
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Family-level stereoselective synthesis and biological evaluation of pyrrolomorpholine spiroketal natural product antioxidants†
Alyssa L.
Verano
 a and
Derek S.
Tan
*ab
a and
Derek S.
Tan
*ab
aPharmacology Graduate Program, Weill Cornell Graduate School of Medical Sciences, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, Box 422, New York, NY 10065, USA. E-mail: tand@mskcc.org
bChemical Biology Program and Tri-Institutional Research Program, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, Box 422, New York, NY 10065, USA
First published on 15th March 2017
Abstract
The pyranose spiroketal natural products pollenopyrroside A and shensongine A (also known as xylapyrroside A, ent-capparisine B) have been synthesized by stereoselective spirocyclizations of a common C1-functionalized glycal precursor. In conjunction with our previously reported syntheses of the corresponding furanose isomers, this provides a versatile family-level synthesis of the pyrrolomorpholine spiroketal natural products and analogues. In rat mesangial cells, hyperglycemia-induced production of reactive oxygen species, which is implicated in diabetic nephropathy, was inhibited by pollenopyrroside A and shensongine A with mid-μM IC50 values, while unnatural C2-hydroxy analogues exhibited more potent, sub-μM activity.
Introduction
Pyrrolomorpholine spiroketals are a novel family of natural products that include both pyranose and furanose isomeric forms and both epimeric configurations at the anomeric carbon (Fig. 1). In 2010, three groups contemporaneously isolated acortatarins A (3) and B (6) from the rhizome of Acorus tatarinowii,1 pollenopyrrosides A (1) and B (3) from the bee-collected pollen of Brassica campestris,2 and capparisines A (3) and B (ent-2) from the mature fruit of Capparis spinosa.3,4 Acortatarin A and a pyranose isomer named acortatarin C (stereochemistry not assigned) were also isolated as bitter components of whole wheat bread crust.5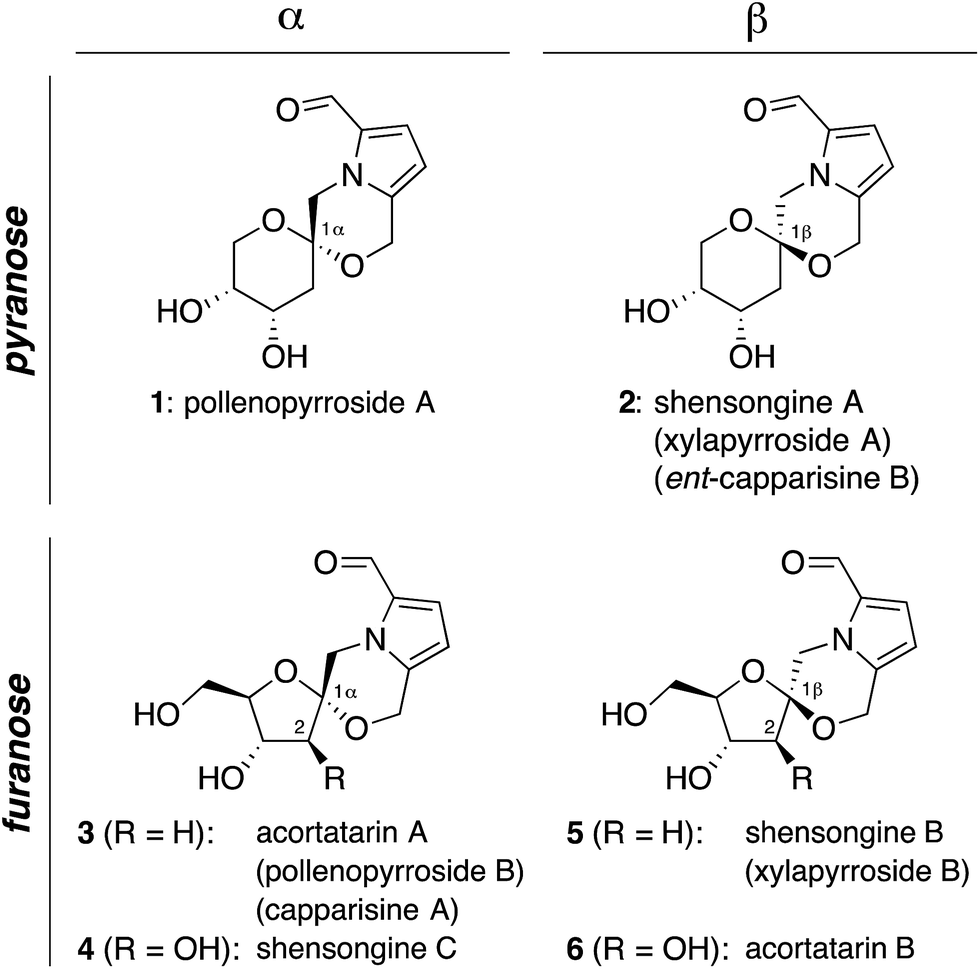 | ||
| Fig. 1 Pyrrolomorpholine spiroketal family of natural products. Structures reflect certain stereochemical revisions made subsequent to the initial isolation reports.4 Identical structures isolated from different natural sources and given different names (parentheses) are referred to herein by the first published name. | ||
More recently, the epimeric β-spiroketals shensongine A (2) and shensongine B (5) were isolated from capsules of the antiarrhythmic Chinese herbal medicine Shensong Yangxin, along with shensongine C (4), a C2-hydroxy congener of acortatarin A, and pollenopyrroside B (3).6 Contemporaneously, the same β-spiroketals xylapyrrosides A (2) and B (5), were isolated from the fungus Xylaria nigripes.7
The plant-derived sources of these natural products have been used in traditional Chinese medicines for the treatment of a variety of diseases.1–3,6,7 Notably, acortatarins A and B exhibit antioxidant activity in a diabetic renal cell model, inhibiting production of reactive oxygen species (ROS) and significantly attenuating hyperglycemia-induced activation of NADPH oxidase and extracellular matrix production,1,8 hallmarks of diabetic nephropathy.9a–c The xylapyrrosides also have moderate antioxidant effects and inhibit t-butyl hydroperoxide-induced cytotoxicity in rat vascular smooth muscle cells.7 Importantly, oxidative stress has been shown to play a critical role in the pathogenesis of a wide variety of disease states,9 due to the ability of excess ROS to inflict direct damage to essential macromolecules, as well as to lead to aberrant cell signaling.10,11 Accordingly, these natural products are of interest based on their therapeutic potential to treat diabetic nephropathy, cardiovascular diseases, and other pathologies in which oxidative stress is implicated.9 However, due to the limited quantities available from the natural sources,12 efficient synthetic access is required for in-depth biological and structure–activity relationship (SAR) studies. As a result, this family of natural products has attracted considerable attention from the synthetic community.7,13 We have previously reported concise, highly stereoselective syntheses of the furanose spiroketals acortatarins A (3) and B (6) and shensongine C (4) using stereoselective spirocyclizations of glycal precursors.14 Herein, we report expansion of this modular approach to a family-level synthesis, providing the naturally-occurring pyranose spiroketals pollenopyrroside A (1) and shensongine A (2, xylapyrroside A, ent-capparisine B), as well as corresponding C2-hydroxy analogues, and evaluation of the in vitro antioxidant activity of these compounds.
