
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic catalysis for C–C and C–X coupling reactions
Dominic R.
Pye
and
Neal P.
Mankad
 *
*
Department of Chemistry, University of Illinois at Chicago, 845 W. Taylor St., Chicago, IL 60607, USA. E-mail: npm@uic.edu
First published on 16th January 2017
Abstract
Bimetallic catalysis represents an alternative paradigm for coupling chemistry that complements the more traditional single-site catalysis approach. In this perspective, recent advances in bimetallic systems for catalytic C–C and C–X coupling reactions are reviewed. Behavior which complements that of established single-site catalysts is highlighted. Two major reaction classes are covered. First, generation of catalytic amounts of organometallic species of e.g. Cu, Au, or Ni capable of transmetallation to a Pd co-catalyst (or other traditional cross-coupling catalyst) has allowed important new C–C coupling technologies to emerge. Second, catalytic transformations involving binuclear bond-breaking and/or bond-forming steps, in some cases involving metal–metal bonds, represent a frontier area for C–C and C–X coupling processes.
 Dominic R. Pye | Dominic studied chemistry at the University of Manchester, where he obtained an MChem in 2010 after completing his final year project with Dr Peter Quayle. He then moved to the University of Bristol where he undertook his PhD on iron-catalysed cross-coupling reactions under the supervision of Professors Robin Bedford and Timothy Gallagher. In 2015 he moved to the University of Illinois at Chicago to work as a post-doctoral research associate investigating heterobimetallic-catalysed carbonylation reactions with Professor Neal Mankad. |
 Neal P. Mankad | Before starting his independent career at the University of Illinois at Chicago (UIC) in 2012, Neal conducted undergraduate research at MIT with Joseph Sadighi, graduate research at Caltech with Jonas Peters, and postdoctoral research at the University of California-Berkeley with Dean Toste. At UIC, his group conducts research on bimetallic catalysis in organic systems, multimetallic catalytic sites in biology, and other topics related to energy and health sciences. Neal's recent awards include an Alfred P. Sloan Research Fellowship, a Thieme Chemistry Journal Award, and the UIC Rising Star Researcher of the Year. He likes exercising, eating, drinking, and traveling (sometimes all on the same day). |
1. Introduction
Coupling reactions that allow for catalytic C–C or C–X bond formation (X = e.g. B, N, O) have revolutionized synthetic chemistry by allowing complex organic structures to be created from simpler building blocks, even at late stages of multistep synthetic sequences.1–5 The dominant paradigm in coupling chemistry is to utilize single-site homogeneous catalysts and tailor reactivity and selectivity patterns using ligand design. For example, much of cross-coupling catalysis involves palladium–phosphine systems that operate by a canonical oxidative addition/reductive elimination cycling mechanism. Decades have been spent designing elaborate phosphine ligands to provide reactivity suitable for modern applications.6–9 Exquisite levels of catalytic activity, regioselectivity, and/or stereoselectivity ultimately have been achieved using this paradigm.Pursuing alternative catalytic paradigms that go beyond this single-site approach has the potential to uncover complementary reactivity and selectivity regimes.10 In addition, in some cases catalytic reactivity can become accessible with inexpensive and earth-abundant metals not typically utilized extensively in cross-coupling catalysis. This perspective highlights one such alternative approach: the use of bimetallic catalysis for C–C and C–X bond forming reactions.11 The focus of the perspective is on recent developments in bimetallic catalysis as applied to catalytic C–C and C–X bond formation in molecular organic systems. All of the included examples involve catalytic amounts of two d-block metals cooperating during catalysis. The following topics are excluded from this perspective: reactions involving d-block metals cooperating with main-group elements (e.g. Na, Al, B, P) during catalysis;12,13 cases with one of the two metals not participating directly in bond-breaking/forming events (e.g. photoredox catalysis,14 Wacker oxidation15); cases with one of the two metals present in stoichiometric quantities; tandem, cascade, or domino reactions16 that do not involve direct communication between codependent catalytic metals; and cluster systems where mechanistic understanding is limited.17 Also excluded from the perspective are catalytic polymerization reactions,18–20 reductions of unsaturated organics (e.g. hydrogenation, hydrosilylation),21–25 and transformations of small-molecule inorganics (e.g. H2, H2O, CO, CO2, N2, O2, etc.),26–30 all of which have had recent advances in their own right using bimetallic approaches.
The contribution of this perspective is timely, as bimetallic catalysis for C–C and C–X coupling is a burgeoning area that stands to make important contributions to the synthetic toolkit. In this perspective, these contributions are categorized into two broad subdivisions, with representative mechanistic schemes shown in Scheme 1. First are bimetallic systems involving catalytic generation of an organometallic nucleophile that undergoes transmetallation with a “traditional” coupling catalyst that operates using its canonical single-site mechanism (Scheme 1a). Here, no binuclear steps are utilized for breaking the bonds of the coupling partners or forming the bonds of the products.31 Second are bimetallic systems that involve binuclear bond activation and/or bond elimination events (Scheme 1b). Here, metal–metal bonds often (though not always) play key roles during catalysis.32
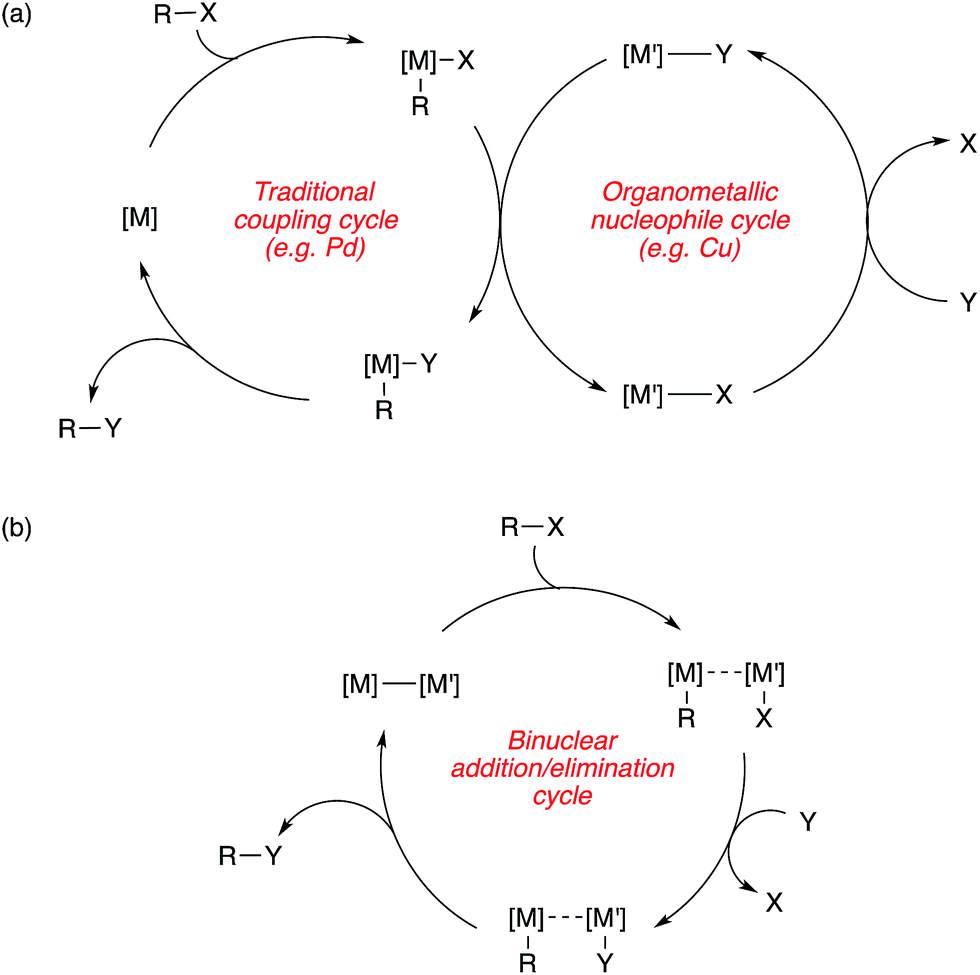 | ||
| Scheme 1 Representative mechanistic schemes for bimetallic catalysis (a) without and (b) with binuclear bond breaking and forming events. | ||
2. Bimetallic catalysis with mononuclear bond breaking & forming mechanisms
2.1 Organocopper nucleophiles
