
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regulating the active species of Ni(OH)2 using CeO2: 3D CeO2/Ni(OH)2/carbon foam as an efficient electrode for the oxygen evolution reaction†
Zhengqing
Liu
a,
Na
Li
a,
Hongyang
Zhao
a,
Yi
Zhang
b,
Yunhui
Huang
bc,
Zongyou
Yin
de and
Yaping
Du
 *a
*a
aFrontier Institute of Science and Technology Jointly with College of Science, State Key Laboratory for Mechanical Behavior of Materials, Xi’an Jiaotong University, 99 Yanxiang Road, Yanta District, Xi’an, Shaanxi Province 710054, China. E-mail: ypdu2013@mail.xjtu.edu.cn
bCollaborative Innovation Center of Intelligent New Energy Vehicle, School of Materials Science and Engineering, Tongji University, Shanghai 201804, P. R. China
cState Key Laboratory of Material Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, Wuhan, Hubei 430074, P. R. China
dResearch School of Chemistry, The Australian National University, Canberra, Australian Capital Territory 2601, Australia
eDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 14th February 2017
Abstract
Three dimensional (3D) N, O and S doped carbon foam (NOSCF) is prepared as a substrate for in situ vertically grown Ni(OH)2 nanosheets. As designed Ni(OH)2/NOSCF possesses strong electrostatic interactions with OH− ions due to many C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups existing in NOSCF, which can facilitate the formation of crucial NiOOH intermediates during the OER process. CeO2 nanoparticles (NPs) of ∼3.3 nm in size are decorated on Ni(OH)2 nanosheets to design a highly efficient CeO2/Ni(OH)2/NOSCF electrocatalyst for the oxygen evolution reaction (OER). The CeO2 NP decorated Ni(OH)2/NOSCF not only exhibits a remarkably improved OER performance with an onset potential of 240 mV, outperforming most reported non-noble metal based OER electrocatalysts, but also possesses a small Tafel slope of 57 mV dec−1 and excellent stability under different overpotentials. The synergistic effect of producing more active species of NiIII/IV and accelerating the charge transfer for Ni(OH)2/NOSCF by the introduction of CeO2 NPs is also investigated. These results demonstrate the possibility of designing energy efficient OER catalysts with the assistance of earth abundant CeO2-based catalysts.
O groups existing in NOSCF, which can facilitate the formation of crucial NiOOH intermediates during the OER process. CeO2 nanoparticles (NPs) of ∼3.3 nm in size are decorated on Ni(OH)2 nanosheets to design a highly efficient CeO2/Ni(OH)2/NOSCF electrocatalyst for the oxygen evolution reaction (OER). The CeO2 NP decorated Ni(OH)2/NOSCF not only exhibits a remarkably improved OER performance with an onset potential of 240 mV, outperforming most reported non-noble metal based OER electrocatalysts, but also possesses a small Tafel slope of 57 mV dec−1 and excellent stability under different overpotentials. The synergistic effect of producing more active species of NiIII/IV and accelerating the charge transfer for Ni(OH)2/NOSCF by the introduction of CeO2 NPs is also investigated. These results demonstrate the possibility of designing energy efficient OER catalysts with the assistance of earth abundant CeO2-based catalysts.
