
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorocarbene, fluoroolefin, and fluorocarbyne complexes of Rh†
Christopher J.
Pell‡
a,
Yanjun
Zhu‡
a,
Rafael
Huacuja§
a,
David E.
Herbert¶
 a,
Russell P.
Hughes
*b and
Oleg V.
Ozerov
*a
a,
Russell P.
Hughes
*b and
Oleg V.
Ozerov
*a
aDepartment of Chemistry, Texas A&M University, College Station, TX 77842, USA. E-mail: ozerov@chem.tamu.edu
bDepartment of Chemistry, Dartmouth College, Hanover, New Hampshire 03755, USA
First published on 16th February 2017
Abstract
The manuscript reports the synthesis, characterization, and analysis of electronic structure in a series of complexes of small perfluorocarbon ligands with the (PNP)Rh fragment (where PNP is a diarylamido/bis(phosphine) pincer ligand). Reactions of (PNP)Rh(TBE) as the source of (PNP)Rh with CHF3 and C2HF5 produced perfluoroalkylidene complexes (PNP)Rh![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2 and (PNP)RhC(F)(CF3). (PNP)RhCF2 could also be obtained via the reaction of (PNP)Rh(TBE) with Me3SiCF3/CsF, with an admixture of (PNP)Rh(C2F4), where TBE = tert-butylethylene. Abstraction of fluoride from these neutral (PNP)RhCxFy complexes was successful, although only abstraction from (PNP)RhCF2 allowed unambiguous identification of the Rh product, [(PNP)Rh
CF2 and (PNP)RhC(F)(CF3). (PNP)RhCF2 could also be obtained via the reaction of (PNP)Rh(TBE) with Me3SiCF3/CsF, with an admixture of (PNP)Rh(C2F4), where TBE = tert-butylethylene. Abstraction of fluoride from these neutral (PNP)RhCxFy complexes was successful, although only abstraction from (PNP)RhCF2 allowed unambiguous identification of the Rh product, [(PNP)Rh![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CF]+. DFT computational studies allowed comparison of relative energies of (PNP)Rh(C2F4) and [(PNP)Rh(C2F3)]+ isomers as well as comparisons between the electronic structure of the CF2, C2F4, and CF+ complexes and their hydrocarbon analogues.
CF]+. DFT computational studies allowed comparison of relative energies of (PNP)Rh(C2F4) and [(PNP)Rh(C2F3)]+ isomers as well as comparisons between the electronic structure of the CF2, C2F4, and CF+ complexes and their hydrocarbon analogues.
Introduction
Organofluorine chemistry's major impact on the world of industrial chemistry has inspired many investigations into the unique properties that are inherent to molecules and materials containing C–F bonds. Transition metal complexes containing perfluorocarbon ligands are an important subset of these studies since they exhibit distinctive bonding properties1 and can mediate perfluoroalkyl–carbon bond forming processes.2 Group 9 perfluoroalkylidenes have garnered interest in the past decade after Hughes developed a simple reductive method for making IrCFR complexes (Fig. 1, top) from iridium-fluoroalkyl precursors.3 These complexes have been analyzed in the context of their potential intermediacy in perfluorolefin metathesis,4 and more recently the Baker group has shown that analogous cobalt perfluorocarbenes (Fig. 1, top)5 are capable of undergoing a [2 + 1] cycloaddition with CF2 (ref. 6) and [2 + 2] cycloaddition with C2F4.7 Analogous chemistry was also reported for a difluorocarbene complex of Ni(0).8 Baker has also shown that cationic cobalt(III) difluorocarbenes could undergo migratory insertion into perfluoroalkyl ligands, possibly providing a blueprint for transition metal catalyzed perfluoroolefin polymerization.9
