
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox non-innocence permits catalytic nitrene carbonylation by (dadi)Ti![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) NAd (Ad = adamantyl)†
NAd (Ad = adamantyl)†
Spencer P.
Heins
a,
Peter T.
Wolczanski
 *a,
Thomas R.
Cundari
*b and
Samantha N.
MacMillan
a
*a,
Thomas R.
Cundari
*b and
Samantha N.
MacMillan
a
aCornell University, Dept. Chemistry & Chemical Biology, Baker Laboratory, Ithaca, NY 14853, USA. E-mail: ptw2@cornell.edu
bUniversity of North Texas, Dept. of Chemistry, CASCaM, Denton, TX 76201, USA. E-mail: t@unt.edu
First published on 6th March 2017
Abstract
Application of the diamide, diimine {–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(1,2-C6H4)N(2,6-iPr2-C6H3)}2m ((dadi)m) ligand to titanium provided adducts (dadi)TiLx (1-Lx; Lx = THF, PMe2Ph, (CNMe)2), which possess the redox formulation [(dadi)4−]Ti(IV)Lx, and 22 πe− (4n + 2). Related complexes containing titanium-ligand multiple bonds, (dadi)TiX (2X; X = O, NAd), exhibit a different dadi redox state, [(dadi)2−]Ti(IV)X, consistent with 20 πe− (4n). The Redox Non-Innocence (RNI) displayed by dadim impedes binding by CO, and permits catalytic conversion of AdN3 + CO to AdNCO + N2. Kinetics measurements support carbonylation of 2NAd as the rate determining step. Structural and computational evidence for the observed RNI is provided.
N(1,2-C6H4)N(2,6-iPr2-C6H3)}2m ((dadi)m) ligand to titanium provided adducts (dadi)TiLx (1-Lx; Lx = THF, PMe2Ph, (CNMe)2), which possess the redox formulation [(dadi)4−]Ti(IV)Lx, and 22 πe− (4n + 2). Related complexes containing titanium-ligand multiple bonds, (dadi)TiX (2X; X = O, NAd), exhibit a different dadi redox state, [(dadi)2−]Ti(IV)X, consistent with 20 πe− (4n). The Redox Non-Innocence (RNI) displayed by dadim impedes binding by CO, and permits catalytic conversion of AdN3 + CO to AdNCO + N2. Kinetics measurements support carbonylation of 2NAd as the rate determining step. Structural and computational evidence for the observed RNI is provided.
Introduction
The diamide, diimine {–CHN(1,2-C6H4)N(2,6-iPr2-C6H3)}2m (m = 0 to −4), i.e. (dadi)m, was previously introduced as a tetradentate chelate ligand capable of at least 5 redox states.1 In the initial study, (dadi)M (M = Cr(THF), Fe) was shown to transfer azide-generated nitrenes into carbon–carbon bonds via aziridination. In the process the dadi2− ligand (20 πe−, 4n) was derivatized to a 22 πe− (4n + 2) ligand (i.e., [RN{CHN(1,2-C6H4)N(2,6-iPr2-C6H3)}2]2−MII (R = 2,6-iPr2C6H3, adamantyl (Ad))) of considerable stability.
Redox non-innocent (RNI) ligands1–20 like dadim can support catalytic activity11–17 by expanding the redox capability of a system, essentially electronically buffering the transition metal center. As Fig. 1 illustrates, a ligand capable of RNI can keep the metal in a stable configuration (Mm+2) by being in a reduced state (Lp → Lp−2), and releasing the electrons to the metal upon oxidative addition of XY (Lp−2Mm+2 → LpMm → LpMm+2XY). Transfer of XY to a substrate, perhaps via a binding step, causes reduction of the metal, but its formal oxidation state is maintained (Mm → Mm+2) by the RNI of the ligand.
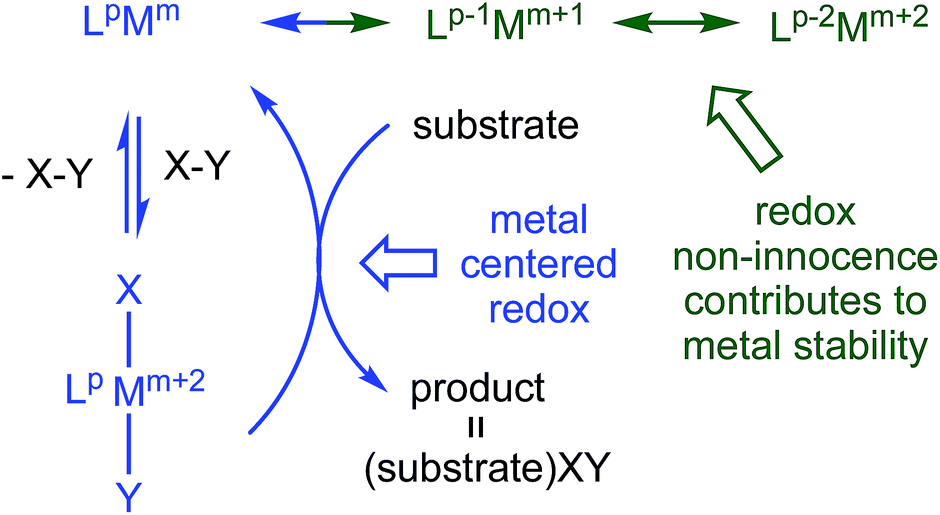 | ||
| Fig. 1 Conventional oxidative addition (blue), and corresponding 1- and 2-e− redox non-innocent (RNI) configurations (green) that can electronically buffer the metal center. The ligand, Lp, π-system can be (4n + 2) → 4n or 4n → (4n − 2) depending on its compatibility with Mm. | ||
In its dianionic (n = −2) state, (dadi)m possessed a 4n π-system that is intrinsically susceptible to redox events,8,9 and redox non-innocence that can potentially support unusual reactivity. Chelation of titanium(II) by (dadi)2− was a target, with the expectation that RNI leading to (dadi)4−Ti(IV) could potentially stabilize the system, as the tetraanion has a 4n + 2 π-system. This report describes (dadi)Ti(X/L) species that manifest RNI, including a rare instance in which carbon monoxide does not affect redox activity at the metal, and does not bind, thus permitting its use as a substrate in catalysis.
Results
Synthesis of (dadi)TiLn (n = 1, L = THF, PMe2Ph; n = 2, L = CNMe)
N(1,2-C6H4)NH(2,6-iPr2-C6H3)}2, i.e., (dadi)H2,1 with 2 equiv. NaN(TMS)2 afforded the disodium salt of (dadi)2− in 94% yield according to Scheme 1. The dianion is bright blue in THF solution, and exhibits a dominant absorption band at 770 nm (ε = 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) that features a ∼1500 cm−1 progression plausible for a diimine.
000 M−1 cm−1) that features a ∼1500 cm−1 progression plausible for a diimine.
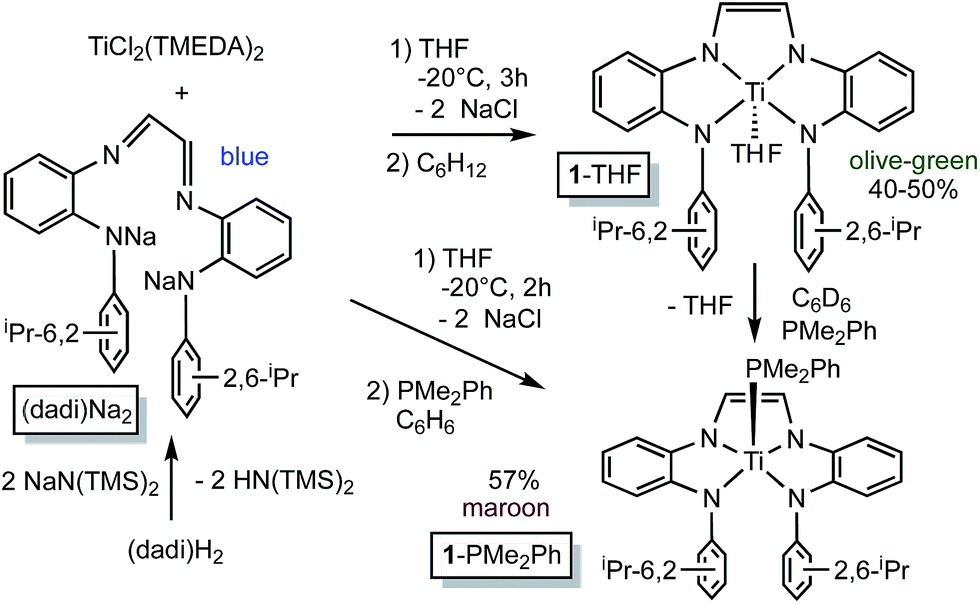 | ||
| Scheme 1 Synthesis of (dadi)Na2 and (dadi)TiL (L = THF, 1-THF; PMe2Ph, 1-PMe2Ph). | ||
Treatment of (TMEDA)2TiCl2 (ref. 21) with (dadi)Na2 in THF at −20 °C for 3 h yielded a green solution. After filtration in cyclohexane, (dadi)Ti(THF) (1-THF) was isolated in ∼40–50% yields as an olive-green powder that contained some residual C6H12. The five-coordinate environment of diamagnetic 1-THF renders the isopropyl groups inequivalent in its 1H NMR spectrum due to hindered rotation of the aryl, and they are observed as doublets in a ∼12:6:6 ratio along with THF resonances.