Introduction
Clearly, using electricity to split water into hydrogen and oxygen (2H2O → 2H2 + O2) is one of the most efficient and attractive methods for the production of renewable energy.1,2 However, large-scale water electrolysis is greatly hindered due to the huge overpotential and significant efficiency loss for the half-cell of the oxygen evolution reaction (OER).3–5 Although noble-metal based materials (e.g. Ir, Pt) are currently regarded as high-efficiency OER catalysts, their low earth abundance and high cost limit their widespread use.6–8 Therefore, in recent years, various efficient and low-cost OER electrocatalysts (such as, Fe, Co, Ni and Mn) with high OER performance (low onset potential, high activity and good stability) in basic electrolytes have been extensively designed and investigated.8–23 Among them, nickel(II) hydroxide (Ni(OH)2)-based materials are attractive electrocatalysts for the OER because of their intrinsic potential for high OER performance and two-dimensional (2D) layered structure.20 Another key reason for the extensive study of Ni(OH)2 is that the high oxidation state of NiIII/IV can serve as an active species for OER catalysts.24 For example, Ye et al. prepared a Ni(OH)2–Au hybrid as an OER catalyst, and a significantly enhanced OER performance was exhibited by enhancing the generation of the NiIII/IV active species. However, the poor kinetics and mass-transferability of Ni(OH)2 as an electrocatalyst for the OER still limit its further development for practical applications.Cerium(IV) oxide (CeO2) is one of the most important rare earth oxides, is stable in alkaline solution, converts easily between the Ce3+ and Ce4+ oxidation states, undergoes reversible oxygen ion exchange (1/2O2 (gas) + 2e− (solid) ↔ O2− (solid)), and has good ionic conductivity and high oxygen-storage capacity (OSC).25–27 The above unique properties enable CeO2 to serve as a cocatalyst to enhance the performance of OER catalysts by improving charge transfer and energy conversion efficiency, which can also solve the poor kinetics and mass-transferability problems of Ni(OH)2 for the OER. However, few studies have focused on the application of CeO2 nanocrystals in the electrocatalytic field. Recently, Li et al. developed an efficient OER electrocatalyst by supporting FeOOH/CeO2 on Ni foam and exhibited enhanced OER performance compared with pure FeOOH.28 They also demonstrated the unique high OSC properties of CeO2, such that CeO2 can straightway absorb the oxygen produced during the OER and accordingly promote the OER. Therefore, the combination of Ni(OH)2 and CeO2 to form a CeO2/Ni(OH)2 hybrid will be an efficient route to improve the electrocatalytic performance of Ni(OH)2via improving the energy conversion efficiency, and thereby promoting the generation of active species of NiIII/IV for enhancing the OER performance.
In order to greatly prevent the Ni(OH)2 nanosheets from aggregation and thus further enhance the OER performance, three dimensional (3D) free-standing carbon foam (CF) is chosen as the substrate for in situ growth of the Ni(OH)2 nanosheets. The advantages of applying such 3D CF as a substrate can be attributed to the interconnected frameworks with large surface area for effective contact with an aqueous electrolyte and rapid interfacial electron charge transfer. Moreover, the obtained CF is doped by N, O and S elements during carbonization without other extra chemicals being added, where the N, O and S elements come from the melamine resin and sodium bisulfite additive of melamine foam (MF) (Fig. S1, ESI†). And N, O and S doped carbon materials are believed to enhance the OER activity.29
Herein, as we expect, Ni(OH)2 nanosheets are successfully grown along the frameworks of N, O and S doped CF (NOSCF) and prevent the undesirable aggregation of Ni(OH)2 nanosheets because of the open cell pores of NOSCF. Then, we prepared uniform CeO2 NPs of ∼3.3 nm in size via a one step colloidal synthesis method, and deposited the surface modified-CeO2 NPs on Ni(OH)2 nanosheets of the as-designed Ni(OH)2/NOSCF to form a self-supported CeO2/Ni(OH)2/NOSCF electrode, as shown in Fig. 1. As a result of the open cell structure of 3D NOSCF for facile electrolyte transport and strong electronic interactions between CeO2 NPs and Ni(OH)2 nanosheets for accelerating the oxidation of NiII to NiIII/IV, the CeO2/Ni(OH)2/NOSCF electrocatalyst delivers an excellent water oxidation performance at a lower onset potential, ranking high among the extensive non-noble electrocatalysts studied for the OER. As we know, this is the first time CeO2 is combined with a functional Ni(OH)2 electrocatalyst, which offers an impressive OER performance, and provides insight into the possibility of enhancing OER catalysis by using rare earth CeO2-based nanomaterials.
