
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGel-based morphological design of zirconium metal–organic frameworks†
Bart
Bueken
 a,
Niels
Van Velthoven
a,
Tom
Willhammar
bc,
Timothée
Stassin
a,
Ivo
Stassen
a,
David A.
Keen
d,
Gino V.
Baron
e,
Joeri F. M.
Denayer
e,
Rob
Ameloot
a,
Sara
Bals
b,
Dirk
De Vos
*a and
Thomas D.
Bennett
*f
a,
Niels
Van Velthoven
a,
Tom
Willhammar
bc,
Timothée
Stassin
a,
Ivo
Stassen
a,
David A.
Keen
d,
Gino V.
Baron
e,
Joeri F. M.
Denayer
e,
Rob
Ameloot
a,
Sara
Bals
b,
Dirk
De Vos
*a and
Thomas D.
Bennett
*f
aCentre for Surface Chemistry and Catalysis, Department of Microbial and Molecular Systems (M2S), KU Leuven, Celestijnenlaan 200F p.o. box 2461, 3001 Leuven, Belgium. E-mail: dirk.devos@kuleuven.be
bEMAT, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium
cDepartment of Materials and Environmental Chemistry, Stockholm University, S-106 91 Stockholm, Sweden
dISIS Facility, Rutherford Appleton Laboratory, Harwell Campus, Didcot, Oxon OX11 0QX, UK
eDepartment of Chemical Engineering, Vrije Universiteit Brussel, Pleinlaan 2, 1050 Brussels, Belgium
fDepartment of Materials Science and Metallurgy, University of Cambridge, 27 Charles Babbage Road, Cambridge CB3 0FS, UK. E-mail: tdb35@cam.ac.uk
First published on 23rd March 2017
Abstract
The ability of metal–organic frameworks (MOFs) to gelate under specific synthetic conditions opens up new opportunities in the preparation and shaping of hierarchically porous MOF monoliths, which could be directly implemented for catalytic and adsorptive applications. In this work, we present the first examples of xero- or aerogel monoliths consisting solely of nanoparticles of several prototypical Zr4+-based MOFs: UiO-66-X (X = H, NH2, NO2, (OH)2), UiO-67, MOF-801, MOF-808 and NU-1000. High reactant and water concentrations during synthesis were observed to induce the formation of gels, which were converted to monolithic materials by drying in air or supercritical CO2. Electron microscopy, combined with N2 physisorption experiments, was used to show that irregular nanoparticle packing leads to pure MOF monoliths with hierarchical pore systems, featuring both intraparticle micropores and interparticle mesopores. Finally, UiO-66 gels were shaped into monolithic spheres of 600 μm diameter using an oil-drop method, creating promising candidates for packed-bed catalytic or adsorptive applications, where hierarchical pore systems can greatly mitigate mass transfer limitations.
Introduction
Metal–organic frameworks (MOFs) continue to attract significant academic interest by virtue of their large structural and chemical diversity.1 These porous solids consist of inorganic nodes based on metal ions, interconnected through coordinatively bonded organic linkers, and have been widely investigated as catalysts,2,3 adsorbents,4–7 drug delivery systems,8,9 proton conductors10 and sensing materials.11,12 Accordingly, significant research has been oriented towards identifying and influencing the relationships between intrinsic MOF properties and targeted applications.The crystallization of MOFs almost exclusively leads to polydisperse microcrystalline powders. While suitable for research purposes, this state limits the applicability of MOFs in industrial settings since the use of fine powders is associated with several technical challenges, including poor handling, dust formation, mass transfer limitations and strong pressure drops in packed beds.13,14 To circumvent these issues, methods for the preparation of MOFs as meso- and/or macroscopically structured objects, preferably with hierarchical pore architectures, are highly sought after. Present approaches aimed at achieving this have mainly focused on structural templating, tuning synthetic conditions to control crystal growth, or on the formation of composite materials.15–19
Recently, zirconium-carboxylate MOFs, such as the Zr-terephthalate UiO-66 ([Zr6O4(OH)4(bdc)6], UiO = Universitetet i Oslo; bdc = 1,4-benzenedicarboxylate) and its isoreticular derivatives,20 have risen to the forefront of the MOF field because of their excellent chemical and thermal stabilities, combined with high porosities and tunable properties.21,22 Macroscale structuring of this subclass of MOFs has to date been achieved following two general strategies. First, Zr-MOFs have been deposited or grown onto support materials such as polymeric or ceramic monoliths, fibers and foams.23–31 These composite materials combine the separation performance of the MOFs and the support's surface texture, but generally possess lower adsorption capacities due to the secondary component. A second approach involves pelletizing single component MOF powders via mechanical compression or extrusion. However, appropriate pelletization pressures should be selected, as in the example reported by Decoste et al. for UiO-66,32 as this avenue can in some cases result in pressure-induced losses of crystallinity and microporosity, and is unsuitable for the introduction of meso- or macroporosity in MOFs.32–36 Binders are sometimes used to mitigate the first of these issues, however they might diminish adsorption capacity, and judicious selection is needed to ensure their compatibility with the targeted application.37–39
A promising new route towards shaped, hierarchically porous, pure MOF materials starts with MOF gels and avoids the microcrystalline powder state altogether. Here, different from amorphous coordination polymer gels,40,41 MOF nanoparticles crystallise during synthesis and aggregate to form a solid network throughout the synthesis solvent.42–46 This results in a gel state of tunable viscosity, which adopts the shape of its container. Subsequent solvent removal results in nanoparticle agglomeration and the formation of monolithic xero- or aerogels. Li et al. for instance applied this strategy to produce a variety of monoliths constructed from Al-MOF nanoparticles.43
Sporadic examples of gel formation during the synthesis of UiO-66-type Zr-MOFs have been reported in the literature.47–51 For example, the synthesis of UiO-66-(COOH)2 from ZrCl4 in water proceeds through the formation of a white gel.48,49 Gelation during the synthesis scale-up of UiO-66, using zirconyl chloride octahydrate (ZrOCl2·8H2O) as precursor, was also reported by Ragon and coworkers.50 Finally, Liu et al. described a MOF gel synthesized from an ethanol–DMF mixture using 2-aminoterephthalic acid and ZrCl4.51 However, detailed investigations into the synthesis or structure of such gels are sparse; there are no reports on formation of crystalline monolithic materials or hierarchically porous architectures from these gels.