For related complexes (dadi)M (M = Fe, Mn),1 the aforementioned, attenuated intraligand (IL) band was split into components at ∼720 nm and ∼940 nm (ε ∼ 5–12000 M−1 cm−1) in the UV-vis spectrum. Blue shifted, low intensity features at 495 (ε ∼ 900 M−1 cm−1) and 590 nm (ε ∼ 700 M−1 cm−1) in 1-THF are remnants of these IL bands according to TDDFT calculations, and are accompanied by a major absorption at 345 nm (ε = 32000 M−1 cm−1). The spectrum suggested a more covalent electronic environment than in the high spin Fe and Mn species, and calculations support a [(dadi)4−](THF)Ti(IV) configuration in which the tetraanionic, tetradentate chelate possesses a 22e− (4n + 2) π-system.
Unlike previous systems,10 the 1H NMR spectroscopic chemical shift of the diimine hydrogens, δ 6.64 in (dadi)Ti(THF) (1-THF), does not distinguish between redox states of (dadi)n. Calculations of pseudo-square planar (dadi)Ti (1) suggest that it would be best considered [(dadi)3−↓]Ti(III)↑, a d1 titanium anti-ferromagnetically coupled to (dadi)3−, but adduct formation causes an electronic reorganization to the [(dadi)4−](L)Ti(IV) configuration.
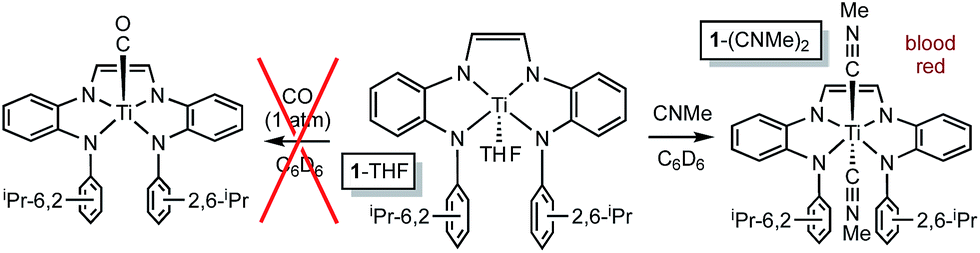 | ||
| Scheme 2 Methylisocyanide binds, but CO does not. | ||
Structural studies of (dadi)TiLn (L = PMe2Ph, L2 = (CNMe)2)
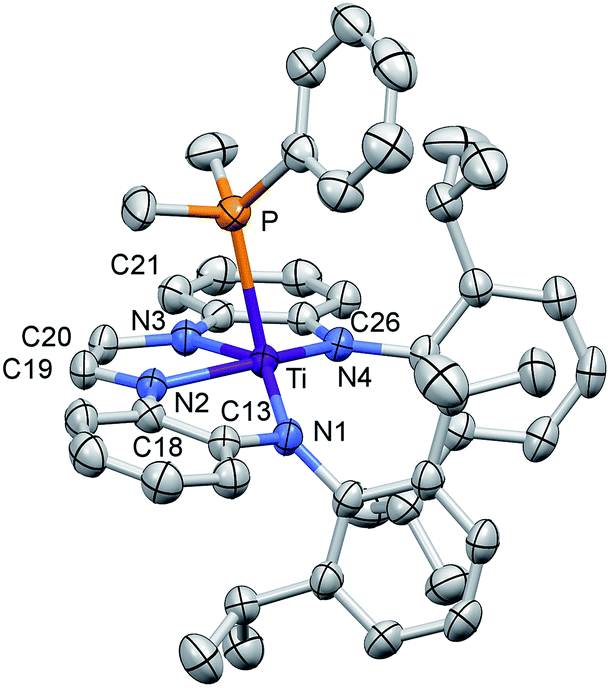 | ||
| Fig. 2 Molecular view of (dadi)TiPMe2Ph (1-PMe2Ph). Interatomic distances (Å) and angles (°): Ti–P, 2.6049(7); Ti–N1, 2.0029(18); Ti–N2, 2.0194(18); Ti–N3, 2.0332(18); Ti–N4, 2.0088(18); N1–C13, 1.411(3); C13–C18, 1.413(3); N2–C18, 1.396(3); N2–C19, 1.379(3); C19–C20, 1.338(3); N3–C20, 1.381(3); N3–C21, 1.396(3); C21–C26, 1.408(3); N4–C26, 1.406(3); P–Ti–N1, 114.64(6); P–Ti–N2, 85.21(5); P–Ti–N3, 85.23(5); P–Ti–N4, 102.35(5); N1–Ti–N2, 78.10(7); N1–Ti–N3, 143.60(7); N1–Ti–N4, 123.37(7); N2–Ti–N3, 73.38(7); N2–Ti–N4, 148.83(7); N3–Ti–N4, 77.14(7). | ||
The crucial d(CNim) of 1.379(3) and 1.381(3) Å are significantly longer than the expected 1.30 Å for a neutral imine,18,19,22 and the d(CimCim) is also anomalous at 1.338(3) Å, a value substantially shorter than a C(sp2)–C(sp2) distance of 1.43 Å.22 Titanium–nitrogen distances to the “imines” are also short, at 2.0194(18) and 2.0332(18) Å, values that reflect greater covalency in the interaction of the tetraamide chelate to the Ti(IV) center. For comparison, the “diimine” Ti–N distances are nearly the same as the titanium arylamide–nitrogen bond lengths of 2.0029(18) and 2.0088(18) Å.
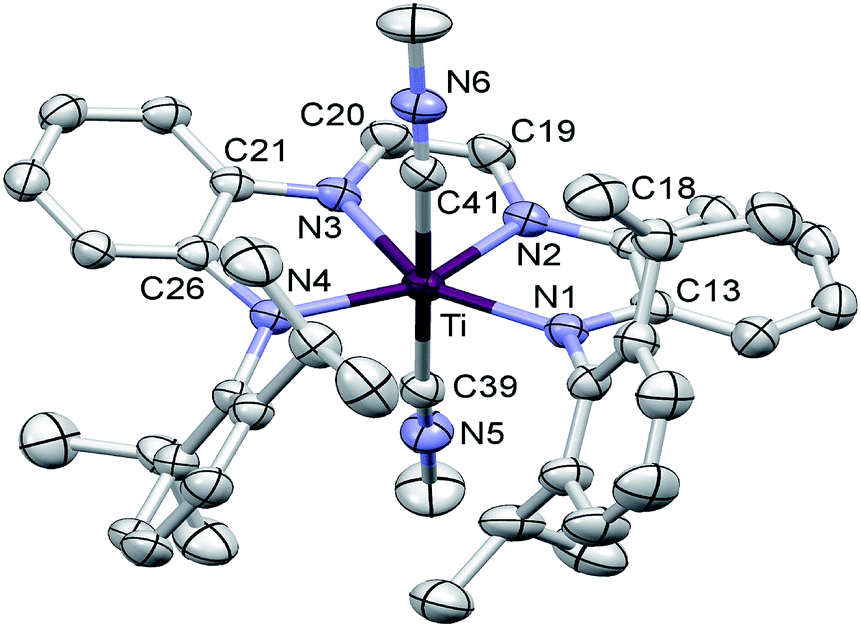 | ||
| Fig. 3 Molecular view of (dadi)Ti(CNMe)2 (1-(CNMe)2). Interatomic distances (Å) and angles (°): Ti–N1, 2.0677(16); Ti–N2, 2.0331(15); Ti–N3, 2.0353(16); Ti–N4, 2.0636(15); Ti–C39, 2.2484(18); Ti–C41, 2.2364(18); N1–C13, 1.396(2); C13–C18, 1.418(3); N2–C18, 1.403(2); N2–C19, 1.373(2); C19–C20, 1.350(3); N3–C20, 1.371(2); N3–C21, 1.401(2); C21–C26, 1.423(2); N4–C26, 1.395(2); C39–N5, 1.141(2); C41–N6, 1.145(2); N1–Ti–N2, 77.04(6); N1–Ti–N3, 149.95(6); N1–Ti–N4, 132.75(6); N2–Ti–N3, 73.96(6); N2–Ti–N4, 150.05(6); N3–Ti–N4, 76.83(6); N1–Ti–C39, 91.41(6); N2–Ti–C39, 84.33(6); N3–Ti–C39, 93.42(6); N4–Ti–C39, 90.98(6); N1–Ti–C41, 91.37(6); N2–Ti–C41, 92.30(6); N3–Ti–C41, 82.13(6); N4–Ti–C41, 90.14(6); C39–Ti–C41, 175.04(7). | ||
Electronic structure of (dadi)Ti(PMe2Ph)
The metric parameters found in X-ray crystal structure of (dadi)TiPMe2Ph (1-PMe2Ph) prompted an electronic structure investigation to assess the proposed RNI. Fig. 4 illustrates a truncated molecular orbital diagram featuring three filled orbitals, with a diimine CC–π-bonding orbital as the HOMO. The unfilled orbitals, predominantly 3d in character, are tightly packed, and well separated from the others, partly as a consequence of DFT calculations.23 Orbitals dxz and dyz possess modest π-character, but are empty, as expected for the (dadi)4− formulation. Overall, the “3d” orbitals are essentially non-bonding, due to the multicomponent mixing intrinsic to low symmetry systems. All filled orbitals shown possess some d-character, so even though dadi is expressed as tetranionic, the metal/dadi admixture reflects the covalence of the system.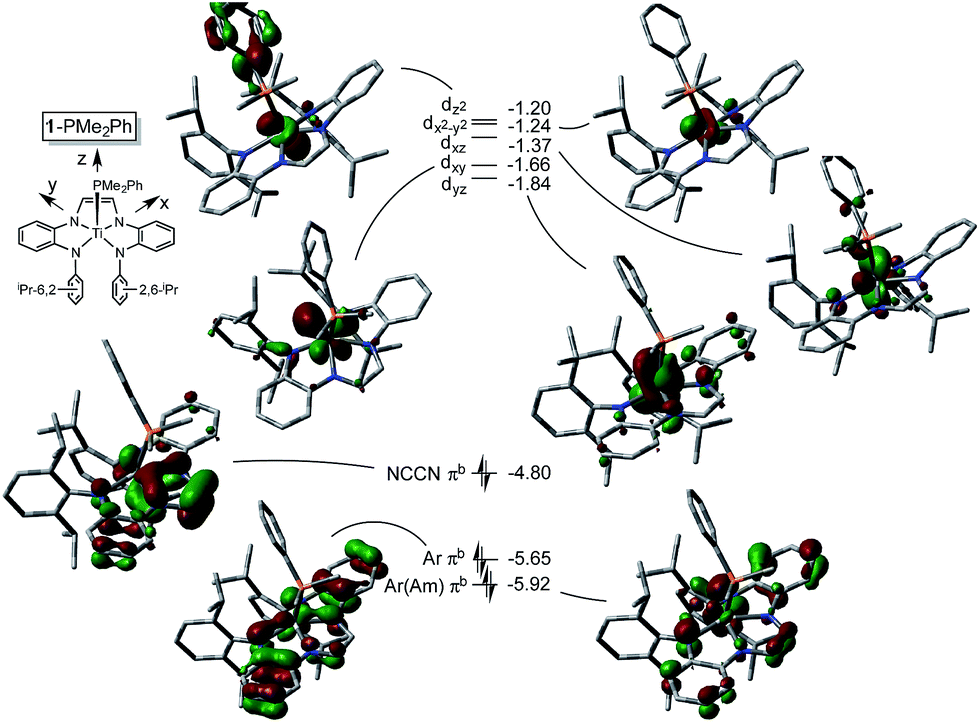 | ||