 | ||
| Fig. 1 Process for the design of a self-supported CeO2/Ni(OH)2/NOSCF electrode and application for the oxygen evolution reaction. | ||
Results and discussion
Design of the CeO2/Ni(OH)2/NOSCF electrocatalyst
N, O and S doped CF was prepared by direct carbonization of melamine foam (MF) in a tube furnace at 700 °C for 1 h under protection of a nitrogen atmosphere. After carbonization, the volume of CF shrunk to about 70% of MF (inset of Fig. 2a). Then, the NOSCF was further oxidized by dipping into a mixture of acids of HNO3 (65%) and H2SO4 (98%) with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 for 6 h, which can produce more CO groups (Fig. S2a, ESI†).29 It has been revealed that the C atoms in the CO groups have a stronger electrostatic interaction with OH− ions than other oxygen-containing groups, which can facilitate the formation of crucial M–OOH intermediates (eqn (1)), and thus accelerate the OER reaction.30,31
:3 for 6 h, which can produce more CO groups (Fig. S2a, ESI†).29 It has been revealed that the C atoms in the CO groups have a stronger electrostatic interaction with OH− ions than other oxygen-containing groups, which can facilitate the formation of crucial M–OOH intermediates (eqn (1)), and thus accelerate the OER reaction.30,31 | (1) |
 | ||
| Fig. 2 (a) A piece of melamine foam derived carbon foam. (b and c) SEM images of Ni(OH)2 nanosheets grown on NOSCF and the corresponding (d) XRD pattern. (e) HRTEM image of synthesized Ni(OH)2 recorded from the edge of the Ni(OH)2/NOSCF. (f) EDS elemental mapping for C, N, S, Ni and O elements recorded on the Ni(OH)2/NOSCF. | ||
The elemental content in the NOSCF was measured by XPS analysis to be about 66.8, 4.3, 24.5 and 0.42 atom% for C, N, O and S, respectively (Fig. S2b, ESI†). Scanning electron microscopy (SEM) images in the inset of Fig. 2a revealed that the as-prepared NOSCF possessed an interconnected network architecture, which could make it an ideal substrate for the growth of some electrocatalysts. Then, by using the framework of NOSCF as a nucleation platform, Ni(OH)2 nanosheets could be uniformly grown in situ along the framework of NOSCF by a simple chemical bath deposition process,32 which could be observed evidently from the SEM images (Fig. 2b and c). The selective growth of Ni(OH)2 nanosheets on the NOSCF could preserve the open-cell structure of the NOSCF (Fig. 2b) and efficiently prevent the aggregation of Ni(OH)2 nanosheets, indicating that it held a large surface area for electrocatalysis. The vertical Ni(OH)2 layers could be clearly observed in an enlarged SEM image of the Ni(OH)2/carbon foam hybrid (Fig. S3, ESI†). Such nanostructured materials can offer a much rougher surface, which reduces the solid–gas interaction, giving rise to a timely release of adhered gas bubbles and thus enhancing the OER performance.
The grown Ni(OH)2 nanosheets had a hexagonal phase (a = b = 0.308 nm, c = 0.234, JCPDS: 38-0715), as confirmed by powder X-ray diffraction (PXRD) analysis (Fig. 2d). As shown in the transmission electron microscopy (TEM) image in Fig. 2e, the grown Ni(OH)2 presented a typical layered structure, and the high resolution (HRTEM) image (inset of Fig. 2e) identified the (101) plane of a hexagonal crystal structure for the Ni(OH)2 nanosheets with an interplanar spacing of 0.23 nm. The corresponding elemental mapping of the designed Ni(OH)2/NOSCF is shown in Fig. 2f; the C, N and S elements were distributed on the whole surface of the frameworks in the NOSCF, and also displayed a very uniform distribution of Ni(OH)2. The loading percentage of Ni(OH)2 in the Ni(OH)2/NOSCF composite was estimated to be ∼63% by thermogravimetric analysis (TGA), as displayed in Fig. S4 (ESI†).
The CeO2 NPs were synthesized by using cerium(IV) ammonium nitrate ((NH4)2Ce(NO3)6) as a precursor in a mixture of solvents of oleylamine and 1-octadecene. As shown in the PXRD pattern in Fig. 3a, the prepared CeO2 samples presented a cubic phase (space group: Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, a = b = c = 5.411 Å, JCPDS: 34-0394). The TEM image in Fig. 3b showed that the as-synthesized CeO2 NPs were relatively monodisperse with an average size of ∼3.3 nm (inset of a histogram of the particle diameters). The good monodispersity of the CeO2 NPs indicates the retention of the used capping ligand (oleylamine) on the surface of CeO2 NPs, as demonstrated by Fourier transform infrared (FTIR) spectroscopy (Fig. S5, ESI†). As seen in Fig. 3c, the HRTEM image of the CeO2 NPs showed clearly crystal lattice fringes with an interplanar spacing of 0.16 nm, which can be ascribed to the (111) crystal plane. The selected area electron diffraction (SAED) pattern shown in Fig. 3d indicated that the synthesized CeO2 NPs were highly crystallized.