Results and discussion
Retrosynthetic analysis of pyrrolomorpholine spiroketal natural products
The isomeric nature of the pyrrolomorpholine spiroketal natural products presents an attractive opportunity to develop a family-level synthesis that would provide access to both furanose and pyranose congeners, as well as both anomeric stereoisomers.15 Our laboratory has a long-standing interest in the stereocontrolled synthesis of spiroketals from glycals, with a particular focus on natural product scaffolds for use in probe and drug discovery.16 Analogous to our approach to the furanose acortatarins,14 we envisioned that syntheses of the corresponding pyranose isomers could be achieved by stereocontrolled spirocyclization of a pyranoglycal intermediate 7 (Fig. 2). Retrosynthetically, this glycal intermediate 7 would originate from coupling of pyrrole-2,5-dicarboxaldehyde (8) with pyranoarabinal (pyranoribal) derivative 9, both of which can be accessed from commercially-available starting materials (10, 11). | ||
| Fig. 2 Retrosynthetic strategy via glycal cyclization precursor 7. | ||
Synthesis of shensongine A via acid-catalyzed spirocyclization of pyranoarabinal intermediate 16
Thus, we synthesized pyrrole-2,5-dicarboxaldehyde (8) as previously reported.14 To access the pyranoglycal coupling partner 14, commercially available 3,4-di-O-acetyl-D-arabinal (11) was deacetylated17 then silylated18 to provide triisopropylsilyl-protected glycal 12 (Fig. 3). This protecting group change was necessary for compatibility with downstream C1-lithiation and also facilitated silica gel purification of intermediates. C1-Formylation19 of glycal 12 followed by aldehyde reduction provided protected C1-hydroxymethyl arabinal 13. By analogy to our use of the corresponding furanose C1-iodomethyl glycal in our synthesis of the acortatarins,14 we proceeded with iodination of 13 to give the pyranose C1-iodomethyl glycal 14a. However, attempted coupling of the iodide 14a with pyrrole 8 under basic conditions led to poor yields of pyrrolomethylglycal 15, due to the susceptibility of the C1-iodomethyl arabinal 14a to decomposition. This is in contrast to the observed stability of the corresponding furanose C1-iodomethyl glycal in our acortatarin syntheses.14 However, we were able to achieve efficient coupling by instead using the more stable mesylate 14b, which provided the desired coupling product 15 in 71% yield. Aqueous base was required for this N-alkylation due to the tendency of pyrrole monomers to dimerize in nonaqueous conditions.20 Monoreduction of the dialdehyde21 with NaBH4 gave the pivotal spirocyclization precursor 16, from which pollenopyrroside A and shensongine A would be accessed. Careful monitoring of the reaction allowed for minimal over-reduction and minimal remaining starting material. | ||
| Fig. 3 Synthesis of pivotal arabinal intermediate 16. DMF = N,N-dimethylformamide; THF = tetrahydrofuran; TIPS = triisopropylsilyl. | ||
Treatment of arabinal 16 with catalytic Brønsted acids led to exclusive formation of the thermodynamic β-spiroketal 17 (Fig. 4). Desilylation furnished shensongine A (2, xylapyrroside A, ent-capparisine B), whose optical rotation, NMR, and high-resolution mass spectral data matched those reported for the natural products.22
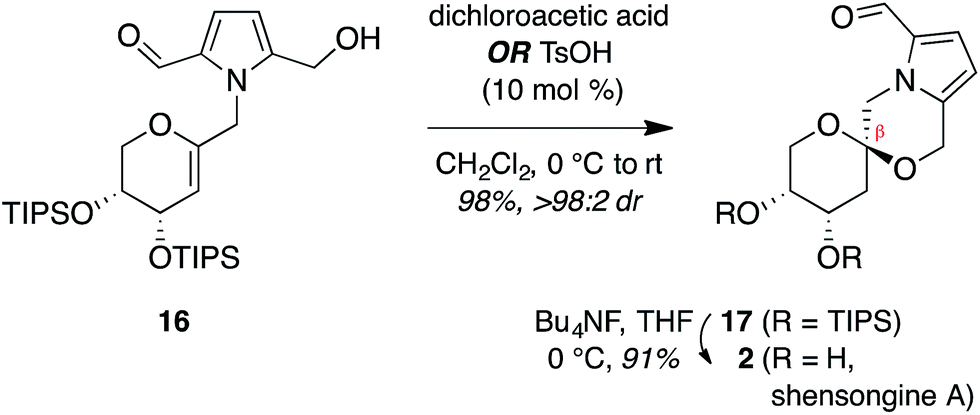 | ||
| Fig. 4 Synthesis of shensongine A (2, xylapyrroside A, ent-capparisine B). | ||
Attempted synthesis of pollenopyrroside A via mercury-mediated spirocyclization of arabinal intermediates 16 and 18
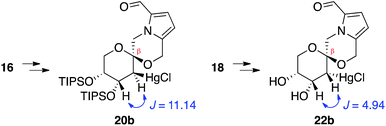