| Fig. 4 Truncated MO diagram of (dadi)TiPMe2Ph (1-PMe2Ph), showing the unfilled orbitals of the d0 species, and three filled dadi-based orbitals (energies in eV). Assignments are tentative due to the low symmetry: M06/6-311+G(d) single point calculations for orbitals at the ONIOM(M06/6-311+G(d)) optimized minimum derived from the X-ray structure as an initial guess. | ||
Synthesis of (dadi)Ti![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) X (X = NAd, O)
X (X = NAd, O)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) NAd.
Organoazides were explored as a means to prepare imide derivatives,23–25 with mixed results.26–28 Use of Me3SiN3 led to a mixture of products, although NMR spectroscopic signals consistent with (dadi)TiNSiMe3 were seen, and 2,6-iPr2C6H3N3 afforded a complex product devoid of symmetry. Treatment of (dadi)Ti(THF) with adamantyl-azide in benzene effervesced immediately, and maroon-red (dadi)TiNAd (2NAd) was isolated in 74% yield, as indicated in Scheme 3. 1H NMR spectra revealed an integrated 6:12:6 ratio of CH(CH3)2 hydrogens, suggestive of a five-coordinate imide complex.
NAd.
Organoazides were explored as a means to prepare imide derivatives,23–25 with mixed results.26–28 Use of Me3SiN3 led to a mixture of products, although NMR spectroscopic signals consistent with (dadi)TiNSiMe3 were seen, and 2,6-iPr2C6H3N3 afforded a complex product devoid of symmetry. Treatment of (dadi)Ti(THF) with adamantyl-azide in benzene effervesced immediately, and maroon-red (dadi)TiNAd (2NAd) was isolated in 74% yield, as indicated in Scheme 3. 1H NMR spectra revealed an integrated 6:12:6 ratio of CH(CH3)2 hydrogens, suggestive of a five-coordinate imide complex.
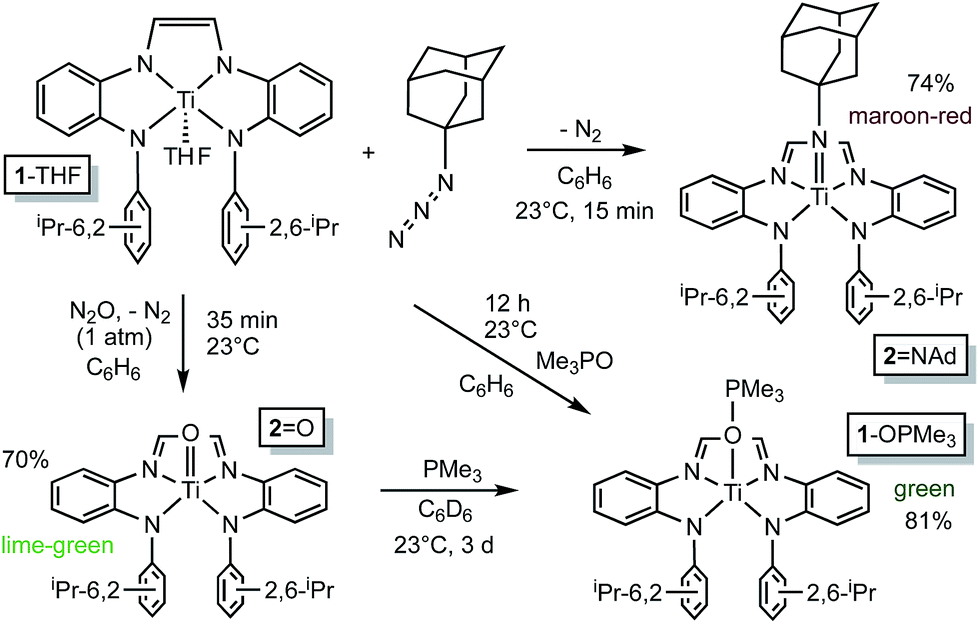 | ||
| Scheme 3 Imide, (dadi)TiNAd (2NAd), and oxo, (dadi)TiO (2O), formation from (dadi)Ti(THF) (1-THF), AdN3 and N2O, respectively. | ||
O.
Generation of the corresponding oxo complex proved to be equally interesting. Initially, common oxygen atom transfer (OAT) agents, such as PhIO, Me3NO, and PyN-O failed, but unexpectedly, nitrous oxide29–34 proved fruitful. Exposure of (dadi)Ti(THF) (1-THF) to N2O (1 atm) in benzene for ∼30 min afforded a lightening of the green solution, and lime green (dadi)TiO (2-O) was eventually precipitated from benzene in 70% yield, as Scheme 3 illustrates. Four sets of isopropyl-methyl doublets were observed in the 1H NMR spectrum in a 6:6:6:6 ratio, and a resonance Raman (λ = 475 nm) spectrum showed an absorption at 1015.4 cm−1 tentatively assigned to the N(TiO).29 OAT from (dadi)TiO (2O) was tested with a variety of substrates (olefins, CO, etc.), but only transfer to PMe3, resulting in green (dadi)Ti–OPMe3 (1-OPMe3), was observed. The latter compound was independently prepared from (dadi)Ti(THF) (1-THF) and 1 equiv. of Me3PO.
Structural study of (dadi)TiO
A molecular view of (dadi)TiO (2O) and accompanying metric parameters are given in Fig. 5. The core O–Ti–N angles average 105.2(4)°, except for ∠O–Ti–N3 = 94.71(5)°, as the oxo tilts slightly toward N3. The diimine bite angle of 71.44(5)°, imine-amide bite angles averaging 75.7(4)°, and the opening N1–Ti–N4 angle of 124.02(5)° describe a slightly distorted square pyramid with basal nitrogens and the apical oxo. The dadi backbone contains d(CN) = 1.299(3) (ave) Å and d(CC) = 1.433(3) Å, consistent with a neutral diimine fragment of a (dadi)2− ligand.18,19,22 The titanium–nitrogen bond lengths to the diimine are 2.1603(13) and 2.1938(12) Å, substantially longer than the electrostatically contracted d(TiN) of the (dadi)4− ligand of (dadi)TiPMe2Ph (1-PMe2Ph) and (dadi)Ti(CNMe)2 (1-(CNMe)2). The d(TiO) of 1.6361(11) Å is typical for the numerous Ti(IV) oxo species that have been characterized (1.63(1) Å ave for 15 5-coord examples), and the remaining amide distances are typical (2.059(7) (ave) Å).