m, a = b = c = 5.411 Å, JCPDS: 34-0394). The TEM image in Fig. 3b showed that the as-synthesized CeO2 NPs were relatively monodisperse with an average size of ∼3.3 nm (inset of a histogram of the particle diameters). The good monodispersity of the CeO2 NPs indicates the retention of the used capping ligand (oleylamine) on the surface of CeO2 NPs, as demonstrated by Fourier transform infrared (FTIR) spectroscopy (Fig. S5, ESI†). As seen in Fig. 3c, the HRTEM image of the CeO2 NPs showed clearly crystal lattice fringes with an interplanar spacing of 0.16 nm, which can be ascribed to the (111) crystal plane. The selected area electron diffraction (SAED) pattern shown in Fig. 3d indicated that the synthesized CeO2 NPs were highly crystallized.
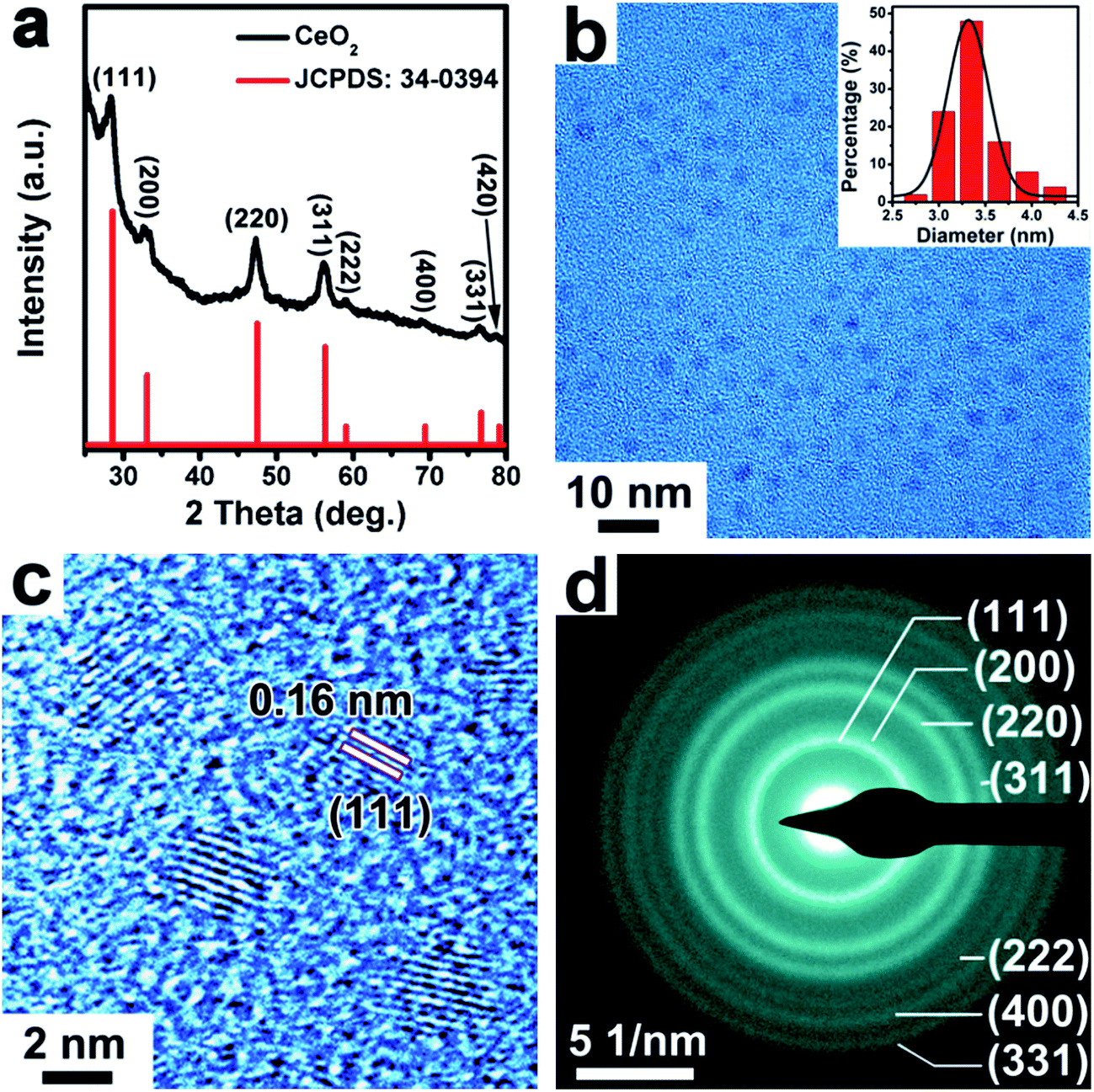 | ||
| Fig. 3 (a) XRD pattern of the obtained CeO2 sample. (b) TEM, (c) HRTEM image and (d) corresponding SAED pattern of synthesized ∼3.3 nm sized CeO2 NPs. Inset of (b): a particle size distribution analysis. | ||
The prepared CeO2 NPs are hydrophobic due to the long carbon chains of oleylamine (OM) used as surfactants for the reaction, and hence cannot directly disperse in water. In order to generate a hydrophilic surface for combining with the Ni(OH)2/NOSCF and testing the OER performance, we employed NaS2 solution to modify the surface of CeO2 NPs (Fig. S5, ESI†).33 As shown in the TEM image in Fig. S6a (ESI†), the CeO2 NPs still kept their particle morphology with high crystallization after surface modification, and could be well dispersed in water (digital photo in Fig. S6b, ESI†). The CeO2 NPs were anchored on the Ni(OH)2/NOSCF using a controllable electrophoretic deposition strategy, the details are shown in the Experimental section.34 All of the diffraction peaks of Ni(OH)2 (JCPDS: 380715) and CeO2 (JCPDS: 34-0394) were detected in CeO2/Ni(OH)2/NOSCF (Fig. 4a). Fig. 4b and c show the representative TEM and HRTEM images of the CeO2/Ni(OH)2 hybrid obtained from CeO2/Ni(OH)2/NOSCF with a deposition duration of 10 min. It can be observed from Fig. 4b that the Ni(OH)2 nanosheets are uniformly decorated with CeO2 NPs. Fig. 4c presents the corresponding HRTEM image with an interplanar spacing of 0.16 nm and 0.23 nm, indexed to the (111) and (101) crystal planes of CeO2 and Ni(OH)2, respectively.
