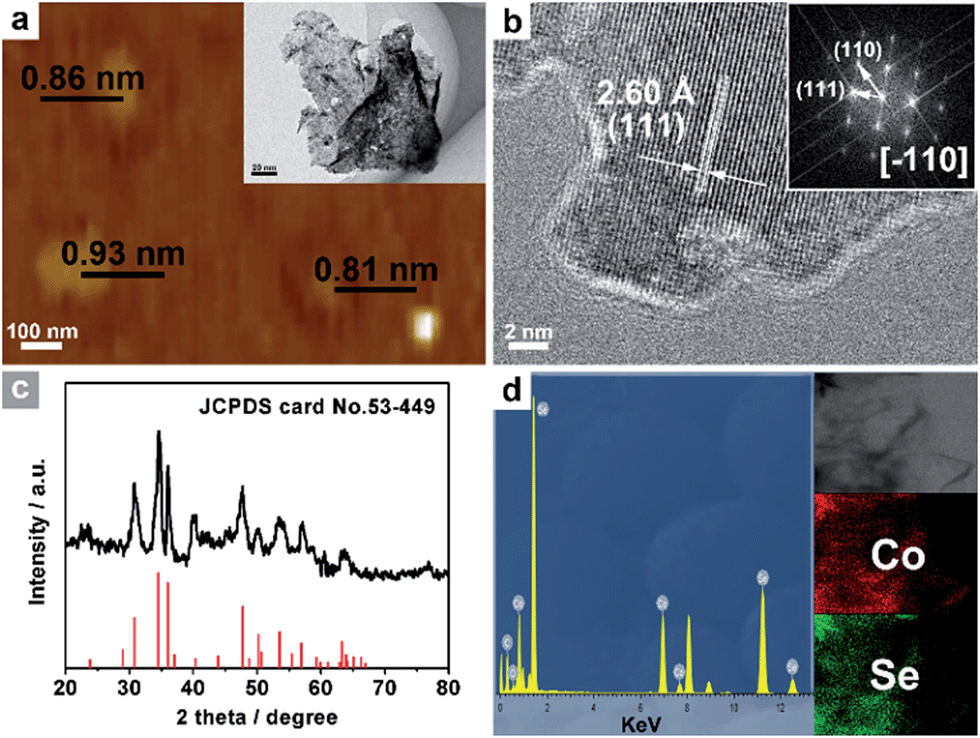 | ||
| Fig. 5 (a) AFM image and TEM image (inset a), and (b) HRTEM images and the associated FFT pattern (inset b) of CoSe2 UPNSs. (c) XRD pattern, (d) EDS spectrum and STEM-EDS elemental mapping images (inset d) of CoSe2 UPNSs. | ||
4. Conclusions
In summary, CoP UPNSs have been successfully synthesized by a handy, robust and efficient chemical transformation strategy of Co3O4 precursors. This facile strategy endows the as-converted samples with four features: ultrathin (two unit cell-thin thickness), porous, a high proportion of exposed {200} facets, and the coexistence of structural distortion and amorphous areas. The products exhibit outstanding performance for HER: low potential (only 56 and 131 mV are required for current densities of 10 and 100 mA cm−2, respectively), small Tafel slope (44 mV per decade), high mass activity (151 A g−1 at an overpotential of 100 mV), nearly 100% faradic efficiency and good stability (at least 20 h). The experimental and theoretical results disclose that the structural distortions and amorphous domains, the high amount of active sites, the facile mass/electron transfer caused by both the porous and ultrathin characteristics, the preferentially exposed active facets, the high utilization efficiency of active sites and the metallic nature are key factors for such excellent performance. Importantly, by using this chemical transformation strategy, other metal selenide and sulfide ultrathin porous nanosheets (e.g. CoSe2, CoS) can be prepared. Our generalized strategy may open a powerful route to synthesize porous ultrathin 2D materials that can't be acquired using the other reported methods. In addition to HER, such novel UPNSs are expected to find other promising energy and catalysis applications (e.g. in water oxidation,28,40 biomass conversion,56,57 and capacitors58).Acknowledgements
We do appreciate the financial support of the National Natural Science Foundation of China (No. 21422104) and the Key Project of Natural Science Foundation of Tianjin City (No. 16JCZDJC30600).Notes and references
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- C. G. Morales-Guio, L.-A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, Nat. Energy, 2016, 1, 16130 CrossRef CAS.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529 CrossRef CAS.
- B. Ni, H. Liu, P.-p. Wang, J. He and X. Wang, Nat. Commun., 2015, 6, 8756 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- S. Zhuo, Y. Xu, W. Zhao, J. Zhang and B. Zhang, Angew. Chem., Int. Ed., 2013, 52, 8602–8606 CrossRef CAS PubMed.
- J. Staszak-Jirkovsky, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K.-C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2016, 15, 197–203 CrossRef CAS PubMed.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Norskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- L. Yu, B. Y. Xia, X. Wang and X. W. Lou, Adv. Mater., 2016, 28, 92–97 CrossRef CAS PubMed.
- P. C. K. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957 CrossRef CAS PubMed.
- L. Liao, S. Wang, J. Xiao, X. Bian, Y. Zhang, M. D. Scanlon, X. Hu, Y. Tang, B. Liu and H. H. Girault, Energy Environ. Sci., 2014, 7, 387–392 CAS.
- H. Lin, Z. Shi, S. He, X. Yu, S. Wang, Q. Gao and Y. Tang, Chem. Sci., 2016, 7, 3399–3405 RSC.
- H. Lin, N. Liu, Z. Shi, Y. Guo, Y. Tang and Q. Gao, Adv. Funct. Mater., 2016, 26, 5590–5598 CrossRef CAS.