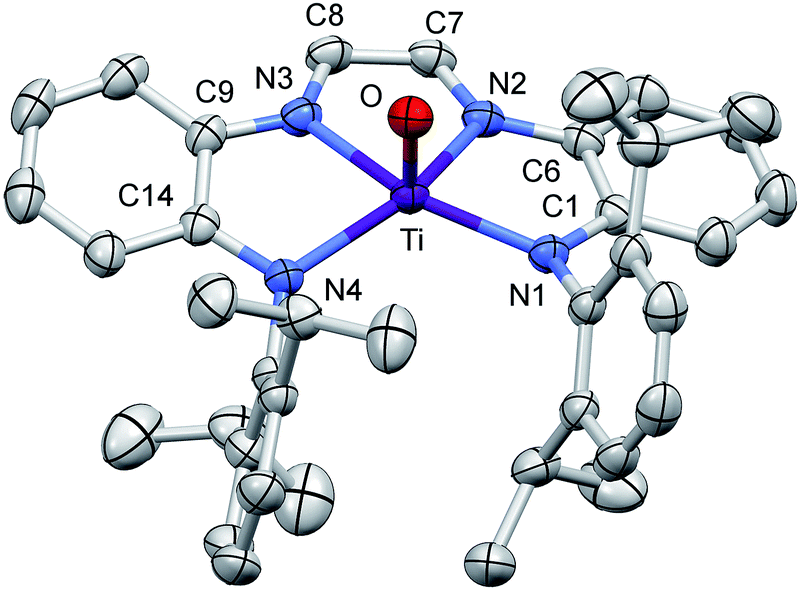 | ||
| Fig. 5 Molecular view of (dadi)TiO (2O). Interatomic distances (Å) and angles (°): Ti–O, 1.6361(11); Ti–N1, 2.0538(13); Ti–N2, 2.1938(12); Ti–N3, 2.1603(13); Ti–N4, 2.0640(12); N1–C1, 1.3770(19); C1–C6, 1.419(2); N2–C6, 1.395(2); N2–C7, 1.301(2); C7–C8, 1.433(2); N3–C8, 1.297(2); N3–C9, 1.388(2); C9–C14, 1.418(2); N4–C14, 1.3750(19); O–Ti–N1, 105.18(5); O–Ti–N2, 104.88(5); O–Ti–N3, 94.71(5); O–Ti–N4, 105.66(5); N1–Ti–N2, 75.40(5); N1–Ti–N3, 144.73(5); N1–Ti–N4, 124.02(5); N2–Ti–N3, 71.44(5); N2–Ti–N4, 136.56(5); N3–Ti–N4, 75.94(5). | ||
Electronic structure of (dadi)TiO
The electronic structures of (dadi)TiO (2O) and (dadi)TiNAd (2NAd) were investigated as examples of species in which the chelate was dianionic. The calculation of 2O is featured herein, since the experimental structure is available for comparison. As Fig. 6 illustrates, the TiO π-bonding orbitals are buried below a group of occupied dadi-π-orbitals, including the HOMO, which has significant Npπ-character, but no metal constituent. These contain components of NCCN bonding, but there are no discrete “diimide” orbitals, although a pair of unoccupied orbitals, including the LUMO, are clearly NCCN π* in character. The diagram is consistent with the claim of a highly delocalized (dadi)2− ligand attached to a titanyl fragment, whose π-bonds are clearly rendered at −8.23 and −8.59 eV.
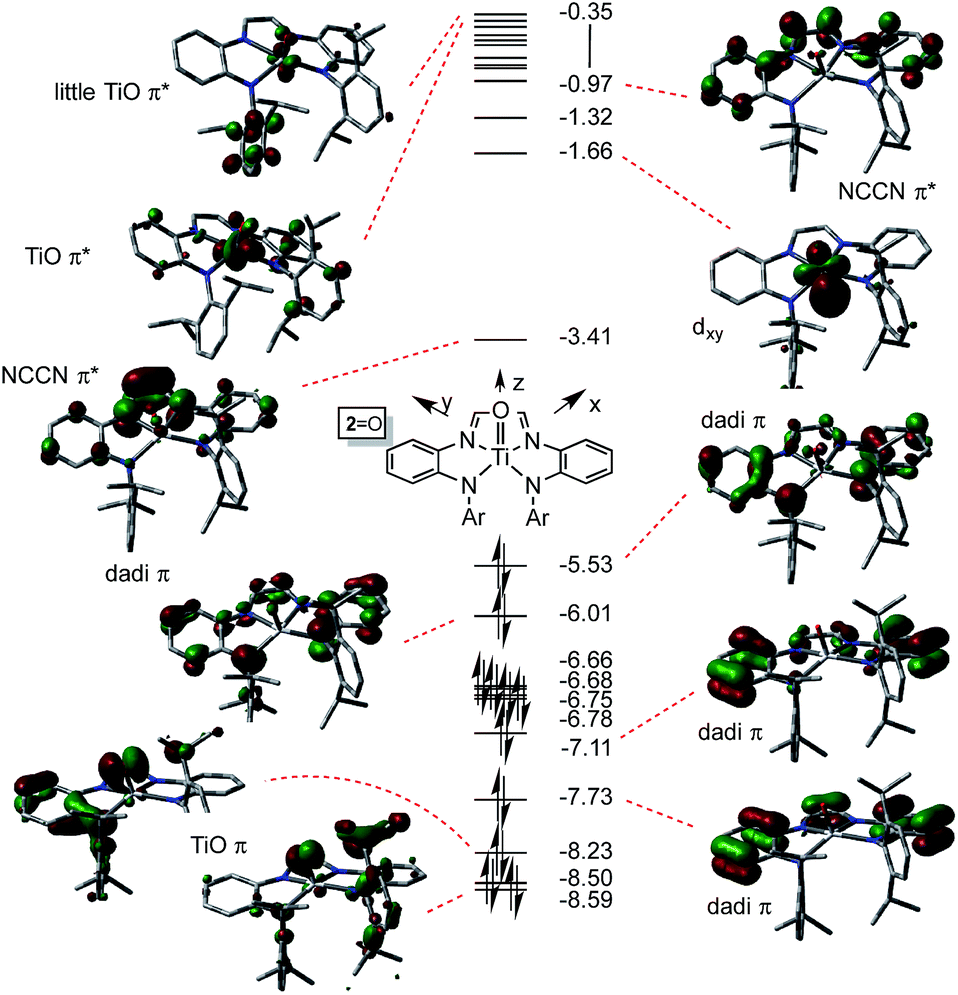 | ||
| Fig. 6 Truncated molecular orbital diagram of (dadi)TiO (2O); orbitals not plotted are either diffuse virtual orbitals or pertain to 2,6-iPr2C6H3. Energies are in eV. | ||
The distortion in (dadi)TiO (2O), reproduced by calculation, appears to be steric in origin, as removal of the 2,6-iPr2-C6H3 groups and re-optimization of the geometry leads to a more symmetric species. Note the similarities in the diagrams of (dadi)TiPMe2Ph (1-PMe2Ph) and 2O, which both show significant gaps between dadi π-system orbitals, an NCCN π*-orbital, and ligand antibonding and metal orbitals. While both diagrams depict Ti(IV), the middle NCCN π*-orbital is empty for 2O, i.e., (dadi)2−, while it is filled for 1-PMe2Ph, i.e., (dadi)4−, thereby revealing disparate redox states for each.
Catalytic carbonylation of adamantyl azide
The RNI of (dadi)n is implicit in the failure of CO to compete with THF in binding to titanium,11–13 rendering it capable of catalyzing carbonylation reactions. Scheme 4 illustrates a plausible mechanism for the carbonylation of the nitrene derived from adamantyl azide (>20 turnovers) as catalyzed by (dadi)TiNAd (2NAd). Insertion of the carbonyl into the titanium–imide bond, perhaps via an initial cis-bonding event, affects formation of an isocyanate adduct, and subsequent substitution by AdN3, dissociative, associative, or interchange, generates an azide adduct that releases N2 to restart the cycle.