 | ||
| Fig. 4 (a) XRD pattern, (b) TEM and (c) HRTEM images of CeO2/Ni(OH)2 hybrid nanostructures obtained from the CeO2/Ni(OH)2/NOSCF. (d) UV-vis absorption spectra of pristine Ni(OH)2 nanosheets, CeO2 NPs and the CeO2/Ni(OH)2 hybrid. A comparison of (e) Ce 3d and (f) Ni 2p high-resolution XPS spectra of CeO2/Ni(OH)2/NOSCF and Ni(OH)2/NOSCF. | ||
To further investigate the strong electronic interactions between Ni(OH)2 nanosheets and CeO2 NPs, UV-vis absorption spectra (Fig. 4d) and X-ray photoelectron spectroscopy (XPS) spectra (Fig. 4e and f) of Ni(OH)2 nanosheets, CeO2 NPs and the CeO2/Ni(OH)2 hybrid were examined. As shown in Fig. 4d, two absorption peaks of the grown Ni(OH)2 nanosheets located at 385 and 670 nm corresponded to the d–d transitions of NiII cations.35 Compared with the pristine Ni(OH)2 nanosheets, the absorption spectrum of CeO2/Ni(OH)2 was obviously red-shifted (∼8 nm), indicating the strong electronic interactions between them.36,37 XPS spectra of Ce 3d and Ni 2p are shown in Fig. 4e and f, respectively. As shown in Fig. 4e, for Ce 3d of CeO2, the peaks located at 920–911 eV and 903–893 eV correspond to Ce 3d3/2, and the peaks located at 877–866 eV correspond to Ce 3d5/2, which demonstrated the coexistence of Ce3+ and Ce4+ in the CeO2 NPs.38 However, after CeO2 NPs were deposited on the Ni(OH)2 nanosheets, the ratio of Ce3+:Ce4+ in the CeO2/Ni(OH)2 hybrid changed compared with pure CeO2 NPs, indicating that the valence states of Ce in the CeO2/Ni(OH)2 hybrid rearranged.28 As shown in Fig. 4f, the XPS spectrum of Ni 2p in Ni(OH)2/NOSCF showed two major peaks at 853.2 and 870.8 eV corresponding to Ni 2p3/2 and Ni 2p1/2, respectively, which were characteristic of the Ni2+ state.39 And some satellite peaks in the Ni 2p region could also be observed in Fig. 4f. Through careful comparison and analysis, we found that the peaks of Ni 2p3/2 and Ni 2p1/2 in the XPS spectrum for CeO2/Ni(OH)2/NOSCF both shifted to lower binding energies of ∼0.5 eV. Therefore, the ratio change of Ce 3d and peak shifts of Ni 2p in the CeO2/Ni(OH)2 hybrid indicate strong electronic interactions between the Ni(OH)2 nanosheets and CeO2 NPs.