In contrast, stereoselective access to the α-anomer, pollenopyrroside A, proved to be more challenging due to the inherent thermodynamic preferences of this [6,6]-spiroketal system. Formation of the contrathermodynamic α-anomer was overwhelmingly disfavored under numerous spirocyclization conditions evaluated.22 By analogy to our previous work in the corresponding furanoribal-derived [6,5]-spiroketal system,14 we first attempted Hg-mediated spirocyclization of pyranoarabinal 16, expecting preferential anti-mercuration to form β-mercurinium intermediate 19, followed by stereoinvertive cyclization to form an α-spiroketal intermediate (not shown), which could be reduced to the desired α-spiroketal 23 (Fig. 5). However, under these conditions, we instead observed exclusive formation of the β-spiroketal 17. We attributed this undesired selectivity to steric conflicts between the bulky 3- and/or 4-O-TIPS groups and the morpholine ring en route to the α-anomer 23. In addition, the 3-O-TIPS protecting group would preclude an intramolecular hydrogen bond with the morpholine oxygen suggested by the crystal structure of pollenopyrroside A,2 which may stabilize the α-configuration. To avoid these issues, we attempted Hg-mediated spirocyclization with the fully deprotected glycal substrate 18. However, the undesired β-anomer 2 was again formed exclusively. We attributed this undesired stereoselectivity to syn-mercuration directed by the free C3-hydroxyl group to form the corresponding α-mercurinium intermediate 21, followed by stereoinvertive cyclization to β-spiroketal intermediate 22a. Isolation and NMR analysis of the intermediate 2-mercurial spiroketals as the corresponding chlorides 20b and 22b provided stereochemical assignments consistent with these mechanistic interpretations.23 Use of other mercury salts and less-hindered protecting groups, including a conformationally restricted cyclic carbonate, did not improve stereoselectivity (Table S1†).22 | ||
| Fig. 5 Attempted synthesis of pollenopyrroside A (1) via mercury-mediated spirocyclization. HMDS = hexamethyldisilazide. | ||
Synthesis of pollenopyrroside A via metal-catalyzed spirocyclization of arabinal intermediate 18
Based on the intramolecular hydrogen bond between the 3-hydroxyl group and morpholine oxygen postulated from the crystal structure of pollenopyrroside A,2 we next pursued a chelation-based approach to favor formation of the desired α-spiroketal (Table 1). Such metal-chelation approaches have been used previously to access contrathermodynamic spiroketals.24 Treatment of β-spiroketal 2 with various metal salts at reflux in CH3CN or dioxane over 24 h had no effect upon the α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β ratio (Table S2†).22 Treatment of the glycal spirocyclization precursor 18 with MgCl2, ZnCl2, or Ti(O-iPr)4 led to no reaction or decomposition (Table 1, entries 1–3). However, treatment of glycal 18 with Sc(OTf)3 led to a 60:40 ratio of diastereomeric spiroketal products, favoring the desired α-anomer 1 (entry 4). We have recently used Sc(OTf)3 in stereoselective spirocyclizations of exo-glycal epoxides to form benzannulated spiroketals.16e In those reactions, solvent played a dramatic role, with the catalyst acting as a Lewis acid in THF but as a mild source of triflic acid in CH2Cl2.25 To assess the mechanistic basis for the observed selectivity in this substrate system, we carried out a control experiment with DTBMP (2,6-di-t-butyl-4-methylpyridine) as an acid scavenger, but no reaction occurred (entry 5), suggesting that the catalyst might, indeed, be serving as a triflic acid source. However, when the reaction was carried out with TfOH alone, we observed complete diastereoselectivity for the undesired β-spiroketal (2) (entry 6). Moreover, no reaction was observed upon treatment with ScCl3 alone (entry 7). In contrast, treatment of glycal 18 with both ScCl3 and TfOH together (entry 8) recapitulated the 60:40 dr observed with Sc(OTf)3 (entry 4). Notably, combination of MgCl2 or ZnCl2 with TfOH also afforded a 50:50 mixture of the two diastereomeric spiroketals 1 and 2. Taken together, these results suggest that the α-spiroketal 1 is formed under kinetic control, and that both a Lewis acid and Brønsted acid are required to overcome the inherent complete selectivity for the β-spiroketal 2. This is in contrast to our previous study with exo-glycal epoxides, in which Sc(OTf)3 served as either a Lewis acid or a Brønsted acid source exclusively, depending upon solvent selection.16e Unfortunately, the diastereomeric mixture was difficult to separate, resulting in only a 25% isolated yield of pollenopyrroside A (1).