It was demonstrated in 1966 by Owsley and co-workers that copper-acetylides react in a stoichiometric fashion with aryl halides.33 The harsh reaction conditions (reflux in pyridine) lead to the formation of side products, such as aryl halide reduction and acetylene dimerization. Monometallic, palladium-catalysed coupling of aryl halides and terminal alkynes was reported by both Cassar34 and Heck35 in 1975; however both catalyst systems still required elevated temperature for high conversion. In 1975 Sonogashira and co-workers reported that the addition of copper salts greatly accelerated the reaction, leading to room temperature reactivity, greater functional group tolerance, and a generally more useful procedure.36 It is now accepted that the role of copper in these reactions is to react with the alkyne in the presence of a base to give a copper acetylide.37 The acetylide moiety then undergoes transmetallation from copper to palladium to give the key arylpalladium(II) acetylide (Scheme 2), which subsequently releases the desired product by reductive elimination. The key step, transmetallation of an organic fragment to palladium, has proven the inspiration for all the copper–palladium bimetallic-catalysis described in this section.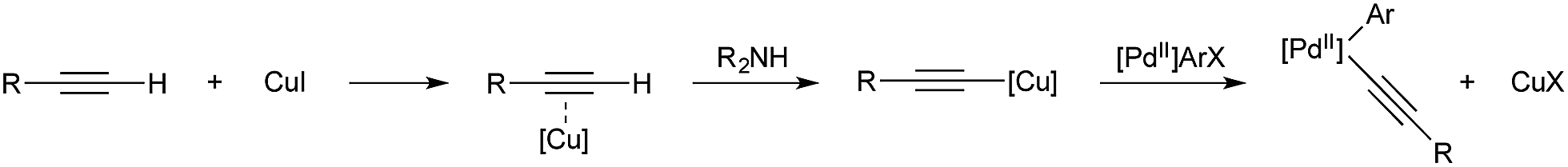 | ||
| Scheme 2 Cu/Pd cooperation in Sonogashira coupling. | ||
An alternative mechanistic paradigm is the catalytic, in situ formation of organometallic species from catalytic amounts of a precatalyst and an organic pro-nucleophile. This avoids the pre-formation and purification of the nucleophilic component, and the organic pro-nucleophiles are generally more stable and readily available than organometallic or main group nucleophiles. One example of this approach is the insertion of unsaturated organic compounds into copper-element bonds (Scheme 3). The product of this insertion is a reactive organo-copper nucleophile ready for further reaction. A further advantage in catalytic formation of the nucleophile is that low concentrations of reactive species leads to fewer undesired side reactions. The functionalisation of an unsaturated bond also leads to further complexity build up in a single step.
 | ||
| Scheme 3 General scheme for organocopper nucleophiles generated by element-cupration. | ||
The insertion of alkenes in to copper–boron bonds has been implicated in many copper-catalysed C–B bond forming processes, such as copper-catalysed hydroboration of alkenes. In 2006 Sadighi proved this unambiguously by the isolation of β-boryl alkyl copper species from the reaction of IPrCu(Bpin) with styrenes (Scheme 4).38
 | ||
| Scheme 4 Borylcupration of styrenes demonstrated by Sadighi. | ||
In 2014 Semba/Nakao39 and Brown40 independently showed that this intermediate could be used as a transmetallating reagent from copper to an arylpalladium(II), which reductively eliminates to form an carboborylated product and Pd(0). The copper boryl species is regenerated through alkoxide assisted transmetallation with B2(pin)2, and palladium(0) undergoes oxidative addition with an aryl bromide to complete two synergistic catalytic cycles. Both groups reported (NHC)copper(I) and Pd(II)/dicyclohexylbiarylphosphine precatalyst mixtures (Scheme 5).
 | ||
| Scheme 5 Cu/Pd catalysis for arylboration developed by the groups of Semba/Nakao and Brown. | ||
Brown and co-workers also showed that when cyclic styrene derivatives (such as 1,2-dihydronapthalene) were employed as substrates, in most cases the reaction gave the trans diastereomer with high selectivity. As the addition of (SIMes)CuB(pin) across the alkene is likely to proceed in a syn-fashion, it was stated that the transmetallation Cu–Pd must proceed with inversion of stereochemistry to give the trans isomer after stereoretentive reductive elimination. Under the described conditions, acyclic, 1,2-disubstituted styrenes gave low diastereoselectivity. Brown later reported modified conditions under which both the syn- and anti-carboboration diastereomers of acyclic disubstituted styrenes could be obtained, selectivity being determined by a change in solvent and ligand (Scheme 6).41 It was found that the use of THF and RuPhos would selectively provide the syn-product, presumably through a stereoretentive transmetallation of the putative syn-Cu-alkyl intermediate, followed by reductive elimination. Diastereoselectivity was found to be reversed if the solvent was changed to toluene, and the Pd ligand to triisobutylphosphine. It should be noted that both the change of solvent and ligand were required; both Pd-RuPhos in toluene and Pd-PiBu3 in THF gave low diastereoselectivity.
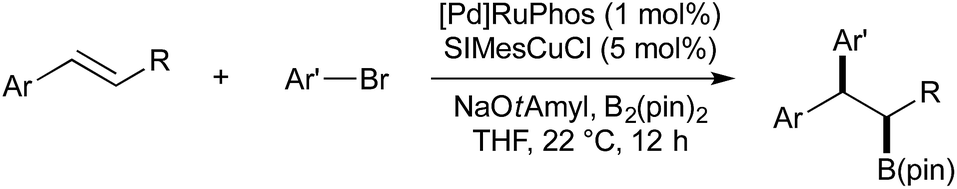 | ||
| Scheme 6 Diastereoselective carboboration developed by Brown. | ||
Liao and co-workers reported an enantioselective variant of the Cu/Pd carboboration reaction in 2015 (Scheme 7).42 Here they use copper(I) acetate with a chiral sulfoxide–phosphine ligand to achieve enantioselective borylcupration of a styrene, followed by transmetallation to a palladium-allyl species generated from the oxidative addition of allyl-tert-butylcarbonates to Pd(0). Products were generated with ee > 90%. When racemic, cyclic allylic carbonates were used as electrophiles, diastereomeric control of the two contiguous chiral centres was achieved. It was also demonstrated that iodobenzene could be used in place of the allylic carbonate to give an enantioselective version of the Nakao–Brown carboboration, albeit in lower yield.
 | ||
| Scheme 7 Enantioselective carboboration developed by Liao. | ||
In 2016 Semba/Nakao reported a procedure for the carboboration of alkenes, this time using nickel rather than palladium to activate the aryl electrophile.43 As a first-row transition metal, nickel is preferable to palladium in terms of cost and earth abundance. It was also found that the Nakao–Brown conditions were not suitable for the use of aryl chlorides, or phenol derived electrophiles (other than triflates). Semba/Nakao demonstrated that styrenes could be converted to their carboborylated product using a precatalyst system of CuCl, Ni(acac)2, and tricyclopentylphosphine, whilst using aryl chlorides or tosylates as the electrophilic carbon source (Scheme 8). Drawbacks, in comparison with the copper/palladium-catalysed systems are the requirement for higher temperatures (80 °C vs. room temperature) and the poor reactivity of β-substituted styrenes (30–40% yield).
 | ||
| Scheme 8 Cu/Ni catalysis developed by Semba/Nakao. | ||
In 2016 Semba/Nakao reported a system wherein borylcupration could be replaced with hydrocupration, thereby furnishing 1,1-diarylalkanes (Scheme 9).44 (NHC)CuH is generated in situ by the reaction of (NHC)CuOtBu with HSi(OEt)3. The use of a deuterium labelled silane gave confirmation of syn-addition across the double bond, and subsequent stereoinversion upon transmetallation. Theoretical calculations suggest an SE2(back) mechanism, which is consistent with stereoinversion.
 | ||
| Scheme 9 Hydroarylation catalysis developed by Semba/Nakao. | ||
Later that year Buchwald used a chiral bisphosphine Cu(I) precatalyst to give an enantioselective Cu/Pd hydroarylation of styrenes (Scheme 10).45 The enantiodetermining step is the addition of a chiral copper hydride across the double bond.