In this contribution, we uncover some of the key parameters controlling gelation of UiO-66, and transpose these across the Zr-MOF family to functionalized UiO-66 materials, UiO-67, MOF-801, MOF-808 and NU-1000. The resulting gels can be easily manipulated and solvent removal by drying in air or under supercritical CO2 leads to the first reported hierarchically porous, monolithic Zr-MOF xero- and aerogels. As a proof-of-principle for the potential of these gels in an industrially relevant shaping process, monolithic UiO-66 spheres are prepared and characterized by oil-drop granulation of a UiO-66 gel.
Experimental
Synthesis
Characterization
Results and discussion
Gel formation
As a prototypical Zr-MOF system, we selected UiO-66 for our initial investigation on gel-based monolith formation, and performed a screening of several synthetic parameters to determine their influence on gelation (Table S1†). For each synthesis, a fixed molar ratio of 1.45 H2bdc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zr was employed and the macroscopic outcome was visually evaluated after two hours of reaction at 100 °C. Three parameters were found to play a crucial role: (1) metal source, (2) reactant concentration and (3) the presence of water. First, the choice of the metal source strongly determined whether or not gelation occurred. Under comparable conditions, syntheses employing ZrOCl2·8H2O formed gels much more readily than those using ZrCl4, which tended to yield microcrystalline precipitates (e.g. Table S1,† entries 6, 8 vs. 21, 26 respectively). Note that, where needed, additional water and HCl were added to compensate for the 8 molar equivalents of crystallization water associated with ZrOCl2·8H2O, and for the additional two equivalents of HCl produced from the hydrolysis of ZrCl4. Secondly, increasing reactant concentrations (Table S1,† entries 1 to 9) afforded progressively more ‘non-flowing’ gels (Fig. 1a). This effect was most pronounced when ZrOCl2·8H2O was employed as metal source. For instance, at a DMF:Zr ratio of ∼1500 (8.7 mM ZrOCl2·8H2O), a microcrystalline precipitate was obtained. Decreasing this ratio to 620 and 388 led to viscous solutions with a gel-like consistency, but which flowed downward upon turning the synthesis vessel upside down. Starting from DMF:Zr ratios below 200, ‘non-flowing’ gels were obtained. For syntheses based on ZrCl4, an increase in reactant concentration alone was insufficient to induce gelation (entries 17, 20, 26 and 30). Rather, a combination between high reactant concentrations and addition of water was decisive for these reaction mixtures, with an increase in the latter clearly steering the syntheses towards more ‘non-flowing’ gels (Table S1,† entries 19–23, 24–25, 26–27, 28–31). However, the concentration of water seemed to have a far less pronounced effect on syntheses starting from ZrOCl2·8H2O. Curiously, acetic acid, often used as a synthesis modulator to facilitate Zr-MOF crystallization,59 did not appear to have any noticeable macroscopic effect on the obtained gel products at the employed acetic acid:Zr ratio of 3.5 (Table S1,† entries 4, 9, 18 vs. 5, 10, 19, respectively).
:Zr was employed and the macroscopic outcome was visually evaluated after two hours of reaction at 100 °C. Three parameters were found to play a crucial role: (1) metal source, (2) reactant concentration and (3) the presence of water. First, the choice of the metal source strongly determined whether or not gelation occurred. Under comparable conditions, syntheses employing ZrOCl2·8H2O formed gels much more readily than those using ZrCl4, which tended to yield microcrystalline precipitates (e.g. Table S1,† entries 6, 8 vs. 21, 26 respectively). Note that, where needed, additional water and HCl were added to compensate for the 8 molar equivalents of crystallization water associated with ZrOCl2·8H2O, and for the additional two equivalents of HCl produced from the hydrolysis of ZrCl4. Secondly, increasing reactant concentrations (Table S1,† entries 1 to 9) afforded progressively more ‘non-flowing’ gels (Fig. 1a). This effect was most pronounced when ZrOCl2·8H2O was employed as metal source. For instance, at a DMF:Zr ratio of ∼1500 (8.7 mM ZrOCl2·8H2O), a microcrystalline precipitate was obtained. Decreasing this ratio to 620 and 388 led to viscous solutions with a gel-like consistency, but which flowed downward upon turning the synthesis vessel upside down. Starting from DMF:Zr ratios below 200, ‘non-flowing’ gels were obtained. For syntheses based on ZrCl4, an increase in reactant concentration alone was insufficient to induce gelation (entries 17, 20, 26 and 30). Rather, a combination between high reactant concentrations and addition of water was decisive for these reaction mixtures, with an increase in the latter clearly steering the syntheses towards more ‘non-flowing’ gels (Table S1,† entries 19–23, 24–25, 26–27, 28–31). However, the concentration of water seemed to have a far less pronounced effect on syntheses starting from ZrOCl2·8H2O. Curiously, acetic acid, often used as a synthesis modulator to facilitate Zr-MOF crystallization,59 did not appear to have any noticeable macroscopic effect on the obtained gel products at the employed acetic acid:Zr ratio of 3.5 (Table S1,† entries 4, 9, 18 vs. 5, 10, 19, respectively).
 | ||
| Fig. 1 Gels and monolithic particles of UiO-66. (a) ‘Non-flowing’ gels of UiO-66 synthesized from ZrOCl2·8H2O and H2bdc (Table S1,† entry 10). (b and c) Optically transparent monolithic xerogel particles obtained by crushing ‘non-flowing’ gels dried in air at 200 °C. Prior to drying, the gels were washed and solvent-exchanged with ethanol. Scale bar = 100 μm (see also Fig. S1†). | ||
Following synthesis, X-ray diffraction patterns were recorded for each of the gels to confirm the formation of UiO-66 (Fig. 2). The diffraction pattern presented in Fig. 2c, which is representative for all formed gels, contains two Bragg reflections centred around 7° and 8.5° 2θ, which correspond to the (111) and (200) reflections of the UiO-66 structure.20 Furthermore, their broadness indicates the presence of UiO-66 nanoparticles, with domain sizes between 10 to 15 nm as determined by the Scherrer equation. As expected, syntheses that did not yield gels produced microcrystalline UiO-66 as a powder precipitate (Fig. 2b).