CeO2/Ni(OH)2/NOSCF electrocatalyst for OER performance
The water oxidation reaction was applied to study the electronic interactions between Ni(OH)2 nanosheets and CeO2 NPs and their effect on the corresponding catalytic activity. The OER performance of the CeO2/Ni(OH)2/NOSCF electrocatalyst was investigated in 1.0 M KOH (pH = 14). As a comparison, Ni(OH)2/NOSCF, CeO2/NOSCF, NOSCF and Ir/C were also tested under the same conditions. It was obvious from the cyclic voltammetry (CV) curves (Fig. 5a) that both the electrocatalysts of Ni(OH)2/NOSCF and CeO2/Ni(OH)2/NOSCF clearly showed redox peaks ranging from 1.2 to 1.6 V, which belong to the NiII/NiIII/IV redox process (Ni(OH)2 + OH− → NiOOH + H2O + e−). And there was an obvious negative shift of the oxidation potential, changed from 1.46 V to 1.41 V for CeO2/Ni(OH)2/NOSCF, indicating that CeO2/Ni(OH)2/NOSCF has higher transfer efficiency from NiII to NiIII/IV and larger charge capacity than Ni(OH)2/NOSCF.40 This conclusion can be further demonstrated by Nyquist plots. As presented in Fig. S7 (ESI†), compared with Ni(OH)2/NOSCF, the CeO2/Ni(OH)2/NOSCF shows a smaller charge transfer resistance (high frequencies) and reduced mass-transfer resistance (low frequencies). The decrease in mass transfer resistance may contribute to the increased number of NiIII/IV active species. | ||
| Fig. 5 (a) CV curves of Ni(OH)2/NOSCF and CeO2/Ni(OH)2/NOSCF. (b) Polarization curves of NOSCF, CeO2/NOSCF, Ni(OH)2/NOSCF, CeO2/Ni(OH)2/NOSCF and the benchmark Ir/C electrocatalyst for comparison. Sweep rate: 5 mV s−1. Inset: the extent of NiII/NiIII/IV transformation for Ni(OH)2/NOSCF and CeO2/Ni(OH)2/NOSCF. (c and d) The corresponding onset potential and Tafel curves for the catalysts derived from (b). (e) Stability test using a continuous OER recorded on the CeO2/Ni(OH)2/NOSCF self-supported electrode under different static potentials (V vs. SCE). | ||
As shown in the polarization curves in Fig. 5b and statistical data in Fig. 5c, the CeO2/Ni(OH)2/NOSCF exhibited a lower onset potential of 240 mV than Ir/C and Ni(OH)2/NOSCF, surpassing most reported non-noble metal based OER electrocatalysts (Table S1, ESI†). In particular, the significant increase of the current density was more obvious when the potential was beyond ∼1.6 V, which could further demonstrate that the OER activity of Ni(OH)2/NOSCF is greatly enhanced when decorated with CeO2 NPs. In addition, we also optimized the loaded mass ratio of CeO2 NPs on Ni(OH)2/NOSCF and found that when the mass ratio of CeO2:Ni(OH)2/NOSCF was 30% (Fig. S8, ESI†), the electrocatalytic activity of CeO2/Ni(OH)2/NOSCF reached its highest level, and therefore this mass ratio was used in the following experiments.
During the OER process, the highly oxidative NiIII/IV cations are believed to serve as active species, which indicates that the enhanced catalytic activity for CeO2/Ni(OH)2/NOSCF observed in our study might be a result of increasing NiII/NiIII/IV transformations. Therefore, we investigated the extent of the NiII/NiIII/IV transformation by integrated oxidation peak areas (inset of Fig. 5b).41,42 When CeO2 NPs were deposited on the Ni(OH)2/NOSCF, the NiII/NiIII/IV extent showed a dramatic increase of about 1.7-fold compared with the Ni(OH)2/NOSCF (inset of Fig. 5b), thus CeO2 NPs potentially facilitated producing more NiIII/IV active species and subsequently led to the improvement of the OER catalytic activity (Fig. 5b).