 | ||
| Scheme 10 Enantioselective hydroarylation developed by Buchwald. | ||
Riant and co-workers have described an asymmetric reduction/allylation of α,β-unsaturated ketones using a copper–palladium bimetallic catalyst system, a silane, and allyl carbonates.46 The products of these reactions are highly valuable, chiral all-carbon quaternary centres. In this instance the enantioselective step is governed by the palladium catalyst, bearing a chiral PHOX ligand, as opposed to a chiral copper hydride vide supra. Copper-catalysed 1,4-reduction of the unsaturated ketone gives a copper-enolate, which will then transmetallate to palladium. Mechanistic studies suggest that this transmetallation can give both the C- and O-bound Pd-enolate, which, after reductive elimination, release both the desired product and an allyl-enol ether. The allyl-enol ether converts to the desired product under the reaction conditions via a palladium-catalysed Cope rearrangement (Scheme 11).
 | ||
| Scheme 11 Reductive allylation of α,β-unsaturated ketones developed by Riant. | ||
The reaction of copper(I) complexes with silyl boranes is known to give copper-silyl species, which Riant and co-workers showed will add across electron deficient alkynes to catalytically generate a vinyl-copper species to be used in palladium-catalysed cross-coupling (Scheme 12).47 In this instance allylic carbonates were used as the electrophiles. The authors found that the regioselectivity of the double bond could be controlled, switching between the Z- and E-isomers by omitting triphenylphosphine from the reaction conditions, and switching from simple copper chloride to NHC ligated IMesCu(DBM) (DBM = dibenzoylmethanate).
 | ||
| Scheme 12 Silylation catalysis developed by Riant. | ||
The effect of triphenyl phosphine on the regioselectivity is ascribed to the relative reactivity of the palladium(II) allyl present, and the steric hindrance of the initial syn-silylcupration product compared to its tautomeric allenolate. In the absence of phosphine, palladium dimer [Pd(allyl)Cl]2 forms. Transmetallation from copper gives an allyl-, vinyl-palladium intermediate from which C–C bond formation occurs via an inner-sphere mechanism. In the presence of excess phosphine, cationic [(Ph3P)nPd(allyl)]+ forms, which does not transmetallate with copper. In this instance the less sterically hindered allenolate reacts with the Pd-allyl complex via an outer-sphere mechanism, giving the products of formal anti-addition.
 | ||
| Scheme 13 Cu/Pd cross-coupling developed by Faul. | ||
The use of tri(organo)silanol nucleophiles in C–C cross-coupling (Hiyama–Denmark coupling) is an attractive alternative to Sn, Mg, Zn, Al, and B based nucleophiles due to the bench stability and low toxicity of silicon compounds. Whilst monometallic systems have been shown to successfully couple C(sp2)–silicon nucleophiles with aryl halides and sulfonates, no general procedure has been developed for the coupling of alkyl-silicon nucleophiles with aryl sulfonates.
The Nakao group have developed aryl[2-(2-hydroxyprop-2-yl)cyclohexyl]dimethylsilanes (Fig. 1) for nickel-catalysed biaryl coupling with aryl sulfonates.50 This reaction, however, suffers from poor tolerance of sensitive functional groups and sterically demanding substrates.
 | ||
| Fig. 1 Alkylsilane reagent developed by Nakao. | ||
In 2016 Nakao published a revised procedure consisting of a bimetallic, Cu/Pd catalyst system which displays much greater functional and steric compatibility, as well as being compatible with alkyl[2-(2-hydroxyprop-2-yl)cyclohexyl]dimethylsilanes (Scheme 14).51
 | ||
| Scheme 14 Cu/Pd variant of Hiyama–Denmark coupling developed by Nakao. | ||
In this reaction the difunctional silyl-alcohol reagent initially coordinates to copper through deprotonation/salt metathesis of the alcohol, followed by intramolecular [2 + 2] σ-bond metathesis to give an organocopper intermediate (Scheme 14). This transmetallates to arylpalladium(II) (from Pd(0) and aryl tosylate). Reductive elimination furnishes the product and closes the dual catalytic cycle.
 | ||
| Scheme 15 Cu/Pd-catalyzed imine synthesis. | ||
2.2 Organogold nucleophiles
In 2009 Blum and co-workers published a gold/palladium-catalysed rearrangement of allyl allenoates to butenolides.54 The reaction proceeds smoothly at room temperature in the presence of an in situ generated gold(I) triflate complex and Pd2(dba)3 (5 mol% each) (Scheme 16). The initial step in the catalytic cycle is coordination of gold to the allene followed by cyclisation to give an activated gold–oxonium species. The allyl moiety is now activated towards oxidative addition by Pd(0) (oxidative addition does not occur in the absence of gold activation). Transmetallation from gold to palladium, followed by reductive elimination, give the butenolide product. | ||
| Scheme 16 Au/Pd catalysis developed by Blum. | ||
Included in the above publication was similar rearrangement of allyl esters onto alkynes to give lactones (Scheme 17). The scope of this rearrangement was expanded, and mechanistic details disclosed, in a 2014 report.55 It is worth noting that, in 2012, Hashmi showed that, when more activated esters are used as substrates (i.e. benzyl or cinnamyl in place of allyl) high yields of the products were obtained under monometallic gold-catalysed conditions.56
 | ||
| Scheme 17 Au/Pd catalysis for lactone formation developed by Blum. | ||
In 2016 Nevado and co-workers published another rearrangement of allenoates; however in this report the gold-catalysed rearrangement is coupled with a palladium-catalysed aryl cross-coupling cycle (Scheme 18).57 Gold-catalysed carbocyclisation of the initial allenoate is followed by transmetallation to Pd(II) aryl and reductive elimination.
 | ||
| Scheme 18 Au/Pd catalysis developed by Nevado. | ||
As part of the mechanistic investigations on allenoate rearrangements, Blum and co-workers noted that cationic oxonium species did not undergo palladium-catalysed cross-coupling, however the neutral analogue would.54 It is the judicious choice of aryl iodides as coupling partners that allow the Nevado reaction to proceed. The iodide formed on oxidative addition of Ar-I to Pd(0) acts as a dealkylating agent, giving iodoethane and a neutral butenolidyl gold species (Scheme 19). This reaction also sequesters iodide from the reaction medium, which is beneficial as Nevado reported that (p-F3CPh)3PAuI is inactive.