 | ||
| Fig. 2 X-ray diffraction patterns of UiO-66 gels and monoliths. (a) Simulated diffraction pattern of UiO-66.20 (b) UiO-66 prepared as microcrystalline powder. (c) UiO-66 prepared as ethanol-exchanged gel (Table S1,† entry 10). (d) Air-dried xerogel monolith prepared from the gel in (c). (e) Aerogel monolith obtained after supercritical CO2 extraction of the gel in (c). | ||
Monolith formation and characterization
We subsequently directed our efforts to transforming the UiO-66 gels into dry, monolithic solids. Prior to solvent evacuation, unreacted linkers were removed from the gel matrices through solvent exchange, by dispersing an as-synthesized gel in an equal volume of fresh DMF, followed by a shear-induced homogenization using a vortex mixer. This readily turned the ‘non-flowing’ system into a volume-expanded, ‘flowing’ gel of lower viscosity, which was allowed to rest overnight. Subsequently, this system could again be converted to a ‘non-flowing’, compacted state through a centrifugation-driven syneresis. The excess solvent was phase-separated from the gel during centrifugation, and could easily be decanted. This whole process was repeated several times using fresh solvent (DMF or ethanol), until after the final step, an ethanol-containing, ‘non-flowing’ state was acquired, with a volume adjusted to be equal to the volume of the as-synthesized gel.Monolithic, optically transparent xerogels were obtained by drying ethanol-exchanged gels in air at 200 °C (Fig. 1b and c and S1†). Alternatively, solvent extraction using supercritical CO2 afforded aerogel monoliths (Fig. S2†). For the latter, no change in volume was observed upon solvent removal, whereas the xerogels underwent significant shrinkage due to the capillary forces exerted during the drying process. Regardless of the drying method, the broadened X-ray diffraction pattern of UiO-66 present in the precursor gels was retained in each case (Fig. 2d and e). While X-ray diffraction only gives information on the average, long-range structure of the monolith, additional information on atomic length scales was obtained by extracting the atomic pair distribution function (PDF) from X-ray total scattering experiments. A PDF provides a distribution of pairwise interatomic distances within a sample, and as such gives more insight into its local structure. Fig. S3† displays the PDFs of a representative air-dried xerogel and a microcrystalline UiO-66 powder recorded under the same conditions. The main peaks, present in both PDFs at interatomic distances of 2.33 Å, 3.75 Å and 4.89 Å, correspond to UiO-66's intracluster Zr–O and Zr–Zr atom pairs, while those at greater interatomic distances can be attributed to correlations between atoms in neighbouring clusters.35,60,61 For the peaks observed below interatomic distances of 1 Å, the quality of lab-recorded total scattering data is typically insufficient to allow for a reliable interpretation.62 The good agreement between both PDFs, as well as with previously reported PDFs for UiO-66,35,60,61 further corroborates the successful crystallization of UiO-66 during gel formation. However, while there are no direct indications that the gels contain additional phases, their presence could not be ruled out entirely based on scattering data alone (both PDF and diffraction data), especially since the nearest-neighbour Zr–Zr and Zr–O distances in ZrO2 are almost identical to those in UiO-66.63
Thus, to gain more insight into the micro/meso-structure and phase-purity of the MOF gels, a typical air-dried xerogel sample (formed using the gel in Table S1,† entry 10) was selected and investigated using electron microscopy (Fig. 3). The annular dark field (ADF) scanning transmission electron microscopy (STEM) micrographs of this sample are shown in Fig. 3b–e, and reveal that the monoliths are made up entirely of aggregated nanoparticles of approximately 10 nm in size. Both selected area electron diffraction (SAED) and Fourier transformations from individual nanocrystallites in the ADF-STEM images (Fig. 3d–f) show these particles to have diffraction features with d-values of 11.5–12.1 Å and 10.5 Å. These coincide with the (111) and (200) reflections of the face-centred cubic UiO-66 crystal structure,20 essentially confirming the observations made from powder X-ray diffraction and PDF analysis. Furthermore, no additional phases, such as amorphous or crystalline zirconium oxides, were observed in these experiments.