:β ratio (Table S2†).22 Treatment of the glycal spirocyclization precursor 18 with MgCl2, ZnCl2, or Ti(O-iPr)4 led to no reaction or decomposition (Table 1, entries 1–3). However, treatment of glycal 18 with Sc(OTf)3 led to a 60:40 ratio of diastereomeric spiroketal products, favoring the desired α-anomer 1 (entry 4). We have recently used Sc(OTf)3 in stereoselective spirocyclizations of exo-glycal epoxides to form benzannulated spiroketals.16e In those reactions, solvent played a dramatic role, with the catalyst acting as a Lewis acid in THF but as a mild source of triflic acid in CH2Cl2.25 To assess the mechanistic basis for the observed selectivity in this substrate system, we carried out a control experiment with DTBMP (2,6-di-t-butyl-4-methylpyridine) as an acid scavenger, but no reaction occurred (entry 5), suggesting that the catalyst might, indeed, be serving as a triflic acid source. However, when the reaction was carried out with TfOH alone, we observed complete diastereoselectivity for the undesired β-spiroketal (2) (entry 6). Moreover, no reaction was observed upon treatment with ScCl3 alone (entry 7). In contrast, treatment of glycal 18 with both ScCl3 and TfOH together (entry 8) recapitulated the 60:40 dr observed with Sc(OTf)3 (entry 4). Notably, combination of MgCl2 or ZnCl2 with TfOH also afforded a 50:50 mixture of the two diastereomeric spiroketals 1 and 2. Taken together, these results suggest that the α-spiroketal 1 is formed under kinetic control, and that both a Lewis acid and Brønsted acid are required to overcome the inherent complete selectivity for the β-spiroketal 2. This is in contrast to our previous study with exo-glycal epoxides, in which Sc(OTf)3 served as either a Lewis acid or a Brønsted acid source exclusively, depending upon solvent selection.16e Unfortunately, the diastereomeric mixture was difficult to separate, resulting in only a 25% isolated yield of pollenopyrroside A (1).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Equiv. | dr (α:β)a |
| a Determined by 1H-NMR. b n.r. = no reaction. c Decomp. = decomposition of starting material. d 25% isolated yield of α-spiroketal 1. e 1.0 equiv. DTBMP. f 0.5 equiv. TfOH; DTBMP = 2,6-di-tert-butyl-4-methylpyridine. | |||
| 1 | MgCl2 | 3.0 | n.r.b |
| 2 | ZnCl2 | 3.0 | n.r. |
| 3 | Ti(O-iPr)4 | 2.0 | Decomp.c |
| 4 | Sc(OTf)3 | 3.0 | 60:40d |
| 5 | Sc(OTf)3 + DTBMPe | 3.0 | n.r. |
| 6 | TfOH | 0.2 | 0:100 |
| 7 | ScCl3 | 3.0 | n.r. |
| 8 | ScCl3 + TfOHf | 3.0 | 60:40 |
| 9 | MgCl2 + TfOHf | 3.0 | 50:50 |
| 10 | ZnCl2 + TfOHf | 3.0 | 50:50 |

Synthesis of pollenopyrroside A via methanol-catalyzed kinetic spirocyclization of arabinal intermediate 16
Thus, to provide more stereoselective access to pollenopyrroside A (1), we turned to our previously reported methanol-catalyzed kinetic spirocyclization of glycal epoxides.16a,d While we recognized that this approach would introduce the need for deoxygenation of the resulting 2-hydroxyl group, this was balanced by the much higher stereoselectivity expected with this spirocyclization. In addition, this epoxidation–spirocyclization strategy would provide access to the non-natural 2-hydroxypyranose analogues for biological evaluation, complementing the known 2-hydroxy natural products in the furanose series (4, 6). Accordingly, anti-epoxidation of glycal 16 with DMDO was followed by spirocyclization with inversion of configuration in methanol to give the desired α-spiroketal 24 in >98:2 dr (Fig. 6a). Desilylation of 24 provided a 2-hydroxy analogue of pollenopyrroside A, 25, for biological evaluation below. Alternatively, Barton–McCombie deoxygenation26 of 24 at the 2-position and desilylation provided pollenopyrroside A (1) in 46% yield over four steps from glycal precursor 16.