 | ||
| Scheme 19 Au/Pd mechanistic results from Blum. | ||
2.3 Others
Many other organometallic nucleophile cycles have been combined with Pd catalysis, following upon pioneering early examples.58,59 In 2016 Lee and co-workers demonstrated a rhodium–palladium-catalysed method that uses triazoles as precursors to rhodium-carbenes, which then couple with palladium-allyl complexes generated in situ.60 This procedure is unusual, in that the anion formed in oxidative generation of Pd-allyl is incorporated into the final product, increasing atom economy. Rhodium(II) is known to activate triazoles to give α-iminocarbenoids. This underdoes nucleophilic attack by the carboxylate anion formed on oxidative addition of an allyl ester by Pd(0). The resultant imine-ester coordinates palladium in a seven-membered intermediate, from which rhodium(II) is eliminated. Reductive elimination of the allyl fragment with N-tosyl furnishes the product and regenerates Pd(0) (Scheme 20). | ||
| Scheme 20 Rh/Pd catalysis developed by Lee. | ||
Vanadium(V) esters are known to catalyse the rearrangement of propargylic alcohols via their corresponding vanadium allenolate. These can be trapped by various N-, O- or C-based nucleophiles. Trost and co-workers reported twice in 2011 on the interception of these intermediates with palladium-allyl complexes to give α-allyl-α,β-unsaturated ketones in a vanadium–palladium dual-catalysed process (Scheme 21). The first report details conditions for the use of aromatic propargylic alcohols.61 As they found that this aromatic activation was essential for the initial vanadium-catalysed rearrangement, later than year they expanded the scope to include aliphatic propargylic alcohols with an activating alkoxy group on the alkyne fragment.62
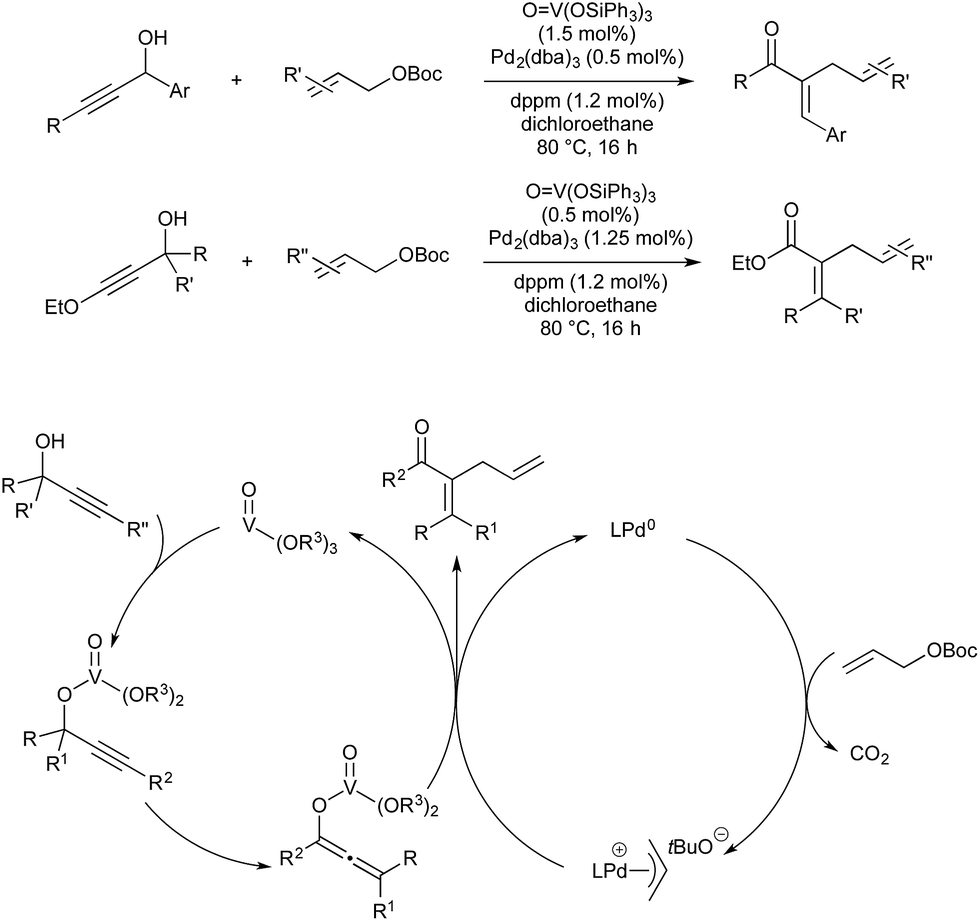 | ||
| Scheme 21 V/Pd catalysis developed by Trost. | ||
Weix and co-workers demonstrated in 2014 that titanium(III)-catalysed radical ring-opening of epoxides could be used in conjunction with reductive nickel catalysis to give an enantioselective coupling of epoxides and aryl bromides.63 The study utilised the fact that Ti(III) is known to catalyse reductive ring opening of epoxides to give α-radical Ti(IV) alkoxides. Weix has shown in previous reductive cross-couplings that carbon centred radicals will react with arylnickel(II) species to give Ni(III) which rapidly undergoes reductive elimination. The resultant Ni(I) reduces Ti(IV) produced in epoxide ring opening to give Ni(II) and regenerate Ti(III), and Ni(II) is in turn reduced to Ni(0) by manganese to complete both catalytic cycles (Scheme 22). The use of a chiral titanium precatalyst bearing Cp ligands derived from menthol allowed enantioselective ring opening of meso-epoxides, giving trans aryl-alcohols in high enantiopurity.
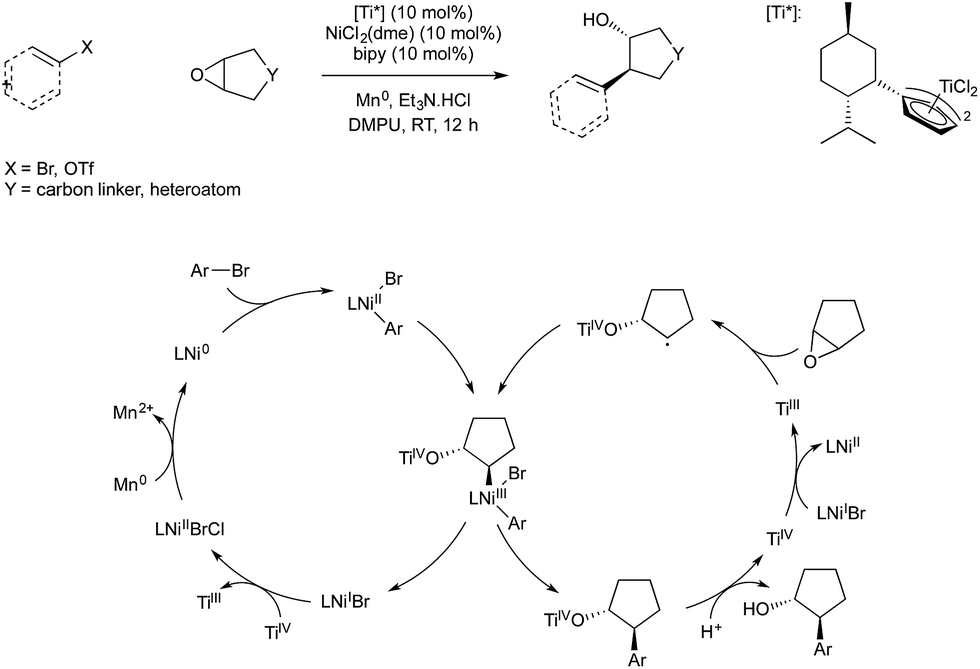 | ||
| Scheme 22 Ti/Ni catalysis developed by Weix. | ||
Weix and co-workers have also used multimetallic catalysis for the reductive coupling of two aryl electrophiles.64 A 4,4′-bipyridine nickel precatalyst was employed for the activation of aryl bromides, and (dppp)PdCl2 for the activation of aryl triflates. Each monometallic catalyst reacts selectively with its intended aryl-electrophile, to give high selectivity for the cross-coupled product over the two possible homo-coupled side products. This selectivity also allows for the use of the electrophiles in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, whereas previous, monometallic, reductive cross-coupling of aryl halides had required excess of one reagent to obtain selectivity. After each metal undergoes oxidative addition of its respective electrophile, arylnickel(II) transmetallates with arylpalladium(II) to give bis(aryl)palladium(II) intermediate, which reductively eliminates the desired product. Zinc metal is the terminal reductant, which regenerates nickel(0) and completes the catalytic cycle (Scheme 23).
:1 ratio, whereas previous, monometallic, reductive cross-coupling of aryl halides had required excess of one reagent to obtain selectivity. After each metal undergoes oxidative addition of its respective electrophile, arylnickel(II) transmetallates with arylpalladium(II) to give bis(aryl)palladium(II) intermediate, which reductively eliminates the desired product. Zinc metal is the terminal reductant, which regenerates nickel(0) and completes the catalytic cycle (Scheme 23).