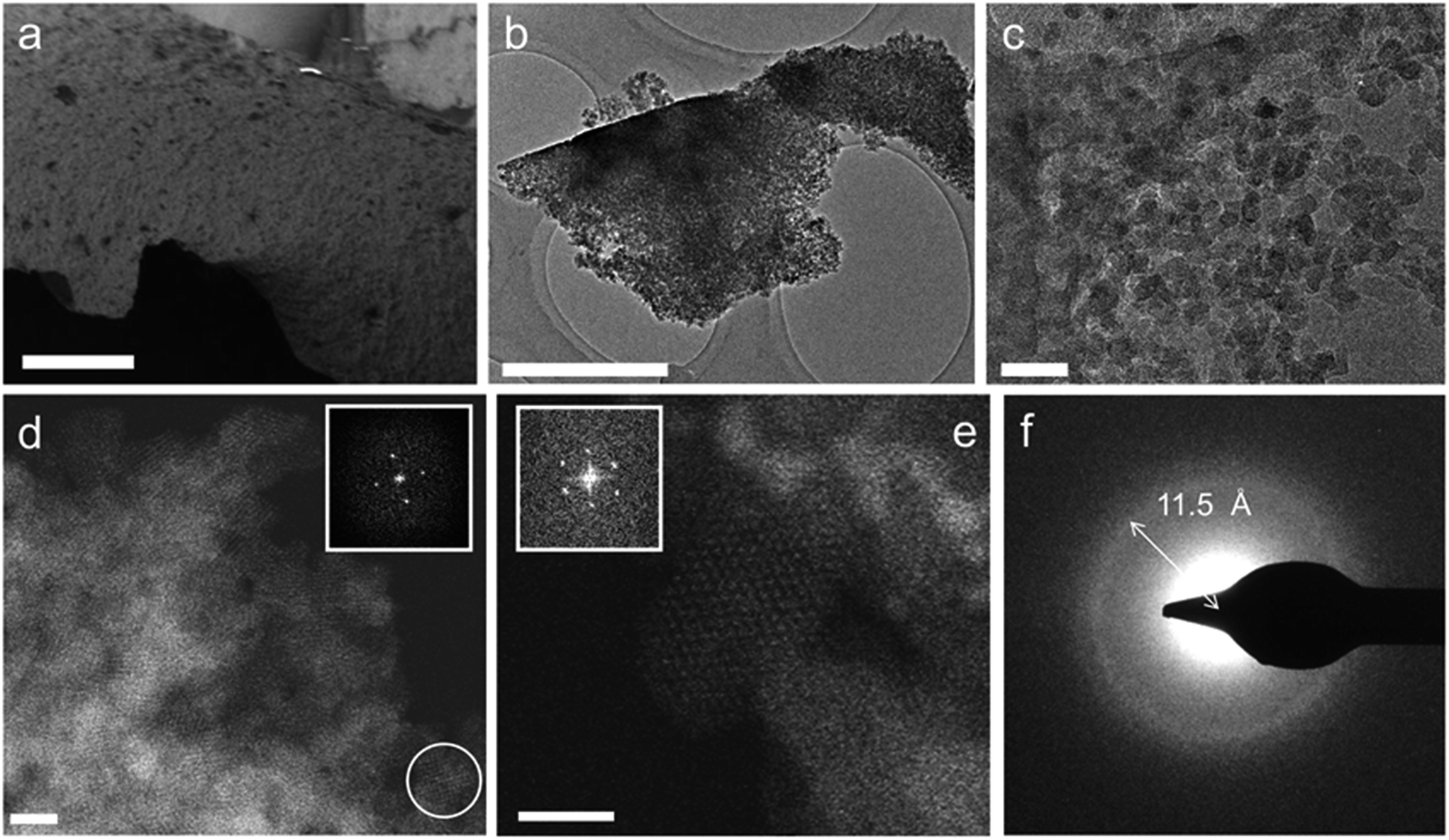 | ||
| Fig. 3 Electron microscopy of monolithic UiO-66 xerogels. (a) Scanning electron microscopy (SEM) image (scale bar = 25 μm) of a xerogel particle. (b and c) TEM images (b, scale bar = 1 μm; c, scale bar = 50 nm) of xerogel particles, which consist of irregularly packed MOF nanoparticles with interparticle voids. (d) ADF-STEM image of the xerogel in (b), illustrating the crystalline nature of the nanoparticles. Bright contrast corresponds to areas of high density (scale bar = 10 nm). The inset shows the Fourier transform of the nanoparticle circled in white, consistent with the face-centered cubic lattice of UiO-66 viewed along the [100] direction. (e) ADF-STEM image of the sample in (c), showing an individual UiO-66 nanoparticle oriented along [110]. Each bright spot corresponds to a single column of [Zr6O4(OH)4(R–COO)12] clusters (scale bar = 10 nm). The Fourier transform of this nanoparticle (inset) features reflections that can be indexed as the (111) and (200) reflections of UiO-66, corresponding to d-spacings of 12.1 Å and 10.5 Å, respectively. (f) SAED pattern of the aggregate particle in (b). The diffraction ring corresponds to a d-spacing of ∼11.5 Å, consistent with UiO-66's (111) reflection (12.0 Å). | ||
From the images in Fig. 3a–d, it is clear that the randomly packed crystallites generate mesoporous interparticle voids throughout the monolithic structure. An electron tomographic reconstruction enabled the 3-dimensional (3D) visualization of these voids in a single aggregate xerogel particle of ∼100 nm wide (Fig. 4a and b), illustrating how mesopores of 10–30 nm diameter permeate throughout the entire particle. Macroscopically, the porosity of the monoliths was confirmed by N2 physisorption experiments (Fig. 4c, Table 1). For both the xero- (red) and aerogel (blue), the isotherms feature a steep increase in N2 uptake at low p/p0, signifying adsorption in the micropores of the UiO-66 framework; at high p/p0, the behaviour is reminiscent of a IUPAC type IV isotherm, as indicated by the hysteretic desorption isotherm characteristic of mesoporous materials.57 For the air-dried UiO-66 xerogel considered above, a total pore volume of 2.09 cm3 g−1 was measured, with a calculated Brunauer–Emmett–Teller (BET) surface area of 1459 m2 g−1 and a micropore area of 844 m2 g−1, as determined by the t-plot method. Applying the Barrett–Joyner–Halenda (BJH) method revealed fairly uniform mesopores of approximately 15 nm in diameter in the xerogel (Fig. S4a†), which matches the range observed by electron microscopy. The micropore size distribution (Fig. S4b†) further shows the presence of 6 Å and 8 Å diameter pores, consistent with the cages present in the UiO-66 structure.20 Similar results were obtained for the aerogel, with a total pore volume of 1.66 cm3 g−1, and BET and micropore surface areas of 1255 m2 g−1 and 847 m2 g−1, respectively. We ascribe this slightly lower total pore volume to incomplete pore evacuation of the aerogel, as indicated by the thermogravimetric analysis (TGA) traces for the samples (Fig. S5†). The BJH pore size distribution however is much broader than for the xerogel, and is centred on 24 nm, consistent with the absence of capillary forces during supercritical CO2 drying. Overall, the porosity of the UiO-66 monoliths far exceeds that of bulk UiO-66 powder by a factor of 3–4 (Table 1). The somewhat lower micropore surface area of the gels compared to that of a typical bulk UiO-66 powder (1085 m2 g−1) likely finds its origin in the crystallite size; as size decreases, micropore surface area and volume is lost relative to the external surface area.64,65 These values are in agreement with the results obtained by Taddei et al.,66 who reported micropore areas of 966 m2 g−1 and 730 m2 g−1 for UiO-66 nanoparticles of 25 and 10 nm, respectively.