 | ||
| Fig. 6 (a) Synthesis of pollenopyrroside A via MeOH-catalyzed kinetic spirocyclization followed by 2-deoxygenation. (b) Synthesis of 2-hydroxy analogue of shensongine A via Ti(O-iPr)4-mediated kinetic cyclization. (c) Acid equilibration experiments indicate that β-spiroketal shensongine A (2) is thermodynamically favored over α-epimer pollenopyrroside A (1) in aqueous acid (pD ≤ 4) while 2-hydroxy analogues 25 and 28 do not equilibrate down to pD 1. DMDO = dimethyldioxirane; AIBN = azobisisobutyronitrile. | ||
For completeness, we also carried out a stereocomplementary spirocyclization of the glycal epoxide derived from 16 using our previously established Ti(O-iPr)4-catalyzed kinetic spirocyclization with retention of configuration16b to provide the corresponding β-spiroketal 27 with complete β-diastereoselectivity (Fig. 6b). Desilylation then provided the 2-hydroxy analogue of shensongine A, 28, for biological evaluation below.
Acid equilibration studies of natural products 1 and 2 and 2-hydroxy analogues 25 and 28
Acid equilibration experiments under aqueous conditions showed that pollenopyrroside A (1) was kinetically stable at pD ≥ 5, but underwent complete conversion to its C1-epimer shensongine A (2) at pD ≤ 4. Conversely, shensongine A was stable down to pD 1, indicating that this β-spiroketal is thermodynamically favored (Fig. 6c). In the 2-hydroxypyranose series, both 25 and its C1-epimer 28 were kinetically stable down to pD 1. Accordingly, epimerization would not be expected for any of these compounds in biological assays under physiologic conditions at pH 7.4.Evaluation of antioxidant activity of pyrrolomorpholine spiroketals
Previous reports have demonstrated the antioxidant activity of acortatarin A against high glucose-induced oxidative stress in rat mesangial cells1,8 suggesting that this scaffold may have therapeutic potential for the treatment of diabetic nephropathy. Thus, we evaluated the antioxidant activity of the complete D-enantiomeric family of furanose and pyranose pyrrolomorpholine spiroketal natural products and analogues. Intracellular ROS levels were measured using the cell-permeable 2′,7′-dichlorofluorescein diacetate (DCFH-DA), which is converted to the highly fluorescent 2′,7′-dichlorofluorescein (DCF) upon deacetylation by intracellular esterases and oxidation.27,28 Rat mesangial cells are highly sensitive to hyperglycemic conditions, and a significant increase in ROS was observed after 3 h exposure to 30 mM D-glucose.22 Inhibition of high glucose-induced oxidative stress was evaluated by concomitant treatment with the pyrrolomorpholine spiroketals, or with 1 mM N-acetylcysteine, a precursor of the antioxidant glutathione, as a positive control.29Consistent with the previous report,1 acortatarins A (3) and B (6) reduced high glucose-induced ROS generation in a dose-dependent manner, with IC50 values of 4.6 and 11 μM, respectively, returning ROS levels to that of normal glucose conditions (Table 2, entries 2, 5). Compared to acortatarin A (3), the β-spiroketal shensongine B (5) was 4-fold less potent (entries 2, 3), suggesting that the spiroketal stereocenter may be important for activity. Consistent with this trend, the α-spiroketal shensongine C (4) was also more potent than its β-spiroketal congener acortatarin B (6) (entries 4, 5).
| Entry | Compound | IC50 (μM) [s.d. log IC50]b | Maximum % ROS inhibitionc |
|---|---|---|---|
| a Cells were treated with compound (0–3 mM) under normal (5.6 mM) or high (30 mM) glucose conditions and overall ROS levels were measured using the fluorescent probe DCFH-DA (50 μM).22 b Data are expressed as geometric mean IC50 (antilog [mean log IC50]) of three independent experiments, each performed in triplicate, with the standard deviation of the log IC50 shown in brackets. c Statistical significance relative to untreated cells under high-glucose conditions was assessed using a two-tailed unpaired Student t-test with 95% confidence intervals; ***p ≤ 0.001. d N-Acetylcysteine (1 mM) was used as a positive control. | |||
| 1 | N-Acetylcysteined | n.a. | 100%*** |
| 2 | Acortatarin A (3) | 4.6 [0.15] | 100%*** |
| 3 | Shensongine B (5) | 19 [0.05] | 100%*** |
| 4 | Shensongine C (4) | 4.8 [0.07] | 100%*** |
| 5 | Acortatarin B (6) | 11 [0.07] | 100%*** |
| 6 | Pollenopyrroside A (1) | 17 [0.01] | 100%*** |
| 7 | Shensongine A (2) | 11 [0.05] | 100%*** |
| 8 | 2-OH pollenopyrroside A (25) | 0.52 [0.04] | 80%*** |
| 9 | 2-OH shensongine A (28) | 0.27 [0.04] | 60%*** |
In the pyranose spiroketal series, both pollenopyrroside A (1) and shensongine A (2) also reduced ROS in a dose-dependent manner, with comparable IC50 values of 17 and 11 μM, respectively, returning ROS levels to that of normal glucose conditions (entries 6, 7). Notably, the corresponding 2-hydroxy analogues 25 and 28 were much more potent inhibitors, with IC50 values of 0.52 and 0.27 μM, respectively, albeit affording only partial rescues (80% and 60% maximum inhibition, respectively). Surprisingly, these analogues exhibited U-shaped dose–response curves with ROS levels actually increasing at higher concentrations (≥100 μM and ≥30 μM, respectively),22 possibly due to as yet undetermined feedback, off-target, or non-specific mechanisms.30 None of the other compounds exhibited U-shaped dose–response curves up to the maximum concentration tested (3000 μM), however, as these compounds have 9- to 70-fold higher IC50 values than 25 and 28, such effects cannot be ruled out at even higher concentrations.