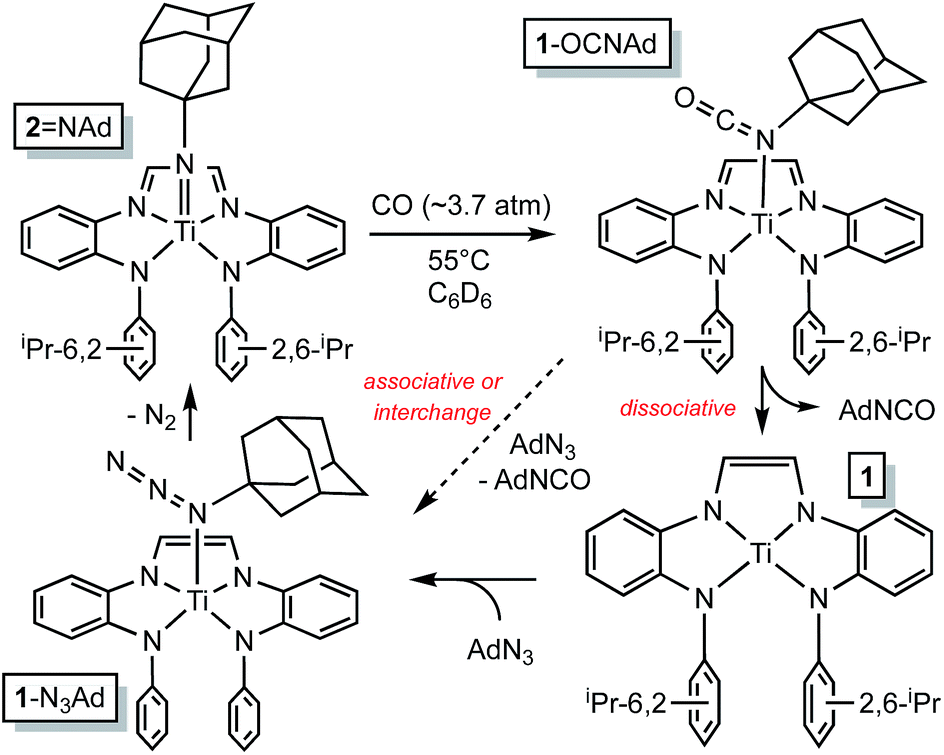 | ||
| Scheme 4 Plausible mechanisms for the catalyzed conversion of AdN3 + CO → AdNCO + N2 by the imide, (dadi)TiNAd (2NAd). | ||
Imide (dadi)TiNAd (2NAd) is the only metal-containing species observed when the catalysis is monitored by 1H NMR spectroscopy, and its concentration diminishes during the course of catalysis. The catalysis was modeled by a two-step process in which the imide is clipped from 2NAd by CO, and renewed by the reaction of (dadi)Ti (1) with AdN3, as shown in Scheme 5. A degradation path was added to account for loss of catalyst over time. Initial kinetics experiments confirmed an order in [CO], and while the concentration of CO in solution was only ∼3.5 times that of 2NAd, the reaction was slow enough such that CO (∼60 fold excess) was replenished from the gas phase. As a consequence CO was a pseudo first-order reagent, and the phenomenological rate (−d[AdN3]/dt = kobs; kobs = kCO[2NAd][CO]) was zero-order since the catalyst concentration also does not change, aside from the degradation. Factoring out [CO] and [2NAd] afforded a second-order rate constant for carbonylation as 3.13(24) × 10−3 M−1 s−1 (ΔG‡ (55 °C) = 23.0(1) kcal mol−1), as listed in Table 1.
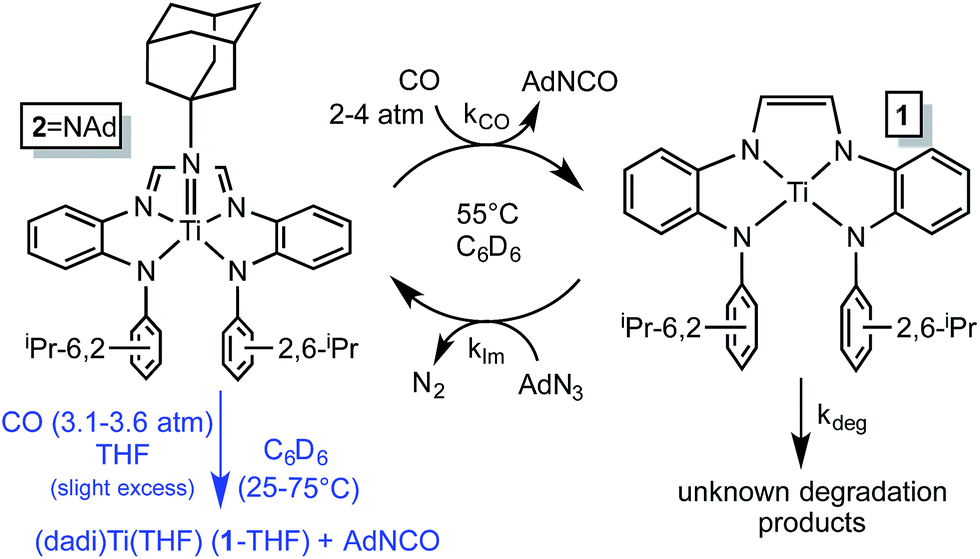 | ||
| Scheme 5 Kinetic model for AdN3 carbonylation catalysis by 2NAd (black) and stoichiometric study of imide carbonylation (blue). | ||
NAd carbonylation, stoichiometric and catalytic, in C6D6
|
2NAd (M) |
COa (M) | T (°C) | k CO × 103 (M−1 s−1) | ΔG‡ (kcal mol−1) |
|---|---|---|---|---|
|
a Calculated from Henry's law and corrected for temperature dependence.
b Rate constants obtained from an average of three trials; from a weighted Eyring plot: ΔH‡ = 9.6(9) kcal mol−1, ΔS‡ = −39.9(28) eu, ΔG‡ (55 °C) = 22.6(9) kcal mol−1. An unweighted Eyring plot gives: ΔH‡ = 9.3(4) kcal mol−1, ΔS‡ = −40.7(13) eu, ΔG‡ (55 °C) = 22.6(6) kcal mol−1.
c To determine order in CO; kobs = 4.85(55) × 10−5 s−1 at [CO] = 0.0105 M; kobs = 9.67(95) × 10−5 s−1 at [CO] = 0.023 M; kobs = 1.52(8) × 10−4 s−1 at [CO] = 0.028 M.
d Catalytic rate obtained from fitting disappearance of [AdN3] and [2NAd] according to Scheme 5 (3 trials): p(CO) = 2.58 atm, total mol CO = 1.82 × 10−4, [AdN3] = 0.123 M, kdeg = 3.2(3) × 10−5 Ms−1. See ESI for details.
|
||||
| 0.047b | 0.023 | 25.0(5) | 1.25(6) | 21.4(1) |
| 0.048b | 0.025 | 35.0(5) | 1.83(8) | 21.9(1) |
| 0.047b | 0.026 | 45.0(5) | 3.58(15) | 22.2(1) |
| 0.047b,c | 0.028 | 55.0(5) | 5.44(42) | 22.7(1) |
| 0.047b | 0.030 | 65.0(5) | 7.95(86) | 23.1(1) |
| 0.047b | 0.033 | 75.0(5) | 14.0(11) | 23.4(1) |
| 0.047c | 0.0105 | 55.0(5) | 4.62(69) | 22.8(1) |
| 0.046c | 0.023 | 55.0(5) | 4.20(45) | 22.8(1) |
| 0.0063d | 0.022 | 55.0(5) | 3.13(24) | 23.0(1) |
A stoichiometric study supported carbonylation as the rate-determining elementary step in the catalysis. Treatment of 2NAd with an excess of CO (pseudo first-order, 3.1–3.6 atm) in C6D6 (with THF present) at 25–75 °C afforded AdNCO, and (dadi)Ti(THF) (1-THF), presumably resulting from trapping of (dadi)Ti (1). A first-order dependence on [CO] was found (−d[2NAd]/dt = kCO[2NAd][CO]), and an Eyring analysis of the second order rate constants listed in Table 1 yielded a ΔH‡ of 9.6(9) kcal mol−1, and a ΔS‡ of −39.9(28) eu. The calculated ΔG‡ at 55 °C is 22.6(9) kcal mol−1, a value consistent with that obtained during catalysis, supporting the claim of carbonylation as the rate-determining step. The modest enthalpy of activation thus reflects the balance between NC bond-formation and TiN bond-breaking in the transition state, while the large negative entropy of activation value is consistent with a 2nd-order process.
Calculated free energy profile of AdN3 carbonylation
Fig. 7 illustrates the calculated free energy profile for the catalytic carbonylation of AdN3 to AdNCO + N2, a reaction that is exergonic by −72.8 kcal mol−1. Formation of the CN bond in AdNCO is −72.3 kcal mol−1 enthalpically more favorable than the corresponding azide NN bond in terms of free energy, and constitutes the driving force for catalysis.