 | ||
| Fig. 4 Hierarchical porosity in UiO-66 monoliths. (a) Electron tomographic reconstruction of a single mesoporous monolithic xerogel particle (approx. 100 nm wide). Matter is represented in red. (b) Slice through the 3D reconstruction in (a). Bright contrast corresponds to matter, revealing intraparticle mesoporous voids. (c) Nitrogen physisorption isotherms (77 K) for a microcrystalline UiO-66 powder (black circles) and xerogel (red diamonds) and aerogel (blue triangles) monoliths (full symbols = adsorption branch; open symbols = desorption branch). The hysteretic desorption above p/p0 = 0.8 is attributed to capillary condensation in the mesopores. The inset shows a logarithmic representation of the adsorption branch at low p/p0. The two-step profile corresponds to the uptake of N2 in the smaller tetrahedral (6 Å) and larger octahedral cages (8 Å), respectively. | ||
| UiO-66 | a BET m2 g−1 | a micro m2 g−1 | a ext m2 g−1 | V pore cm3 g−1 | V micro cm3 g−1 |
|---|---|---|---|---|---|
| Powder | 1167 | 1085 | 82 | 0.47 | 0.40 |
| Xerogel | 1459 | 844 | 615 | 2.09 | 0.34 |
| Xerogel sphere | 1127 | 403 | 724 | 1.90 | 0.17 |
| Aerogel | 1255 | 847 | 408 | 1.66 | 0.33 |
| Nanoparticles (10 nm)66 | 1181 | 730 | 451 | — | 0.29 |
To further investigate the presence of meso- and macroporosity within the monolithic samples, mercury intrusion measurements were performed (Fig. S6 and S7†). A pore volume of 0.016 cm3 g−1 in the 4–100 μm pore diameter range was observed, which can be attributed to intrusion into defects in the monolithic samples (e.g. cracks), as well as in voids between the monolithic particles. The xerogel monoliths thus possess very few macropores. At these intrusion pressures, an apparent density of 0.3862 g cm−3 was measured for the xerogel monolith. Looking at higher Hg intrusion pressures (0.1–200 MPa), a cumulative pore volume of 1.935 cm3 g−1 was measured, which is in excellent agreement with the pore volume determined from N2 physisorption. Hg intrusion at these pressures was however found to be completely irreversible. Following the intrusion experiment, a significant reduction in monolith particle size was seen. Hence, Hg intrusion was not able to directly probe the 5–20 nm pores observed by N2 physisorption, but rather gradually and irreversibly compressed the monolithic particles by mesopore collapse, starting at approximately 5–6 MPa. Although no pore size distribution could be derived from this measurement, it gives an accurate representation of the volume of the pores. From helium pycnometry, a skeletal density of 1.969 (±0.009) g cm−3 was found.
Both xero- and aerogels exhibit excellent thermal stability based on thermogravimetric analysis. As illustrated in Fig. S5,† both materials feature a single decomposition step starting at ∼450 °C (O2 atmosphere), corresponding to the disintegration of the framework's bdc linkers. From these data, an average of approximately 11 linkers per Zr6-cluster was found for the xerogel, which is in line with the excess of H2bdc employed to synthesize the gels.67 For the aerogel, an average of 11.8 linkers per cluster, and an additional mass loss step between 100 °C and 200 °C was observed, suggesting the presence of residual DMF and/or H2bdc not removed by the post-treatment. While maintaining the same thermal stability as bulk UiO-66,20,67 the hierarchical nature of the gels results in a lower mechanical stability relative to ‘pristine’ UiO-66, as was seen from the compression under Hg intrusion experiments. For the air-dried xerogel sample, an elastic modulus (E) between 9.3 (±0.3) GPa and 10.5 (±0.5) GPa was further determined by nanoindentation experiments on polished monolithic particles (Fig. S8 and S9†). While difficult to compare with literature data due to the limited number of reports and the defect-dependence of UiO-66's mechanical properties, at least one computational study indicates this to correspond to about 20–33% of the value determined for UiO-66 with a similar number of missing linkers.68
Oil-drop granulation of mesoporous MOF spheres
To illustrate the potential of Zr-MOF gels to be shaped into a variety of monolithic objects, we aimed to prepare spherical, monodisperse monoliths of UiO-66 based on the industrially employed oil-drop granulation process,69 which prepares spherical silica or alumina granules by dripping sol droplets into an immiscible hot oil, followed by in situ gelation. A similar oil-drop setup was constructed in-house (see ESI†) to dispense a UiO-66 gel. While it was equally possible to perform the oil-drop shaping directly from the gel's synthesis solution, as in the conventional process, we found that using a preformed gel allowed for a more controlled shaping process and significantly simplified the washing procedures following bead formation, since the starting gel was already extensively solvent exchanged with DMF to remove excess reactants.A regular UiO-66 gel (Table S1,† entry 10) was synthesized, and solvent exchanged five times with fresh DMF to remove any unreacted linkers. After the final washing iteration, the volume of the gel was again expanded, in this case with 30 wt% of fresh DMF, yielding a mildly ‘flowing’ gel of which droplets could be dispensed by a perfusor pump into a flow of an immiscible, hot silicone oil. The spherical gel droplets subsequently underwent a temperature-induced syneresis to a xerogel over the course of 10 minutes at 150 °C. After collection and separation from the oil phase, the monolithic spheres were washed several times with dichloromethane to remove any oil residues, followed by a final annealing step (200 °C, air, 1 h). The resulting spheres (Fig. 5a and b) were uniform in size, which could be varied by changing the diameter of the dispensing needle. For instance, xerogel spheres with ∼600 μm in diameter were obtained by using a 1.2 mm diameter dispensing needle, which produced gel droplets of 2 mm in diameter. Thus, the droplets underwent a ∼37-fold volume shrinkage while transitioning to the xerogel spheres. X-ray diffraction (Fig. 5e) confirmed that the UiO-66 nanoparticles maintained their structure. The internal architecture of the spheres was investigated by scanning electron microscopy (Fig. 5c and d). Similar to the xerogel samples prepared by drying the UiO-66 gel in air, interparticle mesopores could be seen on the surface and in cross-sections of the spheres due to an irregular nanoparticle packing. The micro/mesoporous N2 physisorption isotherm (Fig. 5f; Table 1) corroborated the presence of mesopores, narrowly distributed around a diameter of 14.7 nm, and a total pore volume and BET surface area of respectively 1.9 cm3 g−1 and 1127 m2 g−1 were found, essentially matching those found for the air-dried xerogel.