Conclusions
In conclusion, we have developed an efficient, stereoselective, family-level synthesis of the pyrrolomorpholine spiroketal natural products, providing access to pollenopyrroside A (1) and shensongine A (2), as well as the corresponding 2-hydroxy analogues (25, 28), from the common pyranoarabinal intermediate 16. Pollenopyrroside A was synthesized in 11 steps and 15% overall yield from 3,4-di-O-acetyl-D-arabinal, and shensongine A was accessed in 9 steps and 28% overall yield. Complete diastereoselectivity was achieved for the key spiroketal-forming steps toward both natural products, as well as the 2-hydroxy congeners. This compares favorably to previous syntheses,7,13f,g,31 and provides practical access to the natural products and a variety of analogues. Indeed, while the synthesis of pollenopyrroside A involved an epoxidation–cyclization strategy that required subsequent C2-deoxygenation to afford the natural product, this route also provided convenient access to the corresponding 2-hydroxy analogues, which have not yet been described in the pyranose series but are known as natural products in the isomeric furanose series (i.e.: 4, 6). Evaluation of antioxidant activity against hyperglycemia-induced ROS in rat mesangial cells indicated similar activities for the anomeric natural products pollenopyrroside A (1) and shensongine A (2). Strikingly, the 2-hydroxy analogues 25 and 28 exhibited 30- to 40-fold lower IC50 values compared to the natural products, albeit with incomplete ROS inhibition due to U-shaped dose response curves. Notably, in the furanose series, the α-spiroketals acortatarin A (3) and shensongine C (4) were considerably more potent than the β-anomers, shensongine B (5) and acortatarin B (6), although this trend did not extend to the pyranose series (cf.1, 2). Overall, this family-level synthesis allowed the first direct comparison of the antioxidant activities of the entire D-enantiomeric series of furanose and pyranose isomers, and led to the discovery of novel 2-hydroxy analogues with potent, sub-μM IC50 values. These more potent analogues may be useful for mechanistic studies, including target identification efforts. Further investigation into the mechanisms of action of these compounds and additional analogues are ongoing and will be reported in due course.Acknowledgements
We thank Prof. Anthony Sauve (Weill Cornell Medicine) for helpful discussions and access to cell culture facilities, Dr Kristin Kirschbaum (University of Toledo) for helpful discussions, and Dr George Sukenick and Dr Rong Wang, (MSK) for expert NMR and mass spectral support. Financial support from the NIH (P41 GM076267 to D. S. T.; T32 CA062948-Gudas to A. L. V., CCSG P30 CA008748 to C. B. Thompson) and PhRMA Foundation (predoctoral fellowship to A. L. V.) is gratefully acknowledged.Notes and references
- X. Tong, L. Zhou, Y. Wang, C. Xia, Y. Wang, M. Liang, F. Hou and Y. Cheng, Org. Lett., 2010, 12, 1844–1847 CrossRef CAS PubMed.
- J. Guo, Z. Feng, Y. Yang and Z. Zhang, Chem. Pharm. Bull., 2010, 58, 983–985 CrossRef CAS PubMed.
- T. Yang, C. Wang, G. Chou, T. Wu, X. Cheng and Z. Wang, Food Chem., 2010, 123, 705–710 CrossRef CAS.
- Several of the original stereochemical assignments have been revised based on total synthesis and/or X-ray crystallographic analysis using CuKα radiation, which enables assignment of absolute configuration. The revised structures based on current understanding are shown in Fig. 1. See ESI,† Section F for a complete discussion and relevant references.
- D. Jiang and D. G. Peterson, Food Chem., 2013, 141, 1345–1353 CrossRef CAS PubMed.
- B. Ding, Y. Dai, Y.-L. Hou and X.-S. Yao, J. Asian Nat. Prod. Res., 2015, 17, 559–566 CrossRef CAS PubMed.
- M. Li, J. Xiong, Y. Huang, L.-J. Wang, Y. Tang, G.-X. Yang, X.-H. Liu, B.-G. Wei, H. Fan, Y. Zhao, W.-Z. Zhai and J.-F. Hu, Tetrahedron, 2015, 71, 5285–5295 CrossRef CAS.
- Z. Zhao, L. Zhou, X. Chen, Y. Cheng, F. Hou and J. Nie, Chin. Med. J., 2013, 126, 1230–1235 CAS.