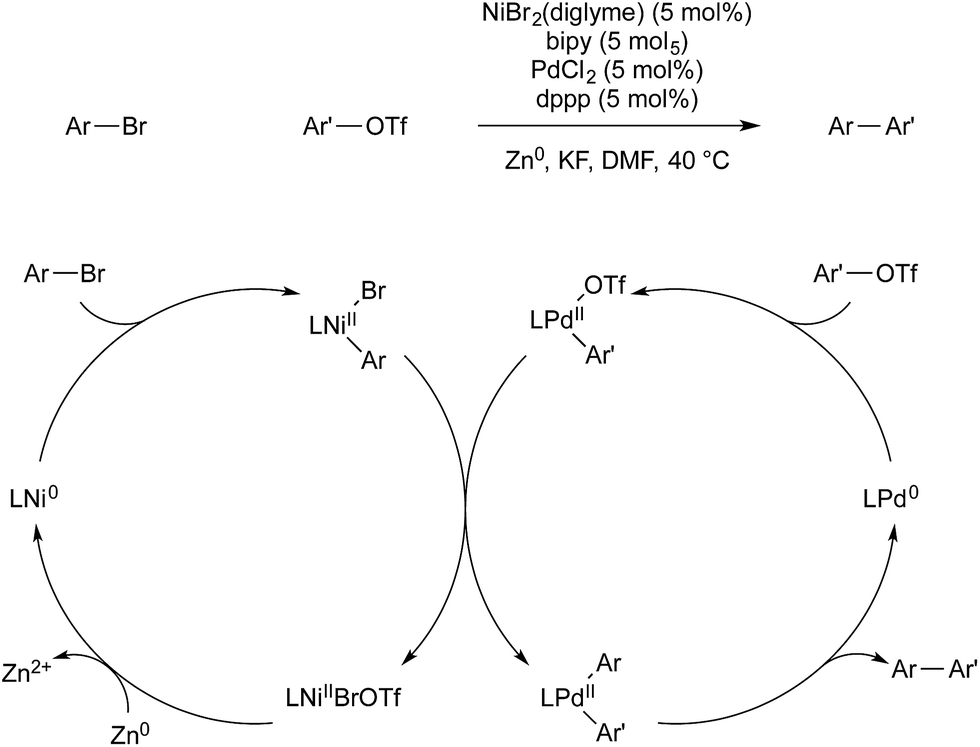 | ||
| Scheme 23 Ni/Pd catalysis developed by Weix. | ||
In 2010, Goossen and co-workers reported a silver–palladium-catalysed decarboxylative biaryl coupling (Scheme 24).65 Here, an organosilver intermediate is generated by silver-catalysed decarboxylation of a benzoic acid, and then employed as the nucleophilic partner for palladium-catalysed biaryl-coupling. This report is an extension of previous, analogous copper/palladium-catalysed reactions.66 Silver salts had previously been found to be more efficient in mediating the decarboxylation of benzoate salts, however they were found to be incompatible with cross-coupling conditions, as silver halide salts formed during the reaction would precipitate and prevent catalytic turnover. Goossen found that if aryl triflates were used as coupling partners, this salt precipitation was avoided and turnover achieved. Due to the greater efficiency of silver over copper to mediate decarboxylation, reaction temperatures could be lowered from 170 to 120 °C. Shen and coworkers have reported related Ag/Pd catalysis for difluoromethylation of aryl electrophiles.67
 | ||
| Scheme 24 Ag/Pd catalysis developed by Goossen. | ||
In 2011 Chen and Ma published an iron/palladium-catalysed cyclisation/cross-coupling of allenoates with allyl electrophiles, which they claim is the first example of transmetallation of an organic fragment from iron to palladium (Scheme 25).68 The mechanism is similar to the gold/palladium-catalysed cyclisation/cross-coupling of allenoates published by Blum and Nevado; however in this instance iron(III) chloride acts as the Lewis acidic metal, giving rise to formation of a metallo-lactone. Oxidative addition of allylic bromides to palladium(0) give electrophilic Pd(II), which receives the lactone fragment from iron, and reductive elimination gives the allylic lactone as the product.
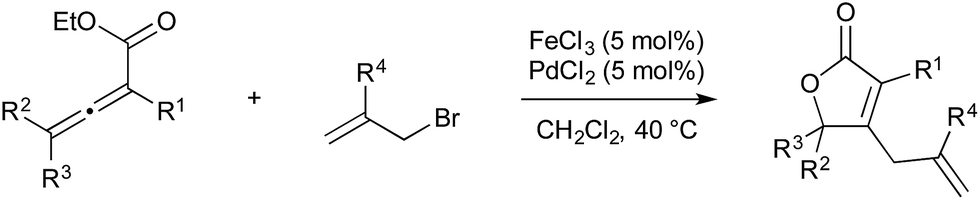 | ||
| Scheme 25 Fe/Pd catalysis developed by Chen and Ma. | ||
3. Bimetallic catalysis with binuclear bond breaking & forming mechanisms
3.1 Serendipitous bimetallic catalysis
While the dominant paradigm in homogeneous catalysis is to design reactions based on single-site mechanisms, in some cases a post hoc analysis of these reactions can reveal that two catalyst sites actually cooperate under the catalytic conditions even when bimetallic chemistry was unintended. Ideally, the elucidation of such serendipitous bimetallic reactions can be leveraged towards the rational design of bimetallic catalysts for useful transformations. | ||
| Scheme 26 (a) Asymmetric epoxide ring-opening catalysis developed by Jacobsen; (b) heterobimetallic catalyst design of Shibasaki. | ||
 | ||
| Scheme 27 Oxidative catalysis involving dipalladium(III) intermediates identified by Ritter. | ||
Furthermore, similar binuclear intermediates have come to be proposed in various coinage metal-catalyzed C–X bond forming oxidations, again raising the possibility of binuclear catalytic mechanisms being operative under certain conditions. First, in 2010, Ritter proposed that Ag(I)-catalyzed C–F coupling under oxidative conditions involves the intermediacy of bimetallic Ag(II)–Ag(II)–F species capable of binuclear C–F elimination,82 thus avoiding the invocation of high-valent Ag(III)–F intermediates (Scheme 28). Indirect evidence for this bimetallic mechanism was obtained from the observed rate acceleration of C–F elimination from AgOTf additive during reaction of Selectfluor with isolated arylsilver(I) species.
 | ||
| Scheme 28 Bimetallic mechanism for Ag-catalyzed fluorination proposed by Ritter. | ||
Contemporaneously, Zhang, Toste, Nevado, Gouverneur, Hashmi, Lloyd-Jones, and others developed oxidative alkene heteroarylation reactions using Au(I) catalysts combined with a stoichiometric oxidant, commonly Selectfluor. These reaction are typically proposed to involve single-site Au(I)/Au(III) redox cycling, as was reviewed recently by Gouverneur.83 Among these various studies, both mononuclear and binuclear gold catalysts were employed. In one study, Toste found that the binuclear dppm(AuBr)2 catalyst exhibited superior performance when compared to mononuclear Ph3PAuBr.84 Furthermore, this effect disappeared when dppm was replaced with a longer and more flexible dppb tether. These observations led Toste and workers to propose a revised mechanism involving Au(II)–Au(II) intermediates capable of C–C coupling with arylboronic acid nucleophiles (Scheme 29).85 The bimetallic mechanism had support from computational studies. Additionally, cyclic voltammetry measurements were used to show that binuclear Au(I)⋯Au(I) aggregates featuring aurophilic interactions facilitated by bridging ligands are more readily oxidized than single-site Au(I) species lacking bridging ligands. Presumably the difference arises from stabilization of oxidized Au(II)–Au(II) species in comparison to high-valent Au(III) species. Thus, under oxidative conditions, it is possible that bimetallic Au(II)–Au(II) intermediates assemble during catalysis even in the absence of bridging ligands.