The enhanced OER activity of CeO2/Ni(OH)2/NOSCF was more obvious by comparing the Tafel slopes. As shown in Fig. 5c and d, the Tafel slope of CeO2/Ni(OH)2/NOSCF was 57 mV dec−1, and it was smaller than those of Ir/C (72 mV dec−1), NOSCF (295 mV dec−1), CeO2/NOSCF (136 mV dec−1), and Ni(OH)2/NOSCF (65 mV dec−1). Through the comparison of the Tafel slopes we could demonstrate that depositing CeO2 NPs on Ni(OH)2/NOSCF could facilitate its OER kinetics, and the OER activity of CeO2/Ni(OH)2/NOSCF was comparable to many other non-noble metal OER electrocatalysts in alkaline media (Table S1, ESI†).
We also tested the stability of the designed CeO2/Ni(OH)2/NOSCF by a chronoamperometry method to evaluate the OER performance. As shown in Fig. 5e, the current density of the OER showed no change during 6 h of continuous operation under various potentials of 0.55, 0.60, 0.65, 0.70, 0.75 and 0.80 V, which suggested that the CeO2/Ni(OH)2/NOSCF had excellent stability for the OER process. Thus, the CeO2/Ni(OH)2/NOSCF with its high catalytic activity as well as excellent stability would be a promising candidate for electrochemical water oxidation.
Conclusions
In summary, we have successfully designed a 3D hierarchical Ni(OH)2/NOSCF electrode by growing Ni(OH)2 nanosheets along the carbon frameworks of N, O and S doped CF. The experiments found that Ni(OH)2 nanosheets of Ni(OH)2/NOSCF decorated with ∼3.3 nm sized CeO2 NPs displayed enhanced OER performance. Compared with Ni(OH)2/NOSCF, the onset potential of CeO2 NP decorated Ni(OH)2/NOSCF decreased from 270 to 240 mV, and the Tafel slope reduced from 65 to 57 mV dec−1, much better than the benchmark Ir/C. As confirmed by UV-vis and XPS results, as well as electrochemical analysis, the reasons for the enhanced OER performance result from the synergistic effect between CeO2 NPs and Ni(OH)2 nanosheets by a 1.7-fold enhancement in the generation of NiIII/IV active species and faster charge transfer. The high OER performance of CeO2/Ni(OH)2/NOSCF in the present study makes CeO2 based composites very promising electrocatalysts for water oxidation.Acknowledgements
We gratefully acknowledge financial support from start-up funding from Xi’an Jiaotong University, the Fundamental Research Funds for the Central Universities (2015qngz12), the China National Funds for Excellent Young Scientists (grant no. 21522106), NSFC (grant no. 21371140, 51361130151, 51672098), and PCSIRT (Program for Changjiang Scholars and Innovative Research Team in University, IRT14R18). We also appreciate Prof. Daniel G. Nocera from Harvard University for his kind suggestions.Notes and references
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- J. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Graetzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- J. H. Wang, W. Cui, Q. Liu, Z. C. Xing, A. M. Asiri and X. P. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2011, 111, 5815 CrossRef CAS.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- Z. X. Fan, Z. M. Luo, Y. Chen, J. Wang, B. Li, Y. Zong and H. Zhang, Small, 2016, 12, 3908–3913 CrossRef CAS PubMed.
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869–9921 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- S. Pintado, S. Goberna-Ferrón, E. C. Escudero-Adán and J. R. Galán-Mascarós, J. Am. Chem. Soc., 2013, 135, 13270–13273 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- L. Wu, Q. Li, C. H. Wu, H. Zhu, A. Mendoza-Garcia, B. Shen, J. Guo and S. Sun, J. Am. Chem. Soc., 2015, 137, 7071–7074 CrossRef CAS PubMed.
- K. Y. Niu, F. Lin, S. Jung, L. Fang, D. Nordlund, C. C. L. McCrory, T. C. Weng, P. Ercius, M. M. Doeff and H. Zheng, Nano Lett., 2015, 15, 2498–2503 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- F. Y. Cheng, T. R. Zhang, Y. Zhang, J. Du, X. P. Han and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 2474–2477 CrossRef CAS PubMed.