- H. B. Wu, B. Y. Xia, L. Yu, X. Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS PubMed.
- Y. Xu, R. Wu, J. Zhang, Y. Shi and B. Zhang, Chem. Commun., 2013, 49, 6656–6658 RSC.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- X. Wang, Y. V. Kolen'ko, X. Q. Bao, K. Kovnir and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 8188–8192 CrossRef CAS PubMed.
- Y. Tan, H. Wang, P. Liu, C. Cheng, F. Zhu, A. Hirata and M. Chen, Adv. Mater., 2016, 28, 2951–2955 CrossRef CAS PubMed.
- M. Ledendecker, S. Krick Calderon, C. Papp, H. P. Steinruck, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361–12365 CrossRef CAS PubMed.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- X.-Y. Yu, Y. Feng, B. Guan, X. W. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 1246–1250 CAS.
- M. Caban-Acevedo, M. L. Stone, J. R. Schmidt, J. G. Thomas, Q. Ding, H.-C. Chang, M.-L. Tsai, J.-H. He and S. Jin, Nat. Mater., 2015, 14, 1245–1251 CrossRef CAS PubMed.
- R. Wu, J. Zhang, Y. Shi, D. Liu and B. Zhang, J. Am. Chem. Soc., 2015, 137, 6983–6986 CrossRef CAS PubMed.
- X. Xu, Y. Chen, W. Zhou, Z. Zhu, C. Su, M. Liu and Z. Shao, Adv. Mater., 2016, 28, 6442–6448 CrossRef CAS PubMed.
- L. L. Feng, G. Yu, Y. Wu, G. D. Li, H. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef CAS PubMed.
- Y. Sun, Z. Sun, S. Gao, H. Cheng, Q. Liu, J. Piao, T. Yao, C. Wu, S. Hu, S. Wei and Y. Xie, Nat. Commun., 2012, 3, 1057 CrossRef PubMed.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao, S. Wei, B. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed.
- Y. Sun, S. Gao, F. Lei and Y. Xie, Chem. Soc. Rev., 2015, 44, 623–636 RSC.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, Angew. Chem., Int. Ed., 2015, 54, 8722–8727 CrossRef CAS PubMed.
- X. Long, G. Li, Z. Wang, H. Zhu, T. Zhang, S. Xiao, W. Guo and S. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef CAS PubMed.
- X. Huang, S. Li, Y. Huang, S. Wu, X. Zhou, S. Li, C. L. Gan, F. Boey, C. A. Mirkin and H. Zhang, Nat. Commun., 2011, 2, 292 CrossRef PubMed.
- X. Zhang, Z. Lai, C. Tan and H. Zhang, Angew. Chem., Int. Ed., 2016, 55, 8816–8838 CrossRef CAS PubMed.
- Y. Kuang, G. Feng, P. Li, Y. Bi, Y. Li and X. Sun, Angew. Chem., Int. Ed., 2016, 55, 693–697 CrossRef CAS PubMed.
- H. Fan, X. Huang, L. Shang, Y. Cao, Y. Zhao, L.-Z. Wu, C.-H. Tung, Y. Yin and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 2167–2170 CrossRef CAS PubMed.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef CAS PubMed.
- Y. Liu, J. Goebl and Y. Yin, Chem. Soc. Rev., 2013, 42, 2610–2653 RSC.
- F. Lei, Y. Sun, K. Liu, S. Gao, L. Liang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2014, 136, 6826–6829 CrossRef CAS PubMed.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587 CrossRef CAS PubMed.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251–6254 CrossRef CAS PubMed.
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., 2014, 126, 5531–5534 CrossRef.
- Y. Sun, S. Gao, F. Lei, J. Liu, L. Liang and Y. Xie, Chem. Sci., 2014, 5, 3976 RSC.
- P. Jiang, Q. Liu, C. Ge, W. Cui, Z. Pu, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2014, 2, 14634 CAS.
- J. Kibsgaard, C. Tsai, K. Chan, J. D. Benck, J. K. Norskov, F. Abild-Pedersen and T. F. Jaramillo, Energy Environ. Sci., 2015, 8, 3022–3029 CAS.
- Y. Duan, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 045332 CrossRef.
- D. Zhang, A. B. Wong, Y. Yu, S. Brittman, J. Sun, A. Fu, B. Beberwyck, A. P. Alivisatos and P. Yang, J. Am. Chem. Soc., 2014, 136, 17430–17433 CrossRef CAS PubMed.
- S.-K. Han, C. Gu, M. Gong and S.-H. Yu, J. Am. Chem. Soc., 2015, 137, 5390–5396 CrossRef CAS PubMed.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- P. Roger, Trans. Faraday Soc., 1958, 54, 1053–1063 RSC.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 Search PubMed.
- S. Peng, L. Li, X. Han, W. Sun, M. Srinivasan, S. G. Mhaisalkar, F. Cheng, Q. Yan, J. Chen and S. Ramakrishna, Angew. Chem., 2014, 126, 12802–12807 CrossRef.
- D. Kong, H. Wang, Z. Lu and Y. Cui, J. Am. Chem. Soc., 2014, 136, 4897–4900 CrossRef CAS PubMed.
- M. S. Faber, R. Dziedzic, M. A. Lukowski, N. S. Kaiser, Q. Ding and S. Jin, J. Am. Chem. Soc., 2014, 136, 10053–10061 CrossRef CAS PubMed.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- L. Shen, L. Yu, H. B. Wu, X.-Y. Yu, X. Zhang and X. W. Lou, Nat. Commun., 2015, 6, 6694 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S14 and Tables S1–S3. See DOI: 10.1039/c6sc05687c |
| This journal is © The Royal Society of Chemistry 2017 |