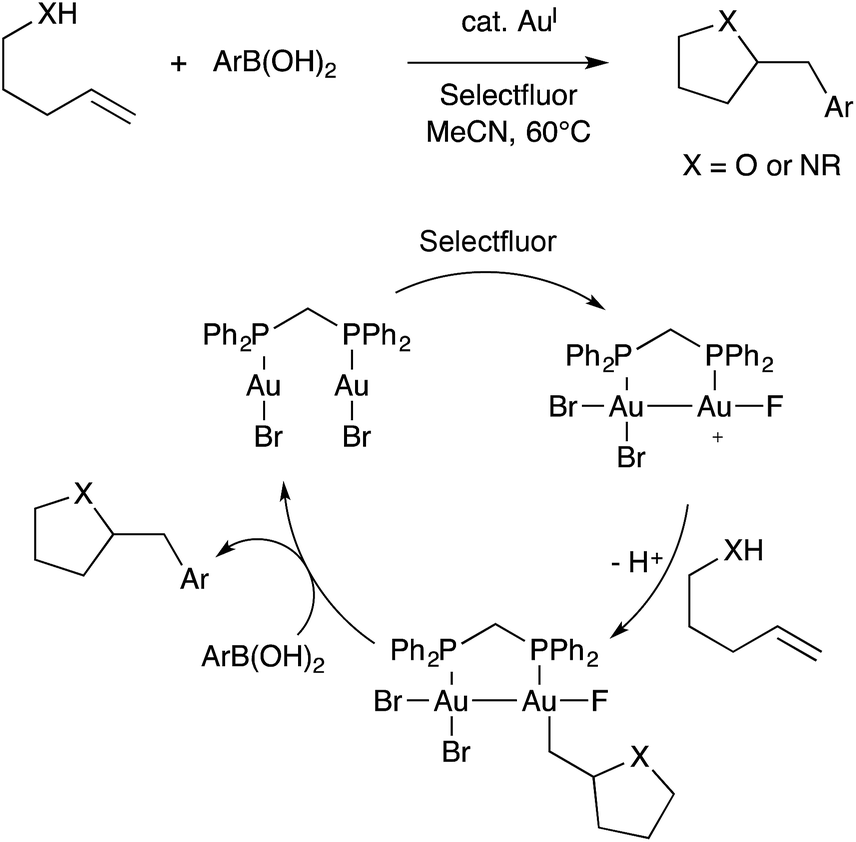 | ||
| Scheme 29 A representative example of oxidative Au catalysis, and the bimetallic mechanism proposed by Toste. | ||
In the Ag-catalyzed fluorination studied by Ritter and the Au-catalyzed heteroarylation studied by Toste, the binuclear coinage metal mechanisms serve to facilitate oxidative catalysis by providing a lower-barrier alternative to the high-potential M(I)/M(III) redox couple. This concept is analogous to the binuclear Pd2 catalysis unveiled by Ritter. This intellectual framework led to the rational design of a binuclear Au(I) catalyst for C(sp3)–C(sp2) coupling with arylboronic acids developed by Toste.86
 | ||
| Scheme 30 Hydroalkylation of alkynes developed by Lalic, and proposed binuclear catalytic mechanism. | ||
3.2 Pre-assembled bimetallic catalysts
The examples in the previous section involve serendipitous bimetallic assembly under catalytic conditions. Beyond such examples, the rational design of bimetallic catalysis involving metal–metal bonds and/or bifunctional metal sites has long been a goal in the organometallic chemistry community. Historically, perhaps the most well-known set of bimetallic catalysts in organic synthesis are the dirhodium(II) carboxylate paddlewheel complexes that catalyze various group transfer reactions that allow for C–C and C–N bond formation.89–91 In addition, the bimetallic species Co2(CO)8 is known to mediate C–C coupling reactions of alkynes such as the Pauson–Khand reaction, although stoichiometric Co is often required.92 Recent developments in these areas highlighted below indicate that pre-assembled bimetallic systems, often featuring metal–metal bonds with or without bridging ligands, are promising candidates for further catalyst development.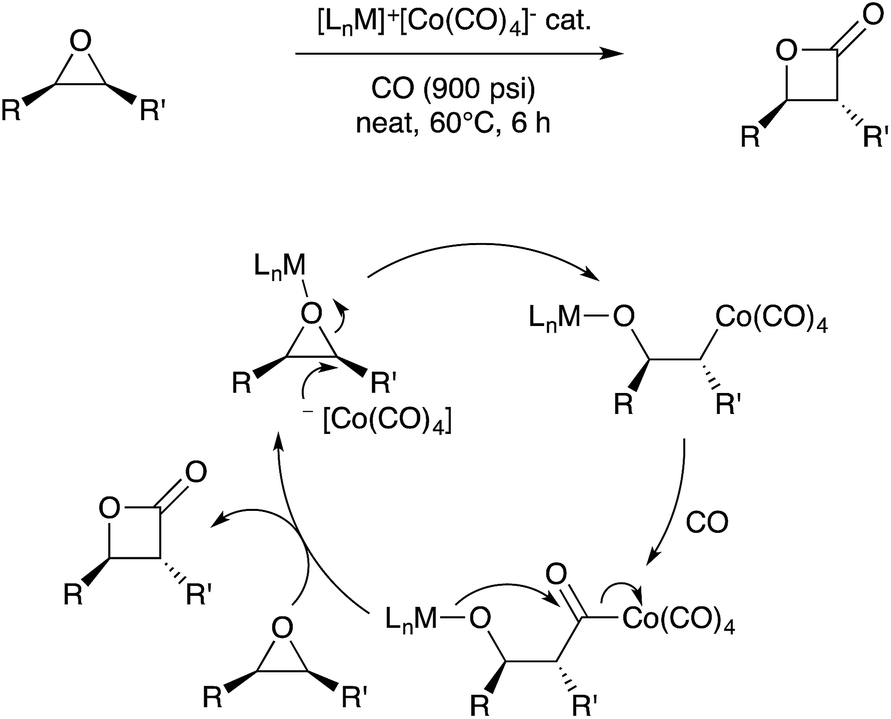 | ||
| Scheme 31 Bifunctional catalysis for epoxide carbonylation developed by Coates. | ||
Beginning in 2013, our group began studying bifunctional catalysts closely related to these heterobimetallic ion pairs. A series of (NHC)Cu–[MCO] catalysts, including derivatives with [MCO] = Co(CO)4, were characterized (NHC = N-heterocyclic carbene).98 Although these complexes do feature Cu–M bonds in the solid state, theoretical analysis indicates that these copper–metal bonds are highly polarized donor/acceptor-type dative interactions. Furthermore, crossover experiments are consistent with equilibrium concentrations of [(NHC)Cu]+[MCO]− pairs forming in solution.99 One derivative, a (NHC)Cu–FeCp(CO)2 species, was shown to catalyse dehydrogenative borylation of unactivated arenes upon photochemical activation (Scheme 32).100 Based on stoichiometric reactivity studies and computational analysis,99 it was proposed that the bifunctional Cu/Fe catalyst reacts with the boron source to generate CpFe(CO)2(Bpin) as the active borylating species (HBpin = pinacolborane). An analogous boryliron complex had previously been shown by Hartwig to mediate stoichiometric C–H borylation.101 The bifunctional Cu/Fe pair is thought to be crucial for cleaving the B–H bond of HBpin in order to generate the active borylating intermediate. Upon C–H borylation, the Cu/Fe catalyst is regenerated through a binuclear H2 elimination that proceeds through a highly polar transition state.102 The bifunctional nature of this series of catalysts is actively being pursued for a variety of other catalytic applications.21,30
 | ||
| Scheme 32 Bifunctional catalysis for C–H borylation developed by Mankad. | ||
 | ||
| Scheme 33 Pd(I)-catalyzed C–X coupling developed by Schoenebeck. | ||
In another Pd-catalyzed C–X coupling process, Nagashima reported that the heterobimetallic cation [Cl2Ti(NtBuPPh2)2Pd(η3-CH2C(CH3)CH2)]+ was capable of extremely efficient allylic amination catalysis.106 Michaelis, Ess, and coworkers conducted a combined experimental/computational mechanistic study on the empirically observed rate acceleration compared to monometallic Pd-allyl cation catalysts.107 A model for rate-determining C–N bond formation was proposed, with the dative Pd → Ti interaction stabilizing Pd(0) character in the transition state and therefore lowering the reductive elimination barrier (Scheme 34). When compared to appropriate monometallic counterparts, rate enhancements of up to 105 were documented in some cases. A Co/Zr-catalyzed Kumada coupling reaction studied by Thomas and coworkers is likely another example of this mechanistic motif.108
 | ||
| Scheme 34 Allylic amination-catalysed by a Pd–Ti heterobimetallic complex, and the proposed rate-determining C–N coupling transition state proposed by Michaelis and Ess. | ||
In part due to the known reactivity of the metal–metal singled bonded Co2(CO)8 towards alkynes and in part due to the potential of using metal–metal multiple bonds to engage unsaturated substrates in pericyclic reactions, bimetallic catalysts have long been examined for catalytic alkyne cyclotrimerisation reactions. This topic was reviewed thoroughly by Mashima recently.109 Among the most exciting advances is a bimetallic Ni catalyst supported by a binucleating naphthyridine-diimine (NDI) ligand that Uyeda and coworkers reported in 2015 is effective at selective and efficient alkyne cyclotrimerisation.110 The metal–metal bonded binuclear catalyst was highly active for coupling terminal alkynes to provide 1,2,4-substituted arene products selectively in preference to 1,3,5-substituted regioisomers or cyclotetramers. On the other hand, related mononuclear Ni catalysts supported by bipyridine, diimine, or pyridylimine ligands were significantly less active and gave complex product mixtures. Based on stoichiometric model reactions, a mechanism was proposed by which alkyne dimerization occurs to yield a nickelacyclopentadiene intermediate, which subsequently couples with a third alkyne equivalent to release arene. The terminal arene-releasing reaction was proposed to be the key selectivity-determining step. Crystallographic and theoretical evidence based on model intermediates was consistent with the cyclic fragment located at the reactive Ni site engaging in a secondary π-interaction with the spectator Ni site. This secondary π-interaction was proposed to stabilise one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and thereby direct the third alkyne substrate towards the uncoordinated CC bond (Scheme 35). Approach of the least hindered carbon centre to this reactive CC moiety thus provides the observed 1,2,4-substituted regioisomer.