- S. Chen and S. Z. Qiao, ACS Nano, 2013, 7, 10190–10196 CrossRef CAS PubMed.
- H. Liang, F. Meng, M. Cabán-Acevedo, L. Li, A. Forticaux, L. Xiu, Z. Wang and S. Jin, Nano Lett., 2015, 15, 1421–1427 CrossRef CAS PubMed.
- J. F. Ping, Y. X. Wang, Q. P. Lu, B. Chen, J. Z. Chen, Y. Huang, Q. L. Ma, C. L. Tan, J. Yang, X. H. Cao, Z. J. Wang, J. Wu, Y. B. Ying and H. Zhang, Adv. Mater., 2016, 28, 7640–7645 CrossRef CAS PubMed.
- W. J. Zhou, X. J. Wu, X. H. Cao, X. Huang, C. L. Tan, J. Tian, H. Liu, J. Y. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2921–2924 CAS.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- G. G. Liu, P. Li, G. X. Zhao, X. Wang, J. T. Kong, H. M. Liu, H. B. Zhang, K. Chang, X. G. Meng, T. Kako and J. H. Ye, J. Am. Chem. Soc., 2016, 138, 9128–9136 CrossRef CAS PubMed.
- Y. Zheng, M. Gao, Q. Gao, H. Li, J. Xu, Z. Wu and S. Yu, Small, 2015, 11, 182–188 CrossRef CAS PubMed.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752–755 CrossRef CAS PubMed.
- K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS PubMed.
- J. X. Feng, S. H. Ye, H. Xu, Y. X. Tong and G. R. Li, Adv. Mater., 2016, 28, 4698–4703 CrossRef CAS PubMed.
- X. Yu, M. Zhang, J. Chen, Y. Li and G. Shi, Adv. Energy Mater., 2015, 150, 1501492 Search PubMed.
- X. Lu, W. L. Yim, B. H. R. Suryanto and C. Zhao, J. Am. Chem. Soc., 2015, 137, 2901–2907 CrossRef CAS PubMed.
- N. Cheng, Q. Liu, J. Tian, Y. Xue, A. M. Asiri, H. Jiang, Y. He and X. Sun, Chem. Commun., 2015, 51, 1616–1619 RSC.
- J. Pu, Y. Tong, S. Wang, E. Sheng and Z. Wang, J. Power Sources, 2014, 250, 250–256 CrossRef CAS.
- A. Nag, M. V. Kovalenko, J. S. Lee, W. Liu, B. Spokoyny and D. V. Talapin, J. Am. Chem. Soc., 2011, 133, 10612–10620 CrossRef CAS PubMed.
- Y. Yang, W. Que, X. Zhang, Y. Xing, X. Yin and Y. Du, J. Hazard. Mater., 2016, 5, 430–439 CrossRef PubMed.
- M. A. Oliver-Tolentino, J. Vázquez-Samperio, A. Manzo-Robledo, R. D. G. González-Huerta, J. L. Flores-Moreno, D. Ramírez-Rosales and A. J. Guzmán-Vargas, J. Phys. Chem. C, 2014, 118, 22432–22438 CAS.
- Y. Shi, J. Wang, C. Wang, T. T. Zhai, W. J. Bao, J. J. Xu, X. H. Xia and H. Y. Chen, J. Am. Chem. Soc., 2015, 137, 7365–7370 CrossRef CAS PubMed.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632–7637 CrossRef CAS PubMed.
- K. Priolkar, P. Bera, P. Sarode, M. Hegde, S. Emura, R. Kumashiro and N. Lalla, Chem. Mater., 2002, 14, 2120–2128 CrossRef CAS.
- H. Li, M. Yu, F. Wang, P. Liu, Y. Liang, J. Xiao, C. Wang, Y. Tong and G. Yang, Nat. Commun., 2013, 4, 1894 CrossRef CAS PubMed.
- P. Oliva, J. Leonardi and J. F. Laurent, J. Power Sources, 1982, 8, 229–255 CrossRef CAS.
- C. Tang, H. S. Wang, H. F. Wang, Q. Zhang, G. L. Tian, J. Q. Nie and F. Wei, Adv. Mater., 2015, 27, 4516–4522 CrossRef CAS PubMed.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, experimental section, XPS spectra, TEM images, FTIR spectra and other electrochemical performance data. See DOI: 10.1039/c6sc05408k |
| This journal is © The Royal Society of Chemistry 2017 |