- Role of oxidative stress in diabetic nephropathy: (a) J. M. Forbes, M. T. Coughlan and M. E. Cooper, Diabetes, 2008, 57, 1446–1454 CrossRef CAS PubMed; (b) N. Kashihara, Y. Haruna, V. K. Kondeti and Y. S. Kanwar, Curr. Med. Chem., 2010, 17, 4256–4269 CrossRef CAS PubMed; (c) D. M. Small, J. S. Coombes, N. Bennet, D. W. Johnson and G. C. Gobe, Nephrology, 2012, 17, 311–321 CrossRef CAS PubMed. Neurodegenerative disease: (d) J. Emerit, M. Edeas and F. Bricaire, Biomed. Pharmacother., 2004, 58, 39–46 CrossRef CAS PubMed; (e) B. Uttara, A. V. Singh, P. Zamboni and R. T. Mahajan, Curr. Neuropharmacol., 2009, 7, 65–74 CrossRef CAS PubMed. Cardiovascular disease: (f) N. R. Madamanchi, A. Vendrov and M. S. Runge, Arterioscler., Thromb., Vasc. Biol., 2005, 25, 29–38 CrossRef CAS PubMed.
- D. Giustarini, I. Dalle-Donne, D. Tsikas and R. Rossi, Crit. Rev. Clin. Lab. Sci., 2009, 46, 241–281 CrossRef CAS PubMed.
- R. A. Roberts, D. L. Laskin, C. V. Smith, F. M. Robertson, E. M. G. Allen, J. A. Doorn and W. Slikker, Toxicol. Sci., 2009, 112, 4–16 CrossRef CAS PubMed.
- A. tatarinowii root (50 kg) yielded 7.3 mg acortatarin A (0.000015 wt%) and 3.4 mg acortatarin B (0.000007 wt%) (ref. 1); B. campestris pollen (15 kg) yielded 6 mg pollenopyrroside A (0.000040 wt%) and 5 mg pollenopyrroside B (acortatarin A) (0.000033 wt%) (ref. 2); C. spinosa powdered fruits (10 kg) yielded 4 mg capparisine A (acortatarin A) (0.000040 wt%) and 5 mg capparisine B (0.000050 wt%) (ref. 3); powdered Shensong Yangxin (2 kg) yielded 8.1 mg shensongine A (0.000405 wt%), 2.8 mg shensongine B (0.000140 wt%), 15.5 mg shensongine C (0.000775 wt%), as well as 14.8 mg pollenopyrroside B (0.000740 wt%) (ref. 6); X. nigripes mycelium (20 kg) yielded 4.0 mg xylapyrroside A (0.000020 wt%) and 21.3 mg xylapyrroside B (0.000107 wt%), as well as 1.2 mg pollenopyrroside A (0.000006 wt%) and 20.1 mg acortatarin A (0.000100 wt%) (ref. 7).
- For syntheses of pyrrolomorpholine spiroketal natural products, see: (a) G. Sudhakar, V. D. Kadam, S. Bayya, G. Pranitha and B. Jagadeesh, Org. Lett., 2011, 13, 5452–5455 CrossRef CAS PubMed; (b) H. M. Geng, J. L. Chen, D. P. Furkert, S. Jiang and M. A. Brimble, Synlett, 2012, 23, 855–858 CrossRef CAS; (c) N. V. Borrero and A. Aponick, J. Org. Chem., 2012, 77, 8410–8416 CrossRef CAS PubMed; (d) T. Teranishi, M. Kageyama and S. Kuwahara, Biosci., Biotechnol., Biochem., 2013, 77, 676–678 CrossRef CAS PubMed; (e) P. Cao, Z.-J. Li, W.-W. Sun, S. Malhotra, Y.-L. Ma, B. Wu and V. S. Parmar, Nat. Prod. Bioprospect., 2015, 5, 37–45 CrossRef CAS PubMed; (f) Z. Cao, Y. Li, S. Wang, X. Guo, L. Wang and W. Zhao, Synlett, 2015, 26, 921–926 CrossRef CAS; (g) J. M. Wood, D. P. Furkert and M. A. Brimble, Org. Biomol. Chem., 2016, 14, 7659–7664 RSC.
- J. M. Wurst, A. L. Verano and D. S. Tan, Org. Lett., 2012, 14, 4442–4445 CrossRef CAS PubMed.
- S. A. Snyder, A. Gollner and M. I. Chiriac, Nature, 2011, 474, 461–466 CrossRef CAS PubMed.
- (a) J. S. Potuzak, S. B. Moilanen and D. S. Tan, J. Am. Chem. Soc., 2005, 127, 13796–13797 CrossRef CAS PubMed; (b) S. B. Moilanen, J. S. Potuzak and D. S. Tan, J. Am. Chem. Soc., 2006, 128, 1792–1793 CrossRef CAS PubMed; (c) G. Liu, J. M. Wurst and D. S. Tan, Org. Lett., 2009, 11, 3670–3673 CrossRef CAS PubMed; (d) J. M. Wurst, G. Liu and D. S. Tan, J. Am. Chem. Soc., 2011, 133, 7916–7925 CrossRef CAS PubMed; (e) I. Sharma, J. M. Wurst and D. S. Tan, Org. Lett., 2014, 16, 2474–2477 CrossRef CAS PubMed; (f) Reviewed in: A. L. Verano and D. S. Tan, Isr. J. Chem., DOI:10.1002/ijch.201600134.