C bond and thereby direct the third alkyne substrate towards the uncoordinated CC bond (Scheme 35). Approach of the least hindered carbon centre to this reactive CC moiety thus provides the observed 1,2,4-substituted regioisomer.
 | ||
| Scheme 35 Dinickel-catalysed alkyne cyclotrimerisation developed by Uyeda, and the proposed bimetallic transition state that steers regioselectivity. | ||
4. Conclusions
In conclusion, bimetallic catalysis allows for novel modes of C–C and C–X bond formation to occur. Rate enhancements, selectivity control, and/or non-precious metal chemistry have all been enabled by bimetallic strategies, as summarized in this perspective. A large portion of the bimetallic catalysis literature as applied to C–C and C–X bond formation involves interfacing classical Pd catalysis with catalytically generated organometallic nucleophiles. Furthermore, at this time the preponderance of such examples involve organocopper nucleophiles and therefore descend from the seminal Sonogashira coupling reaction. Catalytic transformations that employ binuclear bond activation/formation steps, particularly by rational design, are comparatively underdeveloped and ripe for exploration.Acknowledgements
Funding support from the NSF (CHE-1362294), the ACS Green Chemistry Institute (Pharmaceutical Roundtable Grant), and the Alfred P. Sloan Foundation (Research Fellowship to N. P. M.) is gratefully acknowledged.Notes and references
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- R. F. Heck, Acc. Chem. Res., 1979, 12, 146–151 CrossRef CAS.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef CAS PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC.
- J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497–5508 CrossRef CAS.
- A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC.
- E. K. van den Beuken and B. L. Feringa, Tetrahedron, 1998, 54, 12985–13011 CrossRef CAS.
- S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2016, 138, 14699–14704 CrossRef CAS PubMed.
- J. Zhang, S.-C. Sha, A. Bellomo, N. Trongsiriwat, F. Gao, N. C. Tomson and P. J. Walsh, J. Am. Chem. Soc., 2016, 138, 4260–4266 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- M. S. Sigman and E. W. Werner, Acc. Chem. Res., 2012, 45, 874–884 CrossRef CAS PubMed.
- T. L. Lohr and T. J. Marks, Nat. Chem., 2015, 7, 477–482 CrossRef CAS PubMed.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef CAS PubMed.
- J. P. McInnis, M. Delferro and T. J. Marks, Acc. Chem. Res., 2014, 47, 2545–2557 CrossRef CAS PubMed.
- M. R. Radlauer, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2012, 134, 1478–1481 CrossRef CAS PubMed.
- M. K. Karunananda and N. P. Mankad, J. Am. Chem. Soc., 2015, 137, 14598–14601 CrossRef CAS PubMed.
- W. Zhou, S. L. Marquard, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Organometallics, 2013, 32, 1766–1772 CrossRef CAS.
- R. C. Cammarota and C. C. Lu, J. Am. Chem. Soc., 2015, 137, 12486–12489 CrossRef CAS PubMed.
- A. Chakraborty, R. G. Kinney, J. A. Krause and H. Guan, ACS Catal., 2016, 6, 7855–7864 CrossRef CAS.
- T. J. Steiman and C. Uyeda, J. Am. Chem. Soc., 2015, 137, 6104–6110 CrossRef CAS PubMed.
- C. W. Machan, J. Yin, S. A. Chabolla, M. K. Gilson and C. P. Kubiak, J. Am. Chem. Soc., 2016, 138, 8184–8193 CrossRef CAS PubMed.
- R. B. Siedschlag, V. Bernales, K. D. Vogiatzis, N. Planas, L. J. Clouston, E. Bill, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2015, 137, 4638–4641 CrossRef CAS PubMed.
- X.-F. Jiang, H. Huang, Y.-F. Chai, T. L. Lohr, S.-Y. Yu, W. Lai, Y.-J. Pan, M. Delferro and T. J. Marks, Nat. Chem., 2016, 9, 188 CrossRef.
- C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 12023–12031 CrossRef CAS PubMed.
- S. Bagherzadeh and N. P. Mankad, J. Am. Chem. Soc., 2015, 137, 10898–10901 CrossRef CAS PubMed.
- J. M. Lee, Y. Na, H. Han and S. Chang, Chem. Soc. Rev., 2004, 33, 302–311 RSC.
- I. G. Powers and C. Uyeda, ACS Catal., 2016, 936–958 CrossRef.
- C. E. Castro, E. J. Gaughan and D. C. Owsley, J. Org. Chem., 1966, 31, 4071–4078 CrossRef CAS.
- L. Cassar, J. Organomet. Chem., 1975, 93, 253–257 CrossRef CAS.
- H. A. Dieck and F. R. Heck, J. Organomet. Chem., 1975, 93, 259–263 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- K. Sonogashira, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS.
- D. S. Laitar, E. Y. Tsui and J. P. Sadighi, Organometallics, 2006, 25, 2405–2408 CrossRef CAS.
- K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 7567–7570 CrossRef CAS PubMed.
- K. B. Smith, K. M. Logan, W. You and M. K. Brown, Chem.–Eur. J., 2014, 20, 12032–12036 CrossRef CAS PubMed.
- K. M. Logan, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 5228–5231 CrossRef CAS PubMed.
- T. Jia, P. Cao, B. Wang, Y. Lou, X. Yin, M. Wang and J. Liao, J. Am. Chem. Soc., 2015, 137, 13760–13763 CrossRef CAS PubMed.
- K. Semba, Y. Ohtagaki and Y. Nakao, Org. Lett., 2016, 18, 3956–3959 CrossRef CAS PubMed.
- K. Semba, K. Ariyama, H. Zheng, R. Kameyama, S. Sakaki and Y. Nakao, Angew. Chem., 2016, 128, 6383–6387 CrossRef.
- S. D. Friis, M. T. Pirnot and S. L. Buchwald, J. Am. Chem. Soc., 2016, 138, 8372–8375 CrossRef CAS PubMed.
- F. Nahra, Y. Macé, D. Lambin and O. Riant, Angew. Chem., Int. Ed., 2013, 52, 3208–3212 CrossRef CAS PubMed.
- S. Vercruysse, L. Cornelissen, F. Nahra, L. Collard and O. Riant, Chem.–Eur. J., 2014, 20, 1834–1838 CrossRef CAS PubMed.
- J. Huang, J. Chan, Y. Chen, C. J. Borths, K. D. Baucom, R. D. Larsen and M. M. Faul, J. Am. Chem. Soc., 2010, 132, 3674–3675 CrossRef CAS PubMed.
- M. Lesieur, F. Lazreg and C. S. J. Cazin, Chem. Commun., 2014, 50, 8927–8929 RSC.
- S. Tang, S.-H. Li, Y. Nakao and T. Hiyama, Asian J. Org. Chem., 2013, 2, 416–421 CrossRef CAS.
- A. Ohgi, K. Semba, T. Hiyama and Y. Nakao, Chem. Lett., 2016, 45, 973–975 CrossRef CAS.
- F. Rudolphi, B. Song and L. J. Goossen, Adv. Synth. Catal., 2011, 353, 337–342 CrossRef CAS.
- F. Collet, B. Song, F. Rudolphi and L. J. Goossen, Eur. J. Org. Chem., 2011, 6486–6501 CrossRef CAS.