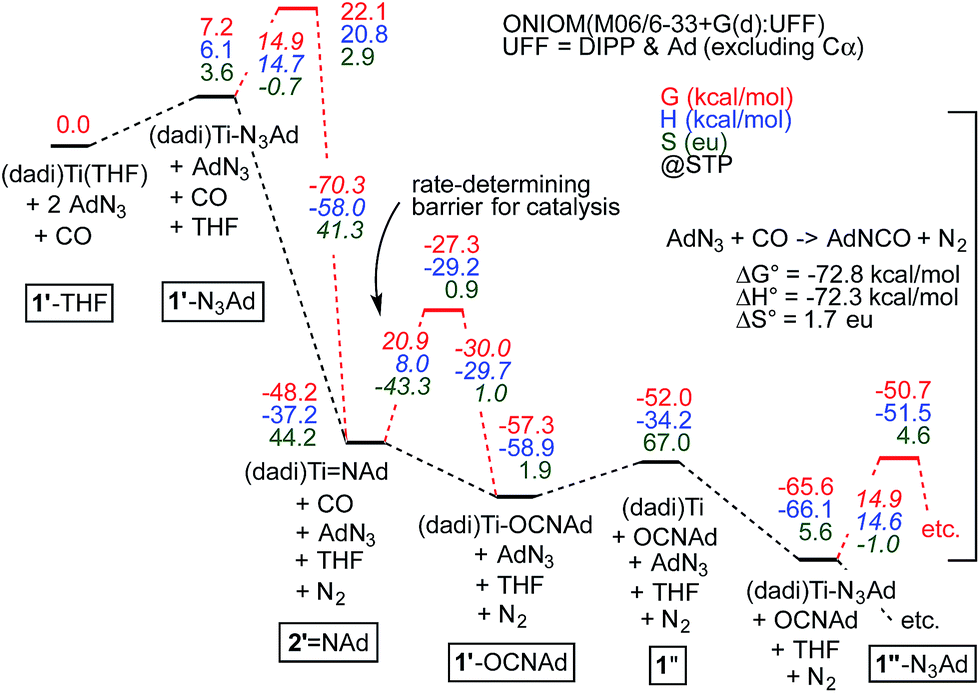 | ||
| Fig. 7 Calculations of Scheme 4, with transition states (primes indicate calculated states; activation energies italicized) pertaining to N2 loss from bound azide, (dadi)Ti(N3Ad) (1-N3Ad) and carbonylation of (dadi)TiNAd (2NAd). Association/dissociation transition states are not included. Small inconsistencies are due to round-off errors. Calculated 1′-L and 1′′-L compounds have (dadi)4− configurations, 2′NAd has a (dadi)2− structure, and 1′′ is computed as [(dadi)3−↓]Ti(III)↑. | ||
The calculated ΔG‡ (298.15 K) for carbonylation of the imide, (dadi)TiNAd (2NAd) is 20.9 kcal mol−1, in decent agreement with experiment (21.5 kcal mol−1 at 25 °C). The slight discrepancy is spread between ΔH‡ (calc'd 8.0 kcal mol−1) and ΔS‡ (calc'd −43.3 eu). The latter is similar to the calculated ΔS‡ = −42.3 eu for the microscopic reverse of N2 extrusion from (dadi)Ti(N3Ad) (1-N3Ad). Formation of (dadi)Ti (1) + AdNCO from 2NAd and CO is only favorable by ΔG° = −3.8 kcal mol−1, and enthalpically unfavorable by ΔH° = 3.0 kcal mol−1, hence carbonylation has modest thermodynamic impetus. With the Bond Dissociation Enthalpy (BDE(calc)) of the AdNCO bond at 81 kcal mol−1, the titanium–imide bond BDE(calc) is ∼84 kcal mol−1.35 Both adducts, 1-N3Ad and 1-OCNAd, are calculated to be most stable as K1-NAd-bound species.
Dinitrogen loss from 1-N3Ad possesses the next highest transition state, at ΔG‡ = 14.9 kcal mol−1. The binding of AdN3 to (dadi)Ti is favorable by only −13.6 kcal mol−1, but the subsequent loss of N2 to form 2NAd is substantial, at −55.4 kcal mol−1, hence there is substantial thermodynamic influence on the dinitrogen loss step. Associative or interchange transition states for AdN3 displacement of isocyanate from 1-OCNAd could not be found. Since dissociation of product OCNAd from 1-OCNAd is calculated to be only ΔG° = 5.3 kcal mol−1, it is unlikely that steps subsequent to carbonylation, and preceding loss of N2 from 1-N3Ad are particularly consequential.
Discussion
Transfers of X (NAd, O) from (dadi)TiX
In the successful transfer of the imido functionality of (dadi)TiNAd (2-Ad) to CO, affording AdNCO, the redox non-innocence of the dadi ligand was critical. Stabilization of presumed transient (dadi)Ti (1) as [(dadi)3−↓]Ti(III)↑, (dadi)TiL (1-L) via (dadi)4−, and 2-Ad through (dadi)2− permits the titanium to exist in favorable, higher oxidation states while catalyzing the carbonylation, providing a textbook example of the RNI in Fig. 1.
The dadi ligand is more oxidizing to the titanium center than one or two bound CO ligands, possessing the 22 πe− (4n + 2) configuration in (dadi)TiL (1-L), but less oxidizing than a nitrene (i.e., NAd), where it resides in its 20 πe− (4n) form. The MO diagrams for (dadi)TiPMe2Ph (1-PMe2Ph) and (dadi)TiO (2O), if taken as analogues for a potential carbonyl species, and (dadi)TiNAd (2-Ad), respectively, support this key electronic rationale. Concerning Fig. 4 (1-PMe2Ph), any backbonding interaction with one of the dπ-orbitals would have to lower it below −4.80 eV in order to depopulate the NCCN π-orbital and render the dadi dianionic. Apparently CO, a potent π-acid toward titanium, is incapable of inducing the necessary change. Note that in Fig. 6 (2O), the related NCCN π-orbital at −3.41 eV is the LUMO in the system, having been depopulated in favor of the oxo π-bonding orbitals of low energy.
The catalytic carbonylation in Scheme 4 produces AdNCO, but fortunately OAT to afford (dadi)TiO (2O) and CNAd is calculated to be only slightly favorable (ΔG° = −1.1 kcal mol−1).36,37 The released CNAd is also favored to scavenge any (dadi)Ti (1) and provide (dadi)Ti(CNAd) with 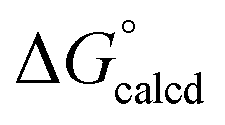 = −22.1 kcal mol−1. It is likely that the barrier for OAT from AdNCO is sluggish compared to dinitrogen loss from (dadi)Ti(N3Ad) (1-N3Ad) to form (dadi)TiNAd (2-Ad), because the latter is exergonic by
= −22.1 kcal mol−1. It is likely that the barrier for OAT from AdNCO is sluggish compared to dinitrogen loss from (dadi)Ti(N3Ad) (1-N3Ad) to form (dadi)TiNAd (2-Ad), because the latter is exergonic by 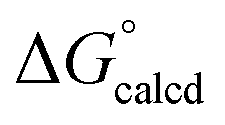 = −69.0 kcal mol−1. Since all adduct formations are expected to be rapid and reversible, OAT is likely not competitive. Note that no IR spectral evidence of isocyanide formation is observed in catalytic runs, even at the end of catalysis.
= −69.0 kcal mol−1. Since all adduct formations are expected to be rapid and reversible, OAT is likely not competitive. Note that no IR spectral evidence of isocyanide formation is observed in catalytic runs, even at the end of catalysis.
Attack by PMe3 on (dadi)TiO (2O) to form (dadi)TiOPMe3 (1-OPMe3) is calculated to be favorable by ΔG° = −22.5 kcal mol−1, but is relatively slow (>24 h, 23 °C), presumably due to known orbital symmetry constraints on OAT.38 The phosphine oxide adduct is more favorable than (dadi)Ti(THF) (1-THF) by 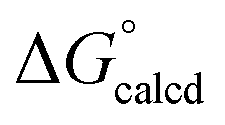 = −17.2 kcal mol−1.
= −17.2 kcal mol−1.
While the ligand redox states are consequential to nitrene transfer, certain atom and group transfers are not so readily explained. For example, the deoxygenation of (dadi)TiO (2O) by CO with 1 equiv. THF present to form (dadi)Ti(THF) (1-THF) and CO2 is calculated to be exergonic by ΔG° = −19.2 kcal mol−1 (ΔH° = −34.6 kcal mol−1, ΔS° = −51.6 eu).36,37 Decomposition is observed, perhaps indicative of additional reactivity from liberated CO2; further study is warranted. In addition, attempts to generate other (dadi)TiNR (2NR) species23–25 led to unanticipated reactivity, hence the adamantyl group protects the imide in (dadi)TiNAd (2-Ad) such that group transfer can be realized.
Attempts to catalyze carbodiimide formation akin to Heyduk's zirconium system12 failed, as treatment of (dadi)TiNAd (2-Ad) with CNMe afforded (dadi)Ti(CNMe)2 (1-(CNMe)2) and only a trace amount of MeNCNAd. Apparently the strong sigma-donation of CNMe, in contrast to the weak donor CO, imparts too much stability to the diisocyanide, as exposure of 1-(CNMe)2 to AdN3 was also ineffective.
Transition state of imide carbonylation
The transition state (TS) for imide carbonylation of (dadi)TiNAd (2NAd) was found via DFT calculations, and views of the TS and its accompanying metrics are illustrated in Fig. 8. The CO attacks the titanium at an angle consistent with its lone pair interacting with dxz/yz, while the imide connection is made via an Npπ → COπ* interaction. Related investigations of early metal nitride carbonylations show initial CO binding to be important,39 and the d(Ti–C) of 1.79 Å shows an interaction with the titanium.