 | ||
| Fig. 5 Monolithic UiO-66 xerogel spheres. (a) Optical image of a single sphere (scale bar = 150 μm). (b) SEM micrograph of a single sphere (scale bar = 150 μm). (c) SEM micrographs of a cross-section of the interior sphere architecture (scale bar = 3 μm). (d) Close-up of (c), highlighting the nanoparticulate structure and mesoporosity (scale-bar = 0.5 μm). (e) X-ray diffraction pattern of UiO-66 monolithic spheres. (f) Nitrogen physisorption isotherm (77 K) of monolithic UiO-66 spheres (full symbols = adsorption branch; open symbols = desorption branch). | ||
Discussion
The observation that high concentrations of water, linker and metal source in the synthesis of UiO-66 induce the formation of a nanocrystalline gel state leads us to suggest a rapid and excessive crystal nucleation to be at the origin of gelation. Indeed, this hypothesis can be rooted in classical crystallization theory, which models the nucleation rate to be exponentially dependent on the reactant supersaturation. Furthermore, a prerequisite for the UiO-66 framework to form is the formation of its [Zr6O4(OH)4]12+ clusters through hydrolysis of the employed Zr-salt.59 The early formation and subsequent retention of inorganic secondary building units during MOF synthesis has been observed previously.70,71 The concentration of water thus greatly influences the crystallization of UiO-66. For instance, Schaate et al. reported how increasing the amount of water present in (diluted) synthesis media yielded progressively smaller UiO-66 crystallites, with sizes down to 14 nm.59 Similarly, Ragon et al. found UiO-66 to crystallize significantly faster in the presence of water and attributed this to an easier formation of the Zr6-clusters.50 The observed differences in gelation between ZrCl4 and ZrOCl2·8H2O can be interpreted in a similar fashion, since ZrOCl2·8H2O already is the primary hydrolysis product of ZrCl4 and occurs as the tetranuclear cluster [Zr4(OH)8(H2O)16]Cl8·12(H2O); the latter can be considered a direct precursor for the eventual Zr6-clusters in UiO-66.72 Thus, syntheses utilizing ZrOCl2·8H2O are likely less sensitive to the addition of extra water due to its more advanced hydrolysis degree.50 Whilst not explored here, others have observed that the addition of modulators of increasing acidity results in smaller particle sizes for UiO-66, which could comprise an alternative route to gelation.59,73,74We propose gelation to be a direct consequence of the high concentration of formed UiO-66 nanoparticles, which aggregate primarily through non-covalent van der Waals interactions, although some degree of coordinative cross-linking or intergrowth between crystallites cannot be ruled out. The resulting colloidal suspension contains a weakly bound network of solids throughout its entire volume, and effectively adopts a gel-like state (Fig. 6). Because of a rapid decrease in reactant concentration, and concomitant increase in solution viscosity, further growth of the UiO-66 crystallites is likely impeded, leading to a kinetically stable state. In more dilute systems, a limited number of nuclei rather continue to grow and precipitate as a microcrystalline MOF powder. While similar gelation mechanisms have been proposed by others for MOF gels based on di- and trivalent cations,42,43,45,46 it should be noted that in several of these gels, additional, non-crystalline phases act as a binder between the MOF nanoparticles,43 or as a scaffold in which they are embedded.45 In case of the UiO-66 gels presented here, the available evidence points to the absence of such phases (Fig. 3).
 | ||
| Fig. 6 Schematic overview of gel formation. (a) Synthesis in dilute conditions, with a limited amount of water leads to microcrystalline particles. (b & c) High reactant and water concentration stimulates formation of the [Zr6O4(OH)4]12+ clusters and nucleation of the Zr-MOF (yellow stars). This leads to gel-like viscous colloidal suspensions of Zr-MOF nanoparticles, in which further crystal growth is hampered. (b) At intermediate nanoparticle concentrations, ‘flowing’ gels can be observed, (c) high nanoparticle concentrations lead to viscoelastic ‘non-flowing’ gels with a network of weakly aggregated particles throughout the entire solvent volume. (d & e) Tuning the nanoparticle concentration interconverts the system between these two states. (f) Solvent removal from the ‘non-flowing’ gels yields monolithic mesoporous xerogels or aerogels, consisting of randomly packed nanoparticles. | ||
Associated with high particle concentrations in the UiO-66 gel is a viscoelastic behaviour, with viscous flow occurring only above a certain yield stress at which sufficient interparticle interactions are overcome. The observed differences between ‘non-flowing’ and ‘flowing’ gels find their origin in a lower viscosity and yield stress for the latter, likely resulting from a lower volume fraction of crystallites and/or larger crystallite sizes, allowing the gel to flow under gravitational forces.13 Post-synthetically manipulating the solid's concentration thus offers a straightforward means to controlling the viscosity and state of the system: applying shear forces, as in vortex mixing, enables the solid network of a ‘non-flowing’ gel to be broken and redispersed in a larger solvent volume; the concentration of particles is lowered, and the viscoelastic properties of the system resemble that of a ‘flowing’ gel. Conversely, centrifugation of a ‘flowing’ gel, followed by removal of excess solvent achieves the opposite transformation (Fig. 6), and allowed us to obtain ‘non-flowing’ gels from as-synthesized ‘flowing’ gels.
Since the inorganic Zr6-cluster is shared by many Zr-MOFs, we hypothesized that a rational choice of synthetic conditions, followed by application of the gelation principles outlined here for UiO-66, would allow the extrapolation of gel formation to other Zr6-cluster systems. Starting from the routine used to synthesize the UiO-66 monoliths (Table S1,† entry 10), ‘non-flowing’ gels of the isoreticular MOFs UiO-66-NO2, UiO-66-NH2, UiO-66-(OH)2, UiO-67 and MOF-801 (ref. 75) were prepared by simply substituting H2bdc for equimolar amounts of their respective linkers (Fig. S10†). MOF-808 (ref. 75) and NU-1000 (ref. 52) are two Zr-MOFs based on respectively 1,3,5-benzenetricarboxylate and 1,3,6,8-tetrakis(p-benzoate)pyrene as linkers and feature topologies that significantly deviate from the face-centered cubic UiO-66 structure. Gels of these MOFs were still readily prepared by both increasing the concentration of reactants and including additional water relative to their original syntheses. In each case, an X-ray diffraction pattern (Fig. S11†) matching that of the desired phase, but with broad reflections indicative of nanosized crystallites, was obtained. Furthermore, each of these gels could be solvent exchanged following the procedure established for UiO-66, and transformed into monolithic xerogels by drying in air (Fig. S10†).
Conclusions
In conclusion, a new method to structure UiO-66 at the mesoscale is presented, by steering the synthesis towards a MOF-nanoparticle based gel state. The UiO-66 gels could be transformed to hierarchically porous monoliths, both as xero- and aerogels, with pore volumes (2.09 cm3 g−1 and 1.66 cm3 g−1, respectively) far exceeding those of bulk UiO-66 powder. The combination of microporosity intrinsic to the UiO-66 structure, with mesoporosity derived from the gel state, is a significant difference in property between the bulk crystalline and monolithic states. Furthermore, the UiO-66 gel state can be exploited to form shaped objects, as exemplified by the mesoporous, binder-free UiO-66 spheres prepared here by an industrially relevant oil-drop granulation. Since gelation is achieved in conditions which enhance the formation rate of the ubiquitous Zr6-clusters, the principles outlined for preparing UiO-66 gels can be extrapolated to form a variety of other Zr6-cluster based MOF gels, as shown for isoreticular analogues of UiO-66, MOF-808 and NU-1000. Hence, the gels and monoliths presented here provide a step towards shaping Zr-MOF materials for applications in catalysis or adsorption. Finally, the optically transparent nature of the xerogels may be of interest in the preparation of transparent films and coatings.Acknowledgements
B. B., T. S. and I. S. acknowledge the FWO Flanders (doctoral and post-doctoral grants). T. W. acknowledges a post-doctoral grant from the Swedish Research Council. T. D. B. acknowledges the Royal Society (University Research Fellowship) and Trinity Hall (University of Cambridge) for funding. S. B. and D. D. V. are grateful for funding by Belspo (IAP 7/05 P6/27) and by the FWO Flanders. D. D. V. further acknowledges funding from the European Research Council (project H-CCAT). S. B. acknowledges financial support from the European Research Council (ERC Starting Grant #335078-COLOURATOMS). The authors acknowledge Arnau Carne and Shuhei Furukawa for assistance with supercritical CO2 extraction, and Charles Ghesquiere for assistance in synthesis.Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974–985 CrossRef CAS PubMed.
- P. Valvekens, F. Vermoortele and D. De Vos, Catal. Sci. Technol., 2013, 3, 1435–1445 CAS.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC.
- B. Van de Voorde, B. Bueken, J. Denayer and D. De Vos, Chem. Soc. Rev., 2014, 43, 5766–5788 RSC.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- M. Giménez-Marqués, T. Hidalgo, C. Serre and P. Horcajada, Coord. Chem. Rev., 2016, 307, 342–360 CrossRef.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994–6010 RSC.
- M. J. Rhodes, Introduction to particle technology, Wiley-VHC, Weinheim, 2008 Search PubMed.
- F. Rezaei and P. Webley, Chem. Eng. Sci., 2009, 64, 5182–5191 CrossRef CAS.
- R. E. Morris, ChemPhysChem, 2008, 10, 327–329 CrossRef PubMed.
- D. Bradshaw, A. Garai and J. Huo, Chem. Soc. Rev., 2012, 41, 2344–2381 RSC.
- D. Bradshaw, S. El-Hankari and L. Lupica-Spagnolo, Chem. Soc. Rev., 2014, 43, 5431–5443 RSC.
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC.
- Y. Lv, X. Tan and F. Svec, J. Sep. Sci., 2016, 40, 272–287 CrossRef PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- Y.-Y. Fu, C.-X. Yang and X.-P. Yan, Chem. Commun., 2013, 49, 7162–7164 RSC.
- M. L. Pinto, S. Dias and J. Pires, ACS Appl. Mater. Interfaces, 2013, 5, 2360–2363 CAS.
- Z. Yan, J. Zheng, J. Chen, P. Tong, M. Lu, Z. Lin and L. Zhang, J. Chromatogr. A, 2014, 1366, 45–53 CrossRef CAS PubMed.
- I. Stassen, M. Styles, T. Van Assche, N. Campagnol, J. Fransaer, J. Denayer, J.-C. Tan, P. Falcaro, D. De Vos and R. Ameloot, Chem. Mater., 2015, 27, 1801–1807 CrossRef CAS.
- E. López-Maya, C. Montoro, L. M. Rodríguez-Albelo, S. D. Aznar Cervantes, A. A. Lozano-Pérez, J. L. Cenís, E. Barea and J. A. R. Navarro, Angew. Chem., 2015, 127, 6894–6898 CrossRef.
- Y. Chen, X. Huang, S. Zhang, S. Li, S. Cao, X. Pei, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2016, 138, 10810–10813 CrossRef CAS PubMed.
- H. Zhu, Q. Zhang and S. Zhu, Chem.–Eur. J., 2016, 22, 8751–8755 CrossRef CAS PubMed.
- H. Zhu, X. Yang, E. D. Cranston and S. Zhu, Adv. Mater., 2016, 28, 7652–7657 CrossRef CAS PubMed.
- J. Zhao, D. T. Lee, R. W. Yaga, M. G. Hall, H. F. Barton, I. R. Woodward, C. J. Oldham, H. J. Walls, G. W. Peterson and G. N. Parsons, Angew. Chem., Int. Ed., 2016, 55, 13224–13228 CrossRef CAS PubMed.
- G. W. Peterson, J. B. DeCoste, T. G. Glover, Y. Huang, H. Jasuja and K. S. Walton, Microporous Mesoporous Mater., 2013, 179, 48–53 CrossRef CAS.
- J. B. Decoste and G. W. Peterson, Chem. Rev., 2014, 114, 5695–5727 CrossRef CAS PubMed.
- B. Van de Voorde, I. Stassen, B. Bueken, F. Vermoortele, D. De Vos, R. Ameloot, J.-C. Tan and T. D. Bennett, J. Mater. Chem. A, 2015, 3, 1737–1742 CAS.
- T. D. Bennett, T. K. Todorova, E. F. Baxter, D. G. Reid, C. Gervais, B. Bueken, B. Van de Voorde, D. De Vos, D. A. Keen and C. Mellot-Draznieks, Phys. Chem. Chem. Phys., 2016, 18, 2192–2201 RSC.
- M. I. Nandasiri, S. R. Jambovane, B. P. McGrail, H. T. Schaef and S. K. Nune, Coord. Chem. Rev., 2016, 311, 38–52 CrossRef CAS.
- M. A. Moreira, J. C. Santos, A. F. P. Ferreira, J. M. Loureiro, F. Ragon, P. Horcajada, K.-E. Shim, Y.-K. Hwang, U.-H. Lee, J.-S. Chang, C. Serre and A. E. Rodrigues, Langmuir, 2012, 28, 5715–5723 CrossRef CAS PubMed.
- S.-N. Kim, Y.-R. Lee, S.-H. Hong, M.-S. Jang and W.-S. Ahn, Catal. Today, 2015, 245, 54–60 CrossRef CAS.
- N. Chanut, A. D. Wiersum, U.-H. Lee, Y. K. Hwang, F. Ragon, H. Chevreau, S. Bourrelly, B. Kuchta, J.-S. Chang, C. Serre and P. L. Llewellyn, Eur. J. Inorg. Chem., 2016, 2016, 4416–4423 CrossRef CAS.
- M. R. Lohe, M. Rose and S. Kaskel, Chem. Commun., 2009, 6056–6058 RSC.
- P. Sutar and T. K. Maji, Chem. Commun., 2016, 52, 8055–8074 RSC.
- P. Horcajada, C. Serre, D. Grosso, C. Boissière, S. Perruchas, C. Sanchez and G. Férey, Adv. Mater., 2009, 21, 1931–1935 CrossRef CAS.
- L. Li, S. Xiang, S. Cao, J. Zhang, G. Ouyang, L. Chen and C.-Y. Su, Nat. Commun., 2013, 4, 1774 CrossRef PubMed.
- W. Xia, X. Zhang, L. Xu, Y. Wang, J. Lin and R. Zou, RSC Adv., 2013, 3, 11007–11013 RSC.
- A. K. Chaudhari, I. Han and J.-C. Tan, Adv. Mater., 2015, 27, 4438–4446 CrossRef CAS PubMed.
- A. Mahmood, W. Xia, N. Mahmood, Q. Wang and R. Zou, Sci. Rep., 2015, 5, 10556 CrossRef PubMed.
- M. Kandiah, S. Usseglio, S. Svelle, U. Olsbye, K. P. Lillerud and M. Tilset, J. Mater. Chem., 2010, 20, 9848–9851 RSC.
- Q. Yang, S. Vaesen, F. Ragon, A. D. Wiersum, D. Wu, A. Lago, T. Devic, C. Martineau, F. Taulelle, P. L. Llewellyn, H. Jobic, C. Zhong, C. Serre, G. De Weireld and G. Maurin, Angew. Chem., Int. Ed., 2013, 52, 10316–10320 CrossRef CAS PubMed.
- F. Ragon, B. Campo, Q. Yang, C. Martineau, A. D. Wiersum, A. Lago, V. Guillerm, C. Hemsley, J. F. Eubank, M. Vishnuvarthan, F. Taulelle, P. Horcajada, A. Vimont, P. L. Llewellyn, M. Daturi, S. Devautour-Vinot, G. Maurin, C. Serre, T. Devic and G. Clet, J. Mater. Chem. A, 2015, 3, 3294–3309 CAS.
- F. Ragon, P. Horcajada, H. Chevreau, Y. K. Hwang, U.-H. Lee, S. R. Miller, T. Devic, J.-S. Chang and C. Serre, Inorg. Chem., 2014, 53, 2491–2500 CrossRef CAS PubMed.
- L. Liu, J. Zhang, H. Fang, L. Chen and C.-Y. Su, Chem.–Asian J., 2016, 11, 2278–2283 CrossRef CAS PubMed.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed.
- A. K. Soper, Tech. Rep. RAL-TR-2011-013, 2011 Search PubMed.
- A. K. Soper and E. R. Barney, J. Appl. Crystallogr., 2011, 44, 714–726 CrossRef CAS.
- D. A. Keen, J. Appl. Crystallogr., 2001, 34, 172–177 CrossRef CAS.
- B. Goris, W. Van den Broek, K. J. Batenburg, H. Heidari Mezerji and S. Bals, Ultramicroscopy, 2012, 113, 120–130 CrossRef CAS.
- J. Rouquerol, F. Rouquerol, and K. S. W. Sing, Absorption by Powders and Porous Solids, Academic Press, London, 1999 Search PubMed.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 2004, 19, 3–20 CrossRef CAS.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- B. Bueken, F. Vermoortele, M. J. Cliffe, M. T. Wharmby, D. Foucher, J. Wieme, L. Vanduyfhuys, C. Martineau, N. Stock, F. Taulelle, V. Van Speybroeck, A. L. Goodwin and D. De Vos, Chem.–Eur. J., 2016, 22, 3264–3267 CrossRef CAS PubMed.
- A. E. Platero-Prats, A. Mavrandonakis, L. C. Gallington, Y. Liu, J. T. Hupp, O. K. Farha, C. J. Cramer and K. W. Chapman, J. Am. Chem. Soc., 2016, 138, 4178–4185 CrossRef CAS PubMed.
- T. D. Bennett, Y. Yue, P. Li, A. Qiao, H. Tao, N. G. Greaves, T. Richards, G. I. Lampronti, S. A. T. Redfern, F. Blanc, O. K. Farha, J. T. Hupp, A. K. Cheetham and D. A. Keen, J. Am. Chem. Soc., 2016, 138, 3484–3492 CrossRef CAS PubMed.
- M. Gateshki, V. Petkov, G. Williams, S. K. Pradhan and Y. Ren, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 224107 CrossRef.
- J. Aguado, D. P. Serrano, J. M. Escola and J. M. Rodríguez, Microporous Mesoporous Mater., 2004, 75, 41–49 CrossRef CAS.
- J. Cravillon, S. Münzer, S.-J. Lohmeier, A. Feldhoff, K. Huber and M. Wiebcke, Chem. Mater., 2009, 21, 1410–1412 CrossRef CAS.
- M. Taddei, K. C. Dümbgen, J. A. van Bokhoven and M. Ranocchiari, Chem. Commun., 2016, 52, 6411–6414 RSC.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- A. W. Thornton, R. Babarao, A. Jain, F. Trousselet and F.-X. Coudert, Dalton Trans., 2016, 45, 4352–4359 RSC.
- F. Schüth and M. Hesse, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VHC, Weinheim, Germany, 2008 Search PubMed.
- S. Surblé, F. Millange, C. Serre, G. Férey and R. I. Walton, Chem. Commun., 2006, 1518–1520 RSC.
- F. Millange, M. I. Medina, N. Guillou, G. Férey, K. M. Golden and R. I. Walton, Angew. Chem., Int. Ed., 2010, 49, 763–766 CrossRef CAS PubMed.
- A. Clearfield and P. A. Vaughan, Acta Crystallogr., 1956, 9, 555–558 CrossRef CAS.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef.
- Z. Hu, I. Castano, S. Wang, Y. Wang, Y. Peng, Y. Qian, C. Chi, X. Wang and D. Zhao, Cryst. Growth Des., 2016, 16, 2295–2301 CAS.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optical microscopy, PDF TGA, Hg intrusion, nanoindentation, gels of various Zr-MOFs, oil-drop process. See DOI: 10.1039/c6sc05602d |
| This journal is © The Royal Society of Chemistry 2017 |