- Y. Shi, K. E. Roth, S. D. Ramgren and S. A. Blum, J. Am. Chem. Soc., 2009, 131, 18022–18023 CrossRef CAS PubMed.
- J. J. Hirner, K. E. Roth, Y. Shi and S. A. Blum, Organometallics, 2012, 31, 6843–6850 CrossRef CAS PubMed.
- A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Ackermann, J. De Buck Becker, M. Rudolph, C. Scholz and F. Rominger, Adv. Synth. Catal., 2012, 354, 133–147 CrossRef CAS.
- P. García-Domínguez and C. Nevado, J. Am. Chem. Soc., 2016, 138, 3266–3269 CrossRef PubMed.
- M. Sawamura, M. Sudoh and Y. Ito, J. Am. Chem. Soc., 1996, 118, 3309–3310 CrossRef CAS.
- S. Ko, C. Lee, M.-G. Choi, Y. Na and S. Chang, J. Org. Chem., 2003, 68, 1607–1610 CrossRef CAS PubMed.
- Z.-S. Chen, L.-Z. Huang, H. J. Jeon, Z. Xuan and S.-G. Lee, ACS Catal., 2016, 6, 7855–7864 CrossRef.
- B. M. Trost and X. Luan, J. Am. Chem. Soc., 2011, 133, 1706–1709 CrossRef CAS PubMed.
- B. M. Trost, X. Luan and Y. Miller, J. Am. Chem. Soc., 2011, 133, 12824–12833 CrossRef CAS PubMed.
- Y. Zhao and D. J. Weix, J. Am. Chem. Soc., 2015, 137, 3237–3240 CrossRef CAS PubMed.
- L. K. G. Ackerman, M. M. Lovell and D. J. Weix, Nature, 2015, 524, 454–457 CrossRef CAS PubMed.
- L. J. Goossen, P. P. Lange, N. Rodríguez and C. Linder, Chem.–Eur. J., 2010, 16, 3906–3909 CrossRef CAS PubMed.
- L. J. Goossen, G. Deng and L. M. Levy, Science, 2006, 313, 662–664 CrossRef CAS PubMed.
- Y. Gu, X. Leng and Q. Shen, Nat. Commun., 2014, 5, 5405 CrossRef CAS PubMed.
- B. Chen and S. Ma, Chem.–Eur. J., 2011, 17, 754–757 CrossRef CAS PubMed.
- E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS PubMed.
- K. B. Hansen and J. L. Leighton, J. Am. Chem. Soc., 1996, 118, 10924–10925 CrossRef CAS.
- B. Wu, J. R. Parquette and T. V. RajanBabu, Science, 2009, 326, 1662 CrossRef CAS PubMed.
- B. Wu, J. C. Gallucci, J. R. Parquette and T. V. RajanBabu, Chem. Sci., 2014, 5, 1102–1116 RSC.
- S. Matsunaga and M. Shibasaki, Chem. Commun., 2014, 50, 1044–1057 RSC.
- S. Matsunaga and M. Shibasaki, Synthesis, 2013, 45, 421–437 CrossRef CAS.
- J. Park and S. Hong, Chem. Soc. Rev., 2012, 41, 6931–7013 RSC.
- S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed.
- D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302–309 CrossRef CAS PubMed.
- D. C. Powers, D. Benitez, E. Tkatchouk, W. A. Goddard III and T. Ritter, J. Am. Chem. Soc., 2010, 132, 14092–14103 CrossRef CAS PubMed.
- D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840–850 CrossRef CAS PubMed.
- D. C. Powers, E. Lee, A. Ariafard, M. S. Sanford, B. F. Yates, A. J. Canty and T. Ritter, J. Am. Chem. Soc., 2012, 134, 12002–12009 CrossRef CAS PubMed.
- G. J. Chuang, W. Wang, E. Lee and T. Ritter, J. Am. Chem. Soc., 2011, 133, 1760–1762 CrossRef CAS PubMed.
- P. Tang, T. Furuya and T. Ritter, J. Am. Chem. Soc., 2010, 132, 12150–12154 CrossRef CAS PubMed.
- M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem.–Eur. J., 2011, 17, 8248–8262 CrossRef CAS PubMed.
- W. E. Brenzovich Jr, D. Benitez, A. D. Lackner, H. P. Shunatona, E. Tkatchouk, W. A. Goddard III and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 5519–5522 CrossRef PubMed.
- E. Tkatchouk, N. P. Mankad, D. Benitez, W. A. Goddard and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 14293–14300 CrossRef CAS PubMed.
- M. D. Levin and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 6211–6215 CrossRef CAS PubMed.
- M. R. Uehling, A. M. Suess and G. Lalic, J. Am. Chem. Soc., 2015, 137, 1424–1427 CrossRef CAS PubMed.
- A. M. Suess, M. R. Uehling, W. Kaminsky and G. Lalic, J. Am. Chem. Soc., 2015, 137, 7747–7753 CrossRef CAS PubMed.
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed.
- H. M. L. Davies and M. S. Long, Angew. Chem., Int. Ed., 2005, 44, 3518–3520 CrossRef CAS PubMed.
- K. P. Kornecki, J. F. Briones, V. Boyarskikh, F. Fullilove, J. Autschbach, K. E. Schrote, K. M. Lancaster, H. M. L. Davies and J. F. Berry, Science, 2013, 342, 351–354 CrossRef CAS PubMed.
- J. Blanco-Urgoiti, L. Añorbe, L. Pérez-Serrano, G. Domínguez and J. Pérez-Castells, Chem. Soc. Rev., 2004, 33, 32–42 RSC.
- D. W. Stephan, Science, 2016, 354, 1248 CrossRef CAS PubMed.
- L. H. Gade, Angew. Chem., Int. Ed., 2000, 39, 2658–2678 CrossRef CAS PubMed.
- J. A. R. Schmidt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 11426–11435 CrossRef CAS PubMed.
- Y. D. Y. L. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1174–1175 CrossRef CAS PubMed.
- M. Mulzer, B. T. Whiting and G. W. Coates, J. Am. Chem. Soc., 2013, 135, 10930–10933 CrossRef CAS PubMed.
- S. Banerjee, M. K. Karunananda, S. Bagherzadeh, U. Jayarathne, S. R. Parmelee, G. W. Waldhart and N. P. Mankad, Inorg. Chem., 2014, 53, 11307–11315 CrossRef CAS PubMed.
- S. R. Parmelee, T. J. Mazzacano, Y. Zhu, N. P. Mankad and J. A. Keith, ACS Catal., 2015, 5, 3689–3699 CrossRef CAS.
- T. J. Mazzacano and N. P. Mankad, J. Am. Chem. Soc., 2013, 135, 17258–17261 CrossRef CAS PubMed.
- K. M. Waltz, X. He, C. Muhoro and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 11357–11358 CrossRef CAS.
- M. K. Karunananda and N. P. Mankad, Organometallics, 2017, 36, 220–227 CrossRef CAS.
- K. J. Bonney, F. Proutiere and F. Schoenebeck, Chem. Sci., 2013, 4, 4434–4439 RSC.
- G. Yin, I. Kalvet and F. Schoenebeck, Angew. Chem., Int. Ed., 2015, 54, 6809–6813 CrossRef CAS PubMed.
- M. Aufiero, T. Sperger, A. S. K. Tsang and F. Schoenebeck, Angew. Chem., Int. Ed., 2015, 54, 10322–10326 CrossRef CAS PubMed.
- H. Tsutsumi, Y. Sunada, Y. Shiota, K. Yoshizawa and H. Nagashima, Organometallics, 2009, 28, 1988–1991 CrossRef CAS.
- W. K. Walker, B. M. Kay, S. A. Michaelis, D. L. Anderson, S. J. Smith, D. H. Ess and D. J. Michaelis, J. Am. Chem. Soc., 2015, 137, 7371–7378 CrossRef CAS PubMed.
- W. Zhou, J. W. Napoline and C. M. Thomas, Eur. J. Inorg. Chem., 2011, 2029–2033 CrossRef CAS.
- K. Yamamoto, H. Nagae, H. Tsurugi and K. Mashima, Dalton Trans., 2016, 45, 17072–17081 RSC.
- S. Pal and C. Uyeda, J. Am. Chem. Soc., 2015, 137, 8042–8045 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |