
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed direct alkylation of heteroarenes†
Cédric
Theunissen
,
Jianjun
Wang
and
Gwilherm
Evano
 *
*
Laboratoire de Chimie Organique, Service de Chimie et PhysicoChimie Organiques, Université libre de Bruxelles (ULB), Avenue F. D. Roosevelt 50, CP160/06, 1050 Brussels, Belgium. E-mail: gevano@ulb.ac.be
First published on 20th March 2017
Abstract
An efficient and broadly applicable process is reported for the direct alkylation of C–H bonds in heteroarenes, privileged scaffolds in many areas of science. This reaction is based on the copper-catalyzed addition of alkyl radicals generated from activated secondary and tertiary alkyl bromides to a wide range of arenes, including furans, thiophenes, pyrroles, and their benzo-fused derivatives, as well as coumarins and quinolinones.
Among the impressive and ever-growing number of chemical transformations, the alkylation of arenes is clearly of prime importance, due to regioselectively alkylated arenes being major starting materials and molecules relevant to most areas of science in which there is a need for small organic molecules. They are indeed used on a daily basis in the pharmaceutical, agrochemical and material sciences, and as building blocks for polymer chemistry or organic electronics. They also have had a profound effect in human health and many other sectors.
The regioselective alkylation of arenes is traditionally performed through the venerable Friedel–Crafts reaction,1 a reaction which requires harsh reaction conditions and often suffers from low chemo- and/or regio-selectivities. This reaction is also hampered by significant limitations in terms of the electronic properties of the arene, and it performs poorly with some classes of alkyl halides. The main alternative strategies for the alkylation of arenes are mostly based on stoichiometric metallation followed by reaction with an electrophile2—a sequence which suffers from limitations in terms of the functional group compatibility due to the requirement for strong bases—or on stepwise pre-functionalization followed by metal-mediated cross-coupling.3
Recent advances in transition-metal catalyzed C–H functionalization have enabled the development of remarkably efficient and reliable tools for the direct alkylation of arenes, which now appear as robust alternatives to the classical processes.4 The most common strategy to ensure high levels of reactivity and selectivity is mostly based on the introduction of a directing group, which will allow for the selective alkylation of a single C–H bond of the starting arene. Following the pioneering work of Murai on the ortho-selective alkylation of arylketones,5 a range of directing groups and alkylating agents have been introduced for the direct ortho- and, much more rarely, meta-alkylation of arenes.6 A complementary strategy, which circumvents the use of directing groups, is based on the innate alkylation of C–H bonds in heteroarenes. A number of efficient reagents and procedures have indeed been designed for the regioselective introduction of alkyl substituents into a variety of heteroarenes, mostly electron-deficient ones7 and azoles possessing acidic C–H bonds.7f,8 In sharp contrast, much less attention has been paid to the direct alkylation of other heteroarenes,6g,h,7f,8c,9 despite the strong synthetic potential of such reactions.
Heteroarenes, such as furans, thiophenes, pyrroles and their benzo-fused derivatives, are privileged scaffolds, and the development of efficient and broadly applicable procedures for their direct alkylation is therefore still highly demanded. The most efficient and general procedures available to date to perform such a task are indeed mostly limited to Ackermann’s alkylation of directing group-containing indoles with alkyl halides,6g,h Zhou’s remarkable alkylation of (benzo)furans, (benzo)thiophenes, pyrroles and indoles possessing an electron-withdrawing group with cyclohexyl iodide,7f Nakao and Hiyama’s alkylation of activated indoles with styrenes,8c Bach’s alkylation of indoles with alkyl bromides9b and Stephenson’s photocatalytic alkylation of indoles, pyrroles and furans with brominated malonates.9a,f While efficient, major limitations still remain with these procedures, such as harsh reaction conditions that can pose a problem for late-stage alkylation, low substrate scope with regards to the nature of the heteroarene and/or alkylating agent, or the need for directing/activating groups on the heteroarene. There is therefore no general and broadly applicable method reported for the direct alkylation of unactivated heteroarenes.
In an attempt to address some of these limitations, and as part of our ongoing program in copper catalysis,10 we report in this manuscript an efficient and broadly applicable copper-catalyzed direct alkylation of heteroarenes with radicals generated from activated secondary and tertiary alkyl bromides (Scheme 1).
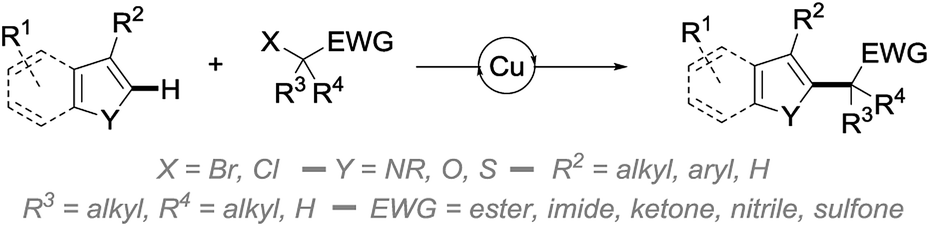 | ||
| Scheme 1 Copper-catalyzed radical direct alkylation of heteroarenes. | ||
Inspired by the recent and remarkable work of Nishikata, using copper catalysts for the generation of tertiary alkyl radicals,11 and the copper-catalyzed atom transfer radical cyclization (ATRC)12 and polymerization (ATRP)13 reactions, we surmised that the right copper complex should be able to promote the generation of alkyl radicals by single electron reduction of the corresponding activated alkyl bromides. Provided that these electron-poor radicals will react with heteroarenes at a sufficient rate,14 and that the oxidized copper-catalyst can promote oxidative re-aromatization, an efficient and broadly applicable system for the direct alkylation of heteroarenes relying on the use of simple reagents and catalyst could potentially be developed.
To test this working hypothesis, benzofuran 1a and ethyl α-bromoisobutyrate 2a were chosen as model substrates. After a brief optimization of the source of copper(I) and the base and solvent that validated our working hypothesis (data not shown, see ESI for details†), the efficiency of a set of representative ligands Lx was evaluated: selected results are shown in Fig. 1. While a NHC (L1) totally inhibited the reaction which was shown to proceed with moderate efficiency in the absence of a ligand, the O,O-bidentate ligand L2 was found to moderately improve the yield. The N,O- and N,N-bidentate ligands L3–L11 were found to be much more efficient, the diimine L7 efficiently promoting the reaction to 89% yield but being difficult to remove from the alkylated product 3a. The tridentate ligands L12–L14 were finally evaluated with L14 (TPMA) being selected for its efficiency (89% yield of 3a obtained as a single regioisomer). The reaction was found to perform equally well with this ligand in dioxane in place of ethanol, an important point for substrates bearing functional groups that would not be compatible with this solvent. We finally attempted to decrease the catalyst loading using L14 as the ligand: a combination of 5 mol% of copper(I) iodide and 10 mol% of L14 resulted in a significant decrease of the yield (50%), which could however be improved to 70% by increasing the reaction time to 72 h. Gratifyingly, the reaction was still found to be operative, although with less efficiency, when further decreasing the catalyst loading to 2 mol% of copper(I) iodide and 4 mol% of L14. The alkylated benzofuran 3a was formed with 58% yield under these conditions.
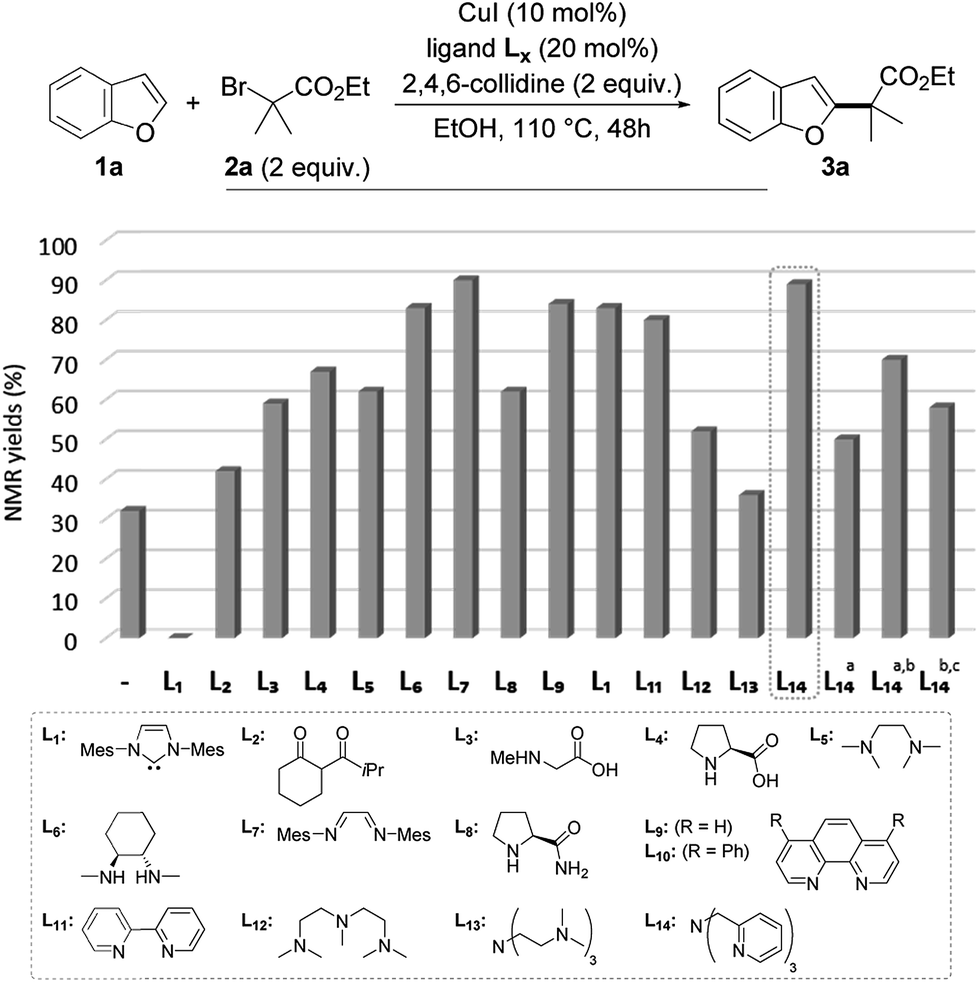 | ||
| Fig. 1 Validation of the working hypothesis and optimization of the reaction (a with 5 mol% of CuI and 10 mol% of L14; b 72 h; c with 2 mol% of CuI and 4 mol% of L14). | ||
Having in hand an efficient optimized catalytic system, we then moved to the study of the scope and limitations of this reaction by first evaluating the reactivity of a series of alkyl bromides 2 for the alkylation of benzofuran 1a (Fig. 2) using 10 mol% of copper(I) iodide and 20 mol% of TPMA in dioxane or ethanol at 110 °C for 2 days. The reaction was found to perform well with tertiary alkyl bromides and provided the corresponding alkylated benzofuran derivatives 3a, 3b, 3h–k, 3o and 3q with good to excellent yields. Secondary alkyl bromides were also gratifyingly found to be efficient alkylating agents, as demonstrated with the alkylation of 1a to 3c–e, 3l, 3m and 3p. However, the presence of an aryl group inhibited the reaction (3f), probably due to the diminished reactivity of the intermediate radical species. In sharp contrast, a primary alkyl bromide only produced the alkylated derivative in low yield (3g). Compared to the only general procedure reported to date to perform such a direct alkylation of benzofurans, developed by Hirano and Miura and relying on a large excess of the starting benzofuran and a much more sensitive Ni(cod)2 catalyst,9d,15 our alkylation typically furnishes the alkylated benzofurans with a 20–90% increase in the yields. Other esters (benzyl 3h, tert-butyl 3i and even the labile phenyl ester 3j) were found to proceed equally well, and this electron-withdrawing group could also be suitably replaced by a ketone (3k), a nitrile (3l), an imide (3m) or a sulfone (3o, p), while a brominated amide (3n) or malonates (data not shown) were found to be respectively inefficient or less reactive. However, this demonstrates the variety of functional groups that can be introduced and used for further functionalization, as evidenced by the reductive desulfonylation of 3o and 3p to their simple alkylated derivatives 4 and 4′, the only synthesis of the former reported to date being based on a low-yielding (18%) metallation of benzofuran with tert-butyllithium followed by alkylation with isopropyl iodide.16 It ought to be mentioned that a secondary alkyl chloride could also be used in place of the corresponding bromide, although with reduced efficiency (3c, 54%), and the alkylation of benzofuran could be performed on a 5 gram (42 mmol) scale with 5 mol% of CuI and 10 mol% of TPMA in refluxing dioxane for 96 h. This yielded 4 grams (41%) of the corresponding alkylated derivative 3a, the reduced efficiency of the alkylation, which still enabled an easy gram-scale synthesis of 3a, being due to the decreased catalyst loading and reduced reaction temperature. Finally, a complex alkylating agent derived from cholesterol could also be used successfully, as exemplified with the preparation of 3q (using reversed stoichiometry).
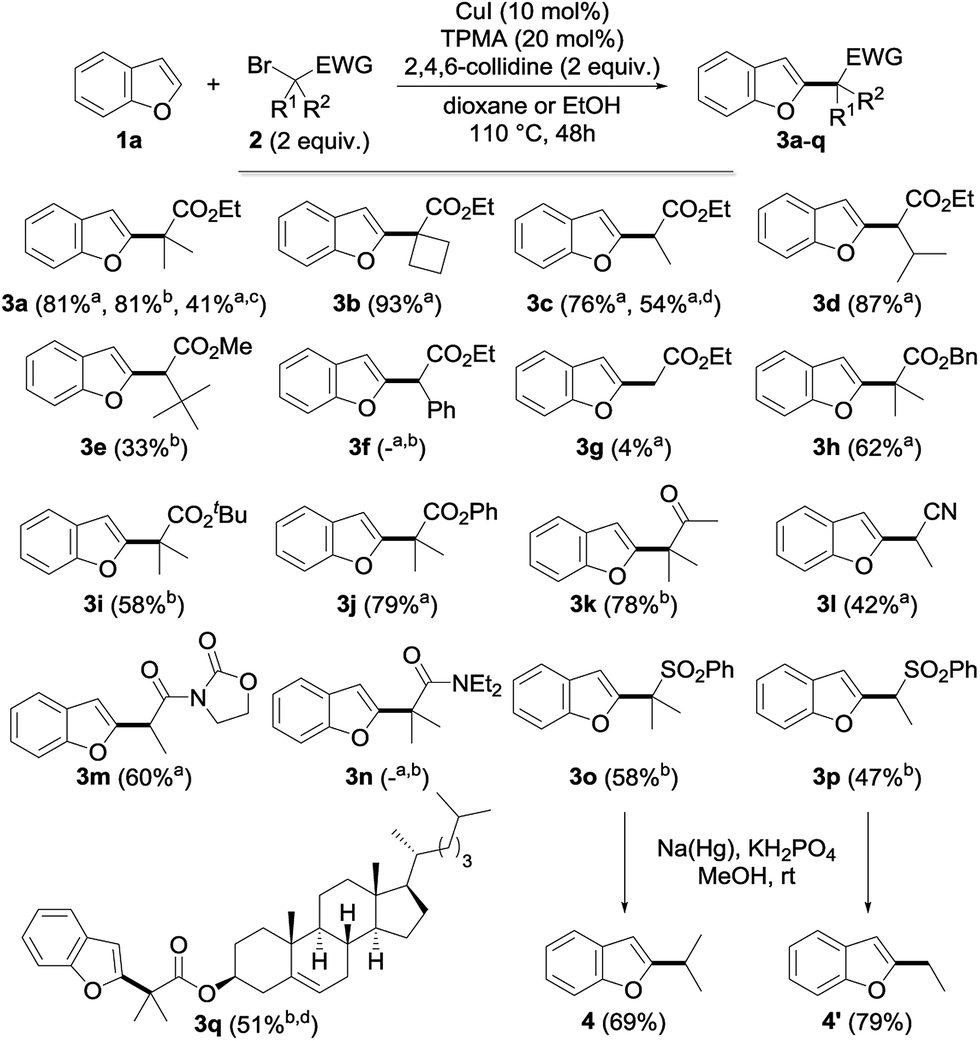 | ||
| Fig. 2 Scope of the copper-catalyzed direct alkylation with representative alkyl bromides (a in dioxane; b in EtOH; c reaction performed on a 5 g scale of 1a with 5 mol% of CuI and 10 mol% of TPMA for 96 h at 101 °C; d starting from the corresponding chloride; e using 2 equiv. of 1a and 1 equiv. of 2). | ||
We then moved to the study of the scope of our process with respect to the other coupling partner and first examined the alkylation of a series of benzofuran derivatives 1 with ethyl α-bromoisobutyrate 2a as the reference alkyl halide. As shown by the results collected in Fig. 3, the reaction was found to be fairly general and tolerates a wide array of substituents—including electron-donating and withdrawing groups—and substitution patterns, including close to the reacting center (3ae). Interestingly, the presence of a halide on the starting benzofuran does not interfere with the reaction, as shown with the alkylation to 3u and 3v in 79 and 76% yields, therefore providing opportunities for further chemical diversification. A range of functional groups were found to be compatible with our procedure, including an unprotected alcohol (3w), a ketone (3aa), an aldehyde (3ab), a nitrile (3ac), an alkyne (3x) and, to a lesser extent, a nitro group (3ad), whereas the presence of an alkene (3y) gave a complex reaction mixture, probably due to its competitive alkylation.11a
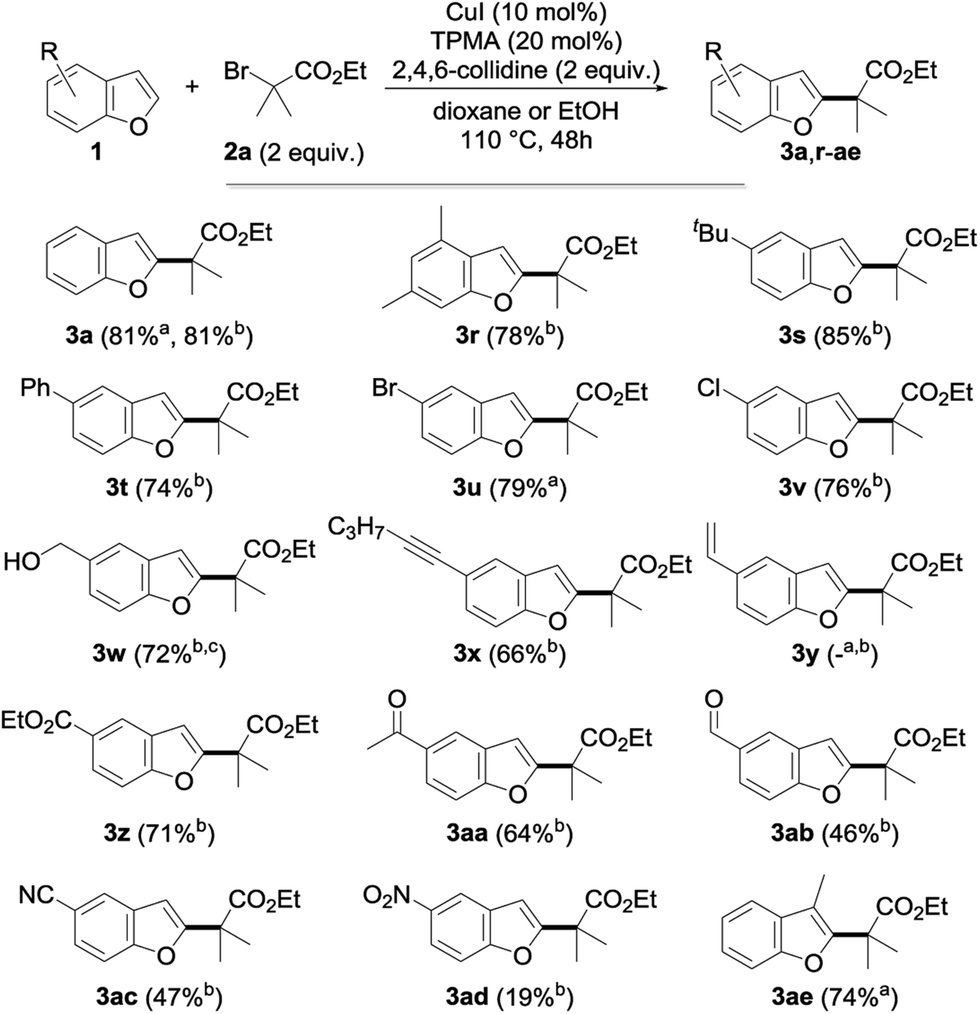 | ||
| Fig. 3 Scope of the copper-catalyzed direct alkylation with representative benzofurans (a in dioxane; b in EtOH; c using 3 equiv. of 2a for 72 h). | ||
Importantly, the extension of the alkylation to other arenes was next undertaken using a set of heteroarenes possessing representative electronic and steric properties with, as in the previous case, ethyl α-bromoisobutyrate 2a as the reference alkyl halide (Fig. 4). The alkylation was pleasingly found to be rather general and efficient with a range of heteroarenes. Furan derivatives could indeed be efficiently and smoothly transformed to their alkylated derivatives 5 in fair to excellent yields (40–93%), and as single C-2 regioisomers. Substitution at C-3 (5h–l) had virtually no effect on the alkylation, and pyridine (5b), ketone (5d), aldehyde (5e), unprotected alcohol (5g) or other aromatic ring functional groups (5h, i, k, l) were well tolerated. The reaction was found to be more substrate-dependent in the absence of a substituent at the C2/C5 position: while ethyl furan-3-carboxylate was cleanly and selectively alkylated to 5m, a mixture of mono- (5n) and bis- (5n′) alkylated furans was obtained starting from 3-phenylfuran. In these two cases, the regioselective functionalization at the C2 position can be reasonably rationalized by the formation of an intermediate radical species stabilized by conjugation with either the phenyl ring or the ester, a stabilization that does not occur when the radical attacks at the C5 position. As for furan itself, it unfortunately could not be alkylated due to its significant volatility.
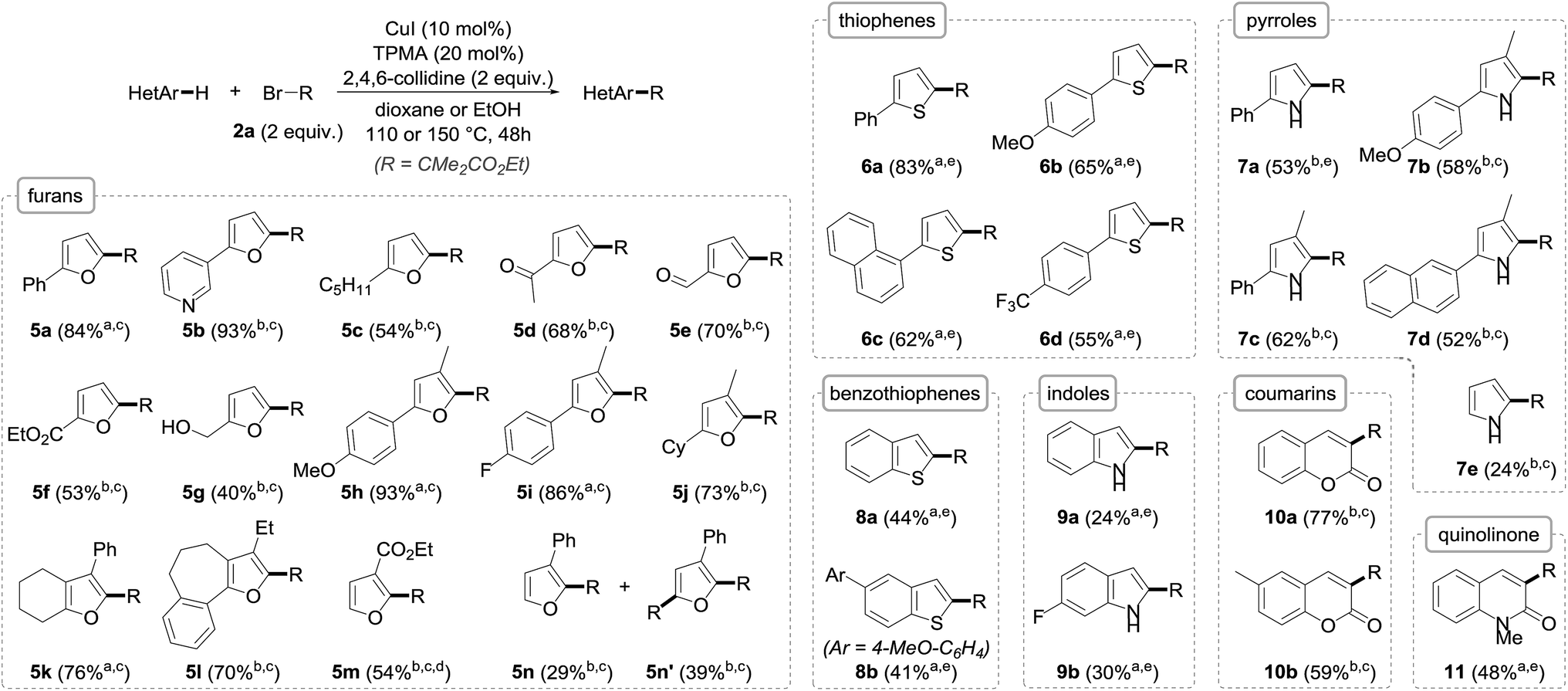 | ||
| Fig. 4 The scope of the copper-catalyzed direct alkylation with representative heteroarenes (R = CMe2CO2Et; a in dioxane; b in EtOH; c at 110 °C; d with 3 equiv. 2a for 72 h; e at 150 °C). | ||
Thiophenes could also be alkylated, although the reaction was found to be more sluggish with these substrates, requiring an increase of the reaction temperature to reach sufficient conversions to their C-2 alkylated derivatives 6, which could be obtained with yields in the 55–83% range.
The alkylation of pyrroles, which represents an important chemical transformation for the design of bioactive molecules, was next studied and found to proceed reasonably well—even if the yields were somehow lower compared to the furan and thiophene series due to incomplete conversions—and did not require protection of the nitrogen atom. Pyrrole itself—whose direct alkylation still remains challenging9f—could be readily alkylated to 7e, although with modest yield, in the absence of competing bis-alkylation.
While the alkylation was shown to be remarkably efficient for the direct functionalization of benzofurans, switching to their sulfur and nitrogen analogues was found to be less efficient despite all attempts made at reoptimization, even if the alkylation of benzothiophenes to 8a and b (41–44%) and indoles to 9a and b (24–30%) was still operative, the mass balances mostly corresponding to the unreacted starting materials. In the case of indoles, the C-2 selectivity, which is complementary to the Friedel–Crafts alkylation occurring at C3, is worth noting. This selectivity, which is in accordance with previously reported results,14 is consistent with the significant HOMO coefficient at C-2 as well as the formation of a stabilized benzylic radical. In the indole series, the N-alkylation of the starting materials was found to be detrimental and resulted in either lower yields or total absence of reactivity, an effect that was also observed with pyrroles.17
In an attempt to further broaden the scope of our copper-catalyzed alkylation, the reactivity of other substrates that might be able to react with the electron-poor radical species was briefly envisioned. While electron-rich benzene derivatives such as 1,3,5-trimethoxybenzene failed to give the corresponding alkylated arenes, coumarins and quinolinone were found to be suitable substrates in our copper-catalyzed alkylation, providing the corresponding functionalized derivatives 10a, b and 11 in fair to good yields.
After an extensive study of the scope of the copper-catalyzed alkylation, we next briefly envisioned the possibility of its extension to an intramolecular version. With this goal in mind, α-bromoacetanilides 12—whose cyclization to the corresponding indolones 13 was previously reported using nickel-18 and iridium-19 based catalysts—were submitted to our optimized reaction conditions. To our delight, the cyclization was found to proceed remarkably well with the cyclized indolones being obtained in 65–84% yields (Fig. 5).
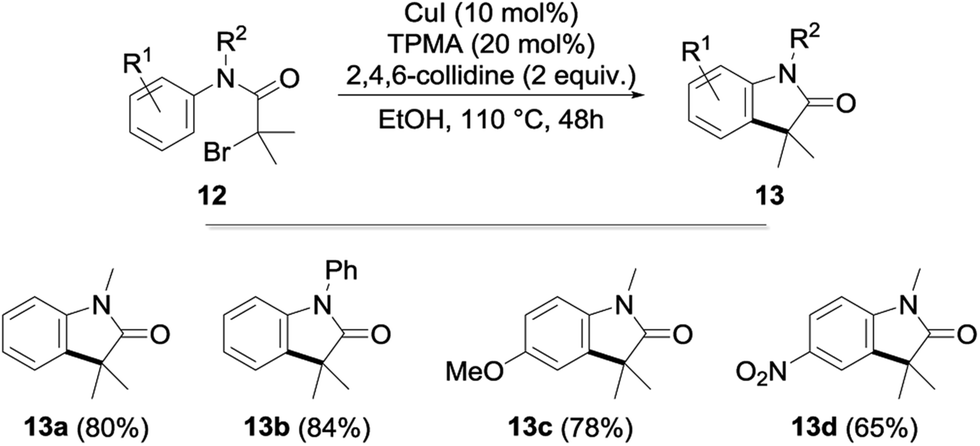 | ||
| Fig. 5 Copper-catalyzed intramolecular alkylation of α-bromoacetanilides to indolones. | ||
Several mechanisms could possibly account for the observed alkylation, including polar and radical pathways. To test the possibility of single electron transfers, and the intermediacy of the radical species, the alkylation of 1a with 2a was performed in the presence of TEMPO 14 (1 equiv.) under our standard conditions (Scheme 2). The alkylation of 1a was totally suppressed in this case, the only product observed at the end of the reaction being the TEMPO adduct 15—a compound that is not formed in the absence of the copper catalyst—which could be isolated in 44% yield.
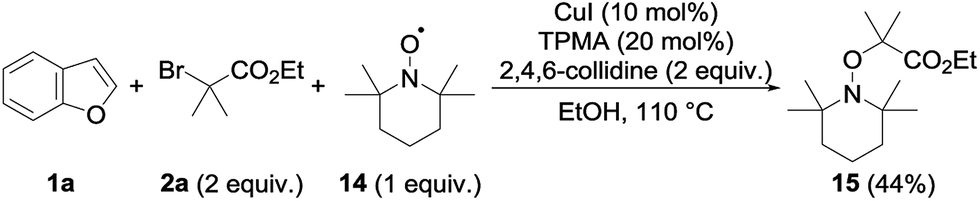 | ||
| Scheme 2 Radical trapping experiment. | ||
A plausible mechanism based on this experiment is shown in Fig. 6. The reaction would be initiated by single electron transfer from the copper(I) catalyst (TPMA)CuII to the σ* orbital of the alkyl halide 2, generating the radical intermediate A and oxidized catalyst (TPMA)CuIIIX. The addition of this intermediate A to the electron-rich heteroarene would afford a transient radical B, which would then be oxidized by (TPMA)CuIIIX to the corresponding carbocation C together with regeneration of the copper(I) catalyst. Rearomatization by the loss of a proton, trapped by 2,4,6-collidine, would finally afford the alkylated heteroarene. Alternatively, the benzylic radical might directly react with (TPMA)CuIIIX to furnish, after elimination of HX, the alkylated heteroarene.
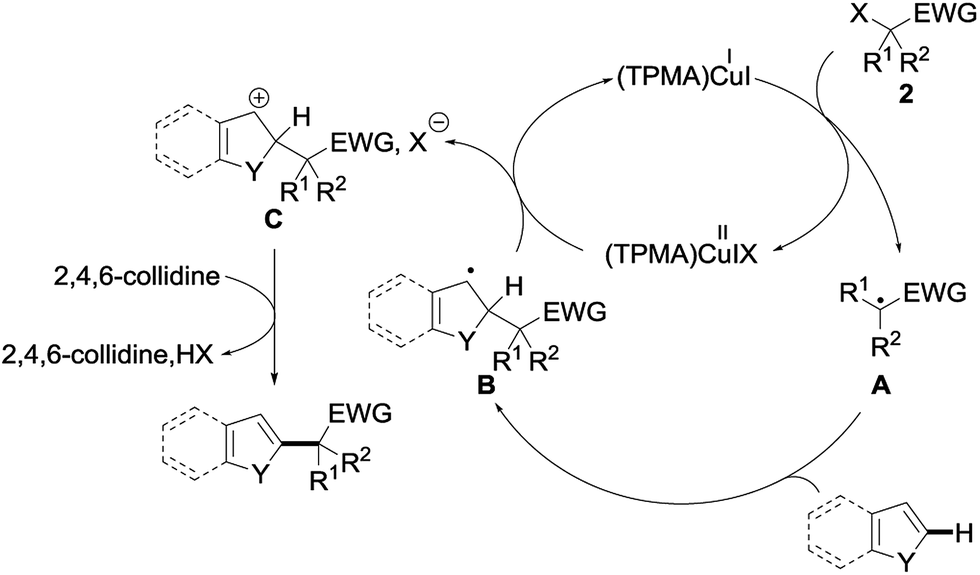 | ||
| Fig. 6 Possible mechanism for the copper-catalyzed direct alkylation of heteroarenes. | ||
Conclusions
In conclusion, we have developed an efficient and broadly applicable process for the direct alkylation of C–H bonds in heteroarenes. These are privileged scaffolds in many areas of science for which there are only a few available methods enabling their direct alkylation. This reaction is based on the copper-catalyzed addition of alkyl radicals, generated from activated secondary and tertiary alkyl bromides, to a wide range of arenes including furans, thiophenes, pyrroles and their benzo-fused derivatives, as well as coumarins and quinolinones. The notable features of this reaction are its wide substrate scope, availability of the catalyst and reagents used, and functional group tolerance.Acknowledgements
Our work was supported by the Université libre de Bruxelles (ULB) and the Fédération Wallonie-Bruxelles (ARC Consolidator 2014-2019). C. T. and J. W. respectively acknowledge the Fonds pour la formation à la Recherche dans l’Industrie et dans l’Agriculture (F.R.I.A.), and the Chinese Scholarship Council for graduate fellowships. The Fonds Emile Defay is gratefully acknowledged for additional financial support.Notes and references
- R. M. Roberts and A. A. Khalaf, Friedel–Crafts Alkylation Chemistry: a Century of Discovery, Marcel Dekker, New York, 1984 Search PubMed.
- R. Chinchilla, C. Nájera and M. Yus, Chem. Rev., 2004, 104, 2667 CrossRef CAS PubMed.
- For a borylation–alkylation sequence, see: D. W. Robbins and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 933 CrossRef CAS PubMed.
- For reviews, see: (a) L. Ackermann, Chem. Commun., 2010, 4866 RSC; (b) S. Messaoudi, J.-D. Brion and M. Alami, Eur. J. Org. Chem., 2010, 6495 CrossRef CAS.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS.
- For recent examples of directed C–H alkylation of arenes, see: (a) L. Ilies, Q. Chen, X. Zeng and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 5221 CrossRef CAS PubMed; (b) M. Schinkel, I. Marek and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 3977 CrossRef CAS PubMed; (c) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308 CrossRef CAS PubMed; (d) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877 CrossRef CAS PubMed; (e) K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 9279 CrossRef CAS PubMed; (f) P. S. Thuy-Boun, G. Villa, D. Dang, P. Richardson, S. Su and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 17508 CrossRef CAS PubMed; (g) B. Punji, W. Song, G. A. Shevchenko and L. Ackermann, Chem.–Eur. J., 2013, 19, 10605 CrossRef CAS PubMed; (h) W. Song, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 2477 CrossRef CAS PubMed; (i) L. Illies, T. Matsubara, S. Ichikawa, S. Asako and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 13126 CrossRef PubMed; (j) R.-Y. Zhu, J. He, X.-C. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 13194 CrossRef CAS PubMed; (k) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2015, 137, 531 CrossRef CAS PubMed; (l) Z. Wang, Y. Kunonibu and M. Kanai, J. Am. Chem. Soc., 2015, 137, 6140 CrossRef CAS PubMed; (m) P.-X. Shen, X.-C. Wang, P. Wang, R.-Y. Zhu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11574 CrossRef CAS PubMed; (n) G. Cera, T. Haven and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 1484 CrossRef CAS PubMed; (o) Z. Ruan, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 3153 CrossRef CAS PubMed; (p) R. Shang, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2016, 138, 10132 CrossRef CAS PubMed.
- For examples, see: (a) J. C. Lewis, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2007, 129, 5332 CrossRef CAS PubMed; (b) Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 13666 CrossRef CAS PubMed; (c) Y. Jujiwara, J. A. Dixon, F. O’Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95 CrossRef PubMed; (d) B. Xiao, Z.-J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 616 CrossRef CAS PubMed; (e) A. P. Antonchick and L. Burgmann, Angew. Chem., Int. Ed., 2013, 52, 3267 CrossRef CAS PubMed; (f) X. Wu, J. W. T. See, K. Xu, H. Hirao, J. Roger, J.-C. Hierso and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13573 CrossRef CAS PubMed; (g) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87 CrossRef CAS PubMed; (h) T. McCallum and L. Barriault, Chem. Sci., 2016, 7, 4754 RSC.
- For examples, see: (a) K. L. Tan, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2002, 124, 13964 CrossRef CAS PubMed; (b) O. Vechorkin, V. Proust and X. Hu, Angew. Chem., Int. Ed., 2010, 49, 3061 CrossRef CAS PubMed; (c) Y. Nakao, N. Kashihara, K. S. Kanyiva and T. Hiyama, Angew. Chem., Int. Ed., 2010, 49, 4451 CrossRef CAS PubMed; (d) T. Yao, K. Hirano, T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 12307 CrossRef CAS PubMed; (e) L. Ackermann, B. Punji and W. Song, Adv. Synth. Catal., 2011, 353, 3325 CrossRef CAS; (f) T. Yao, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 775 CrossRef CAS PubMed; (g) P. Ren, I. Salihu, R. Scopelliti and X. Hu, Org. Lett., 2012, 14, 1748 CrossRef CAS PubMed.
- (a) L. Furst, B. S. Matsuura, J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, Org. Lett., 2010, 12, 3104 CrossRef CAS PubMed; (b) L. Jiao and T. Bach, J. Am. Chem. Soc., 2011, 133, 12990 CrossRef CAS PubMed; (c) C. S. Sevov and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2116 CrossRef CAS PubMed; (d) A. Nakatani, K. Hirano, T. Satoh and M. Miura, Chem.–Eur. J., 2013, 19, 7691 CrossRef CAS PubMed; (e) X.-H. Ouyang, R.-J. Song, M. Hu, Y. Yang and J.-H. Li, Angew. Chem., Int. Ed., 2016, 55, 3187 CrossRef CAS PubMed; (f) E. C. Swift, T. M. Williams and C. R. J. Stephenson, Synlett, 2016, 27, 754 CrossRef CAS.
- For representative contributions from our group on copper-catalyzed reactions, see: (a) M. Toumi, F. Couty and G. Evano, Angew. Chem., Int. Ed., 2007, 46, 572 CrossRef CAS PubMed; (b) A. Coste, M. Toumi, K. Wright, V. Razafimahaléo, F. Couty, J. Marrot and G. Evano, Org. Lett., 2008, 10, 3841 CrossRef CAS PubMed; (c) M. Toumi, F. Couty, J. Marrot and G. Evano, Org. Lett., 2008, 10, 5027 CrossRef CAS PubMed; (d) A. Coste, G. Karthikeyan, F. Couty and G. Evano, Angew. Chem., Int. Ed., 2009, 48, 4381 CrossRef CAS PubMed; (e) A. Coste, F. Couty and G. Evano, Org. Lett., 2009, 11, 4454 CrossRef CAS PubMed; (f) K. Jouvin, F. Couty and G. Evano, Org. Lett., 2010, 12, 3272 CrossRef CAS PubMed; (g) K. Jouvin, A. Bayle, F. Legrand and G. Evano, Org. Lett., 2012, 14, 1652 CrossRef CAS PubMed; (h) A. Pradal and G. Evano, Chem. Commun., 2014, 50, 11907 RSC; (i) A. Nitelet and G. Evano, Org. Lett., 2016, 18, 1904 CrossRef CAS PubMed; (j) C. S. Demmer, E. Benoit and G. Evano, Org. Lett., 2016, 18, 1438 CrossRef CAS PubMed; (k) C. Deldaele and G. Evano, ChemCatChem, 2016, 8, 1319 CrossRef CAS.
- (a) T. Nishikata, Y. Noda, R. Fujimoto and T. Sakashita, J. Am. Chem. Soc., 2013, 135, 16372 CrossRef CAS PubMed. Also see, for a related approach using nickel catalysis: (b) C. Liu, S. Tang, D. Liu, J. Yuan, L. Zheng, L. Meng and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 3638 CrossRef CAS PubMed.
- A. J. Clark, Eur. J. Org. Chem., 2016, 2231 CrossRef CAS.
- K. Matyjaszweski, Macromolecules, 2012, 45, 4015 CrossRef.
- For an example of addition of free alkyl radicals to pyrroles and indoles, see: (a) M. A. Guerro and L. D. Miranda, Tetrahedron Lett., 2006, 47, 2517 CrossRef; (b) Y. M. Osornio, R. Cruz-Almanza, V. Jiménez-Montaño and L. D. Miranda, Chem. Commun., 2003, 2316 RSC . For a photocatalytic radical alkylation of eletron-rich heteroarenes with brominated malonates, see ref. 9a and f.
- For an isolated example of gold-catalyzed direct alkylation of 3-methylbenzofuran with phenyldiazoacetate, see: Y. Xi, Y. Su, Z. Yu, B. Dong, E. J. McClain, Y. Lan and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 9817 CrossRef CAS PubMed.
- N. Ortega, S. Urban, B. Beiring and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 1710 CrossRef CAS PubMed.
- N-Methyl-pyrrole and N-methyl-indole failed to undergo the alkylation, even when forcing the reaction conditions, while N-methyl-2-phenyl-pyrrole gave the corresponding C5-alkylated product in 25% yield after 48 h in dioxane at 150 °C.
- C. Liu, D. Liu, W. Zhang, L. Zhou and A. Lei, Org. Lett., 2013, 15, 6166 CrossRef CAS PubMed.
- J.-Q. Chen, Y.-L. Wei, G.-Q. Xu, Y.-M. Liang and P.-F. Xu, Chem. Commun., 2016, 52, 645 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization and copies of 1H and 13C NMR spectra for all new compounds. See DOI: 10.1039/c6sc05622a |
| This journal is © The Royal Society of Chemistry 2017 |