- A. J. Janczuk, W. Zhang, P. R. Andreana, J. Warrick and P. G. Wang, Carbohydr. Res., 2002, 337, 1247–1259 CrossRef CAS PubMed.
- H. Abe, S. Shut, S. Tamura and A. Matsuda, Tetrahedron Lett., 2001, 42, 6159–6161 CrossRef CAS.
- J. Robertson, P. T. Chovatia, T. F. Fowler, J. M. Withey and D. J. Woollaston, Org. Biomol. Chem., 2010, 8, 226–233 CAS.
- J. M. Muchowski and P. Hess, Tetrahedron Lett., 1988, 29, 777–780 CrossRef CAS.
- Monoreduction of diformylpyrrole: (a) R. Li, A. D. Lammer, G. M. Ferrence and T. D. Lash, J. Org. Chem., 2014, 79, 4078–4093 CrossRef CAS PubMed; (b) Y.-W. Chin, S. W. Lim, S.-H. Kim, D.-Y. Shin, Y.-G. Suh, Y.-B. Kim, Y. C. Kim and J. Kim, Bioorg. Med. Chem. Lett., 2003, 13, 79–81 CrossRef CAS PubMed.
- See ESI† for complete details.
- NMR analysis of the 2-mercurial intermediates, isolated after brine workup prior to NaBH4 reduction, indicated that substrate 16 undergoes anti-mercuration, while 18 undergoes syn-mercuration, presumably due to directing effects of the free hydroxyl groups
 .
. - For syntheses of spiroketals in which metal chelation effects are postulated, see: (a) D. R. Williams, P. A. Jass and R. D. Gaston, Tetrahedron Lett., 1993, 34, 3231–3234 CrossRef CAS; (b) D. A. Evans, B. W. Trotter, P. J. Coleman, B. Côté, L. C. Dias, H. A. Rajapakse and A. N. Tyler, Tetrahedron, 1999, 55, 8671–8726 CrossRef CAS; (c) A. B. Smith, V. A. Doughty, Q. Lin, L. Zhuang, M. D. McBriar, A. M. Boldi, W. H. Moser, N. Murase, K. Nakayama and M. Sobukawa, Angew. Chem., Int. Ed., 2001, 40, 191–195 CrossRef CAS; (d) J. L. Hubbs and C. H. Heathcock, J. Am. Chem. Soc., 2003, 125, 12836–12843 CrossRef CAS PubMed; (e) S. F. Tlais and G. B. Dudley, Org. Lett., 2010, 12, 4698–4701 CrossRef CAS PubMed; (f) M. J. Kurth, E. G. Brown, E. Hendra and H. Hope, J. Org. Chem., 1985, 50, 1115–1117 CrossRef CAS.
- (a) T. C. Wabnitz, J. Yu and J. B. Spencer, Chem.–Eur. J., 2004, 10, 484–493 CrossRef CAS PubMed; (b) D. C. Rosenfeld, S. Shekhar, A. Takemiya, M. Utsunomiya and J. F. Hartwig, Org. Lett., 2006, 8, 4179–4182 CrossRef CAS PubMed; (c) T. T. Dang, F. Boeck and L. Hintermann, J. Org. Chem., 2011, 76, 9353–9361 CrossRef CAS PubMed.
- S. Iacono and J. R. Rasmussen, Org. Synth., 1986, 64, 57–62 CrossRef CAS.
- H. Wang and J. A. Joseph, Free Radical Biol. Med., 1999, 27, 612–616 CrossRef CAS PubMed.
- L. M. Magalhães, M. A. Segundo, S. Reis and J. L. F. C. Lima, Anal. Chim. Acta, 2008, 613, 1–19 CrossRef PubMed.
- K.-Y. Hung, S.-Y. Liu, S.-H. Kao, J.-W. Huang, C.-K. Chiang and T.-J. Tsai, Am. J. Nephrol., 2009, 29, 192–202 CrossRef CAS PubMed.
- The eight pyrrolomorpholine natural products and 2-hydroxy analogues were not similar to known aggregators, as assessed by the Aggregator Advisor database, http://advisor.bkslab.org/; J. J. Irwin, D. Duan, H. Torosyan, A. K. Doak, K. T. Ziebart, T. Sterling, G. Tumanian and B. K. Shoichet, J. Med. Chem., 2015, 58, 7076–7087 CrossRef CAS PubMed.
-
Ref. 7 provided shensongine A in 13.7% yield over 11 steps, with the key spirocyclization proceeding in >98:2 diastereoselectivity to the thermodynamic β-spiroketal. Ref. 13f provided pollenopyrroside A in 2.7% yield and shensongine A in 8.8% yield over 14 steps with the key spirocyclization proceeding in 1:3 α/β dr. Ref. 13g provided pollenopyrroside A in 5.6% yield and shensongine A in 0.79% yield over 11 steps, with the key spirocyclization proceeding in 1.2:1 dr as an inseparable mixture of unassigned diastereomers.

Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc05505b |
| This journal is © The Royal Society of Chemistry 2017 |