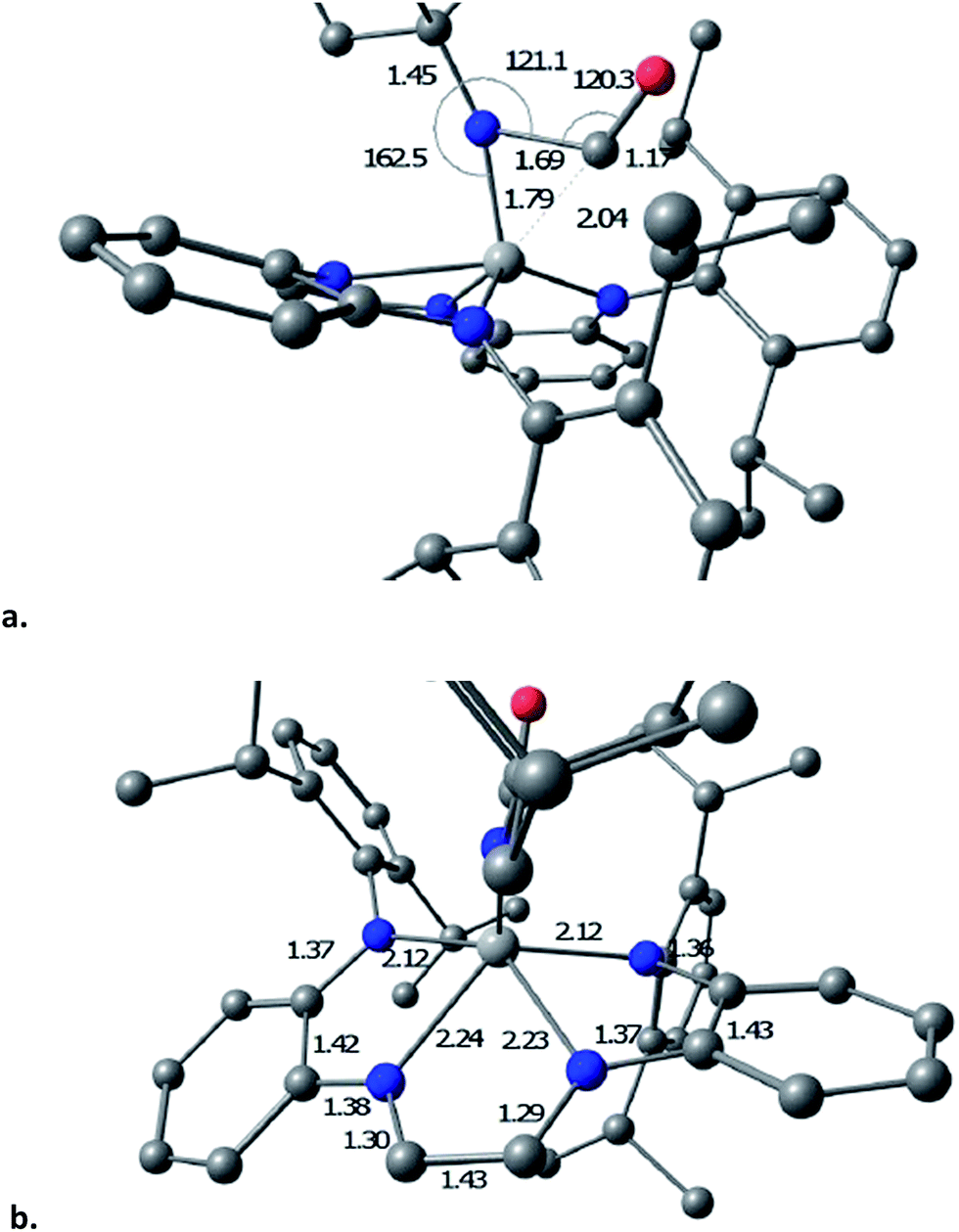 | ||
| Fig. 8 Views of the computed transition state for carbonyl addition to the imide of (dadi)TiNAd (2NAd): (a) metrics (distances (Å), angles (°)) of the imide-carbonyl fragment; (b) distances of the core and dadi ligand. | ||
The TS appears early in the reaction coordinate, as the d(TiN) of 1.79 Å is only 0.11 Å longer than the calculated d(TiN) in 2NAd, the TiNC(Ad) angle is still large (162.5°), and the d(NC) is long at 1.69 Å. The accompanying NCO angle is 120.3°, significantly bent from the 175° in the calculated κ1-N-OCNAd adduct that is the initial product of carbonylation, but there is considerable lengthening of the d(CO) to 1.17 Å. The calculated metrics pertaining to the TS (dadi)Ti core still conform to (dadi)2−, as the 2.12 and 2.14 Å Ti–Nam distances are significantly shorter than the Ti–Nim bond lengths of 2.23 and 2.24 Å. In a late TS, the core geometry would be expected to more closely correspond to (dadi)4−, which is calculated to have similar amide and “imine” d(TiN).
Conclusions
Structural and computational investigations show that dadim is capable of binding as a tetraanion in [dadi4−]Ti(IV)Lx (1-Lx; Lx = THF, PMe2Ph, (CNMe)2) and as a dianion in [dadi2−]Ti(IV)X (2X; X = O, NAd). The redox non-innocence of dadim is crucial in making CO binding to (dadi)Ti essentially devoid of π-backbonding, and uncompetitive with THF adduct formation by ΔG°(calc'd) = 7.7 kcal mol−1. In contrast, formation of oxo or adamantyl-imido multiple bonds is competitive with dadi reduction, ultimately permitting the successful catalytic carbonylation of AdN3 in which the redox changes occur at dadi rather than titanium.
Carbonylation catalysis with titanium is quite unusual, with Buchwald's enone synthesis as the primary example,40 and catalytic nitrene transfer from titanium is limited to Tonks' recent pyrrole synthesis.41 While the former is unlikely to involve formal RNI, the capability of Cp to covalently distribute charge is remarkable;42 a bound pyrrole product in the latter system could act in a redox non-innocent fashion.
Experimental section
Full experimental details are given in the ESI,† including kinetics and computational procedures in addition to syntheses. Brief descriptions of the syntheses, kinetic measurements, and computations are given in the schemes, and figure and table captions. Elemental analyses on all (dadi)Ti-based compounds failed normal standards, despite multiple attempts on crystalline samples, and utilization of several companies. As a consequence, full spectral characterizations, including all pertinent NMR spectra, are provided in the ESI.†Crystal data for 1-PMe2Ph: C46H55N4PTi, M = 742.81, monoclinic, P21/c, a = 18.3497(6), b = 10.3436(3), c = 25.5974(8) Å, β = 108.022(2)°, V = 4620.1(3) Å3, T = 223(2) K, λ = 0.71073 Å, Z = 4, Rint = 0.0524, 40189 reflections, 8152 independent, R1(all data) = 0.0672, wR2 = 0.1340, GOF = 1.048, CCDC-1522529.
Crystal data for 1-(CNMe)2: C47H62N6Ti, M = 758.92, triclinic, P-1, a = 10.2323(6), b = 13.2844(7), c = 16.9919(9) Å, α = 100.164(3)° β = 104.521(2)° γ = 95.151(2)°, V = 2179.0(2) Å3, T = 223(2) K, λ = 0.71073 Å, Z = 2, Rint = 0.0344, 29680 reflections, 8887 independent, R1(all data) = 0.0639, wR2 = 0.1178, GOF = 1.026, CCDC-1252531.
Crystal data for 2O: C49H62N4OTi, M = 770.92, triclinic, P-1, a = 13.1179(8), b = 13.2082(8), c = 13.7748(8) Å, α = 105.738(3)° β = 99.727(3)° γ = 98.022(3)°, V = 2220.5(2) Å3, T = 223(2) K, λ = 0.71073 Å, Z = 2, Rint = 0.0313, 40966 reflections, 9425 independent, R1(all data) = 0.0532, wR2 = 0.1215, GOF = 1.041, CCDC-1522530.
Acknowledgements
Support from the National Science Foundation (CHE-1402149 (PTW); CHE-1531468 (UNT computer facility); CHE-1531632 (NMR facility)), the Department of Energy Office of Basic Energy Sciences (DE-FG02-03ER15387 (TRC)), and Cornell University is gratefully acknowledged. We thank Prof. Kyle M. Lancaster for obtaining a resonance Raman spectrum of 2O.
Notes and references
- S. P. Heins, W. D. Morris, P. T. Wolczanski and E. B. Lobkovsky, Angew. Chem., Int. Ed., 2015, 54, 14407–14411 CrossRef CAS PubMed.
- L. A. Berben, B. de Bruin and A. G. Heyduk, Chem. Commun., 2015, 51, 1553–1554 RSC.
- (a) W. I. Dzik, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Angew. Chem., Int. Ed., 2011, 50, 3356–3358 CrossRef CAS PubMed; (b) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- R. F. Munhá, R. A. Zarkesh and A. F. Heyduk, Dalton Trans., 2103, 42, 3751–3766 RSC.
- P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687–1695 CrossRef CAS PubMed.
- K. G. Caulton, Eur. J. Inorg. Chem., 2012, 435–443 CrossRef CAS.
- K. Ray, T. Petrenko, K. Wieghardt and F. Neese, Dalton Trans., 2007, 1552–1566 RSC.
- V. A. Williams, P. T. Wolczanski, J. Sutter, K. Meyer, E. B. Lobkovsky and T. R. Cundari, Inorg. Chem., 2014, 53, 4459–4474 CrossRef CAS PubMed.
- W. D. Morris, P. T. Wolczanski, J. Sutter, K. Meyer, T. R. Cundari and E. B. Lobkovsky, Inorg. Chem., 2014, 53, 7467–7484 CrossRef CAS PubMed.
- B. A. Frazier, P. T. Wolczanski, I. Keresztes, S. DeBeer, E. B. Lobkovsky, A. W. Pierpont and T. R. Cundari, Inorg. Chem., 2012, 51, 8177–8186 CrossRef CAS PubMed.
- A. F. Heyduk, R. A. Zarkesh and A. I. Nguyen, Inorg. Chem., 2011, 50, 9849–9863 CrossRef CAS PubMed.
- (a) A. I. Nguyen, R. A. Zarkesh, D. C. Lacy, M. K. Thorson and A. F. Heyduk, Chem. Sci., 2011, 2, 166–169 RSC; (b) K. J. Blackmore, N. Lal, J. W. Ziller and A. F. Heyduk, J. Am. Chem. Soc., 2008, 130, 2728–2729 CrossRef CAS PubMed; (c) R. A. Zarkesh, J. W. Ziller and A. F. Heyduk, Angew. Chem., Int. Ed., 2008, 47, 4715–4718 CrossRef CAS PubMed.
- (a) K. J. Blackmore, J. W. Ziller and A. F. Heyduk, Inorg. Chem., 2005, 44, 5559–5561 CrossRef CAS PubMed; (b) M. R. Haneline and A. F. Heyduk, J. Am. Chem. Soc., 2006, 128, 8410–8411 CrossRef CAS PubMed; (c) N. A. Ketterer, H. Fan, K. J. Blackmore, X. Yang, J. W. Ziller, M.-H. Baik and A. F. Heyduk, J. Am. Chem. Soc., 2008, 130, 4364–4374 CrossRef CAS PubMed; (d) A. F. Heyduk, R. A. Zarkesh and A. I. Nguyen, Inorg. Chem., 2011, 50, 9849–9863 CrossRef CAS PubMed; (e) R. F. Munha, R. A. Zarkesh and A. F. Heyduk, Inorg. Chem., 2013, 52, 11244–11255 CrossRef CAS PubMed; (f) S. Hananouchi, B. T. Krull, J. W. Ziller, F. Furche and A. F. Heyduk, Dalton Trans., 2014, 43, 17991–18000 RSC.
- (a) J. M. Hoyt, K. T. Sylvester, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 4862–4877 CrossRef CAS PubMed; (b) K. T. Sylvester and P. J. Chirik, J. Am. Chem. Soc., 2009, 131, 8772–8773 CrossRef CAS PubMed; (c) M. W. Bouwkamp, A. C. Bowman, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13340–13341 CrossRef CAS PubMed; (d) S. K. Russell, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2011, 133, 8858–8861 CrossRef CAS PubMed; (e) J. M. Darmon, S. C. Stieber, K. L. Sylvester, I. Fernández, E. Lobkovsky, S. P. Semproni, E. Bill, K. Wieghardt, S. DeBeer and P. J. Chirik, J. Am. Chem. Soc., 2012, 134, 17125–17137 CrossRef CAS PubMed.
- S. S. Karpiniec, D. S. McGuiness, G. J. P. Britovsek and J. Patel, Organometallics, 2012, 31, 3439–3442 CrossRef CAS.
- R. Sikari, S. Sinha, U. Jash, S. Das, P. Brandäo, B. de Bruin and N. D. Paul, Inorg. Chem., 2016, 55, 6114–6123 CrossRef CAS PubMed.
- (a) A. L. Smith, K. I. Hardcastle and J. D. Soper, J. Am. Chem. Soc., 2010, 132, 14358–14360 CrossRef CAS PubMed; (b) C. A. Lippert, S. A. Arnstein, C. D. Sherrill and J. D. Soper, J. Am. Chem. Soc., 2010, 132, 3879–3892 CrossRef CAS PubMed; (c) C. J. Rolle III, K. I. Hardcastle and J. D. Soper, Inorg. Chem., 2008, 47, 1892–1894 CrossRef PubMed.
- C. C. Lu, E. Bill, T. Weyhermüller, E. Bothe and K. Wieghardt, J. Am. Chem. Soc., 2008, 130, 3181–3197 CrossRef CAS PubMed.
- V. A. Williams, E. B. Hulley, P. T. Wolczanski, K. M. Lancaster and E. B. Lobkovsky, Chem. Sci., 2013, 4, 3636–3648 RSC.
- (a) T. Marshall-Roth and S. N. Brown, Dalton Trans., 2015, 677–685 RSC; (b) L. G. Ranis, K. Werellapatha, N. J. Pietrini, B. A. Bunker and S. N. Brown, Inorg. Chem., 2014, 53, 10203–10206 CrossRef CAS PubMed; (c) A. H. Randolph, N. J. Seewald, R. Karl and S. N. Brown, Inorg. Chem., 2013, 52, 12587–12598 CrossRef CAS PubMed.
- J. J. H. Edema, R. Duchateau, S. Gambarotta, R. Hynes and E. Gabe, Inorg. Chem., 1991, 30, 154–156 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC; R. Stowasser and R. Hoffmann, J. Am. Chem. Soc., 1999, 121, 3414–3420 CrossRef CAS.
- A. R. Fout, U. J. Kilgore and D. J. Mindiola, Chem.–Eur. J., 2007, 13, 9428–9440 CrossRef CAS PubMed.
- C. Lorber, R. Choukroun and L. Vendier, Eur. J. Inorg. Chem., 2006, 4503–4518 CrossRef CAS.
- (a) N. Hazari and P. Mountford, Acc. Chem. Res., 2005, 38, 839–849 CrossRef CAS PubMed; (b) L. Gade and P. Mountford, Coord. Chem. Rev., 2001, 216–217, 65–97 CrossRef CAS; (c) P. Mountford, Chem. Commun., 1997, 2127–2134 RSC.
- S. D. Gray, J. L. Thorman, L. M. Berreau and L. K. Woo, Inorg. Chem., 1997, 36, 278–283 CrossRef CAS.
- S. M. Mullins, A. P. Duncan, R. G. Bergman and J. Arnold, Inorg. Chem., 2001, 40, 6952–6963 CrossRef CAS PubMed.
- (a) T. E. Hanna, E. Lobkovsky and P. J. Chirik, Eur. J. Inorg. Chem., 2007, 2677–2685 CrossRef CAS; (b) T. E. Hanna, I. Keresztes, E. Lobkovsky, W. H. Bernskoetter and P. J. Chirk, Organometallics, 2004, 23, 3448–3458 CrossRef CAS; (c) I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 13379–13389 CrossRef CAS PubMed.
- M. R. Smith III, P. T. Matsunaga and R. A. Andersen, J. Am. Chem. Soc., 1993, 115, 7049–7050 CrossRef.
- W. B. Tolman, Angew. Chem., Int. Ed., 2010, 49, 1018–1024 CrossRef CAS PubMed.
- (a) W. A. Howard, T. M. Trnka, M. Waters and G. Parkin, J. Organomet. Chem., 1997, 525, 95–121 CrossRef; (b) C. C. Cummins, R. R. Schrock and W. M. Davis, Inorg. Chem., 1994, 33, 1448–1457 CrossRef CAS.
- J. T. Groves and J. S. Roman, J. Am. Chem. Soc., 1995, 117, 5594–5595 CrossRef CAS.
- N. A. Piro, M. F. Lichterman, W. H. Harman and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 2108–2111 CrossRef CAS PubMed.
- C. E. Laplaza, A. L. Odom, W. M. Davis, C. C. Cummins and J. D. Protasiewicz, J. Am. Chem. Soc., 1995, 117, 4999–5000 CrossRef CAS.
- J. L. Bennett and P. T. Wolczanski, J. Am. Chem. Soc., 1997, 119, 10696–10719 CrossRef CAS.
- W. Darwish, E. Seikel, R. Käsmarker, K. Harms and J. Sundermeyer, Dalton Trans., 2011, 40, 1787–1794 RSC.
- (a) A. E. Guiducci, C. L. Boyd, E. Clot and P. Mountford, Dalton Trans., 2009, 5960–5979 RSC; (b) A. E. Guiducci, C. L. Boyd and P. Mountford, Organometallics, 2006, 25, 1167–1187 CrossRef CAS.
- A. S. Veige, L. M. Slaughter, E. B. Lobkovsky, P. T. Wolczanski, N. Matsunaga, S. A. Decker and T. R. Cundari, Inorg. Chem., 2003, 42, 6204–6224 CrossRef CAS PubMed.
- (a) A. F. Cozzolino, J. S. Silvia, N. Lopez and C. C. Cummins, Dalton Trans., 2014, 43, 4639–4652 RSC; (b) J. S. Silvia and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 446–447 CrossRef CAS PubMed.
- (a) N. M. Kablaoui, F. A. Hicks and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 4424–4431 CrossRef CAS; (b) F. A. Hicks and S. L. Buchwald, J. Am. Chem. Soc., 1999, 119, 7026–7033 CrossRef.
- Z. W. Gilbert, R. J. Hue and I. A. Tonks, Nat. Chem., 2015, 8, 63–68 CrossRef PubMed.
- P. T. Wolczanski, Organometallics, 2017, 36, 622–631 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1522529–1522531. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05610e |
| This journal is © The Royal Society of Chemistry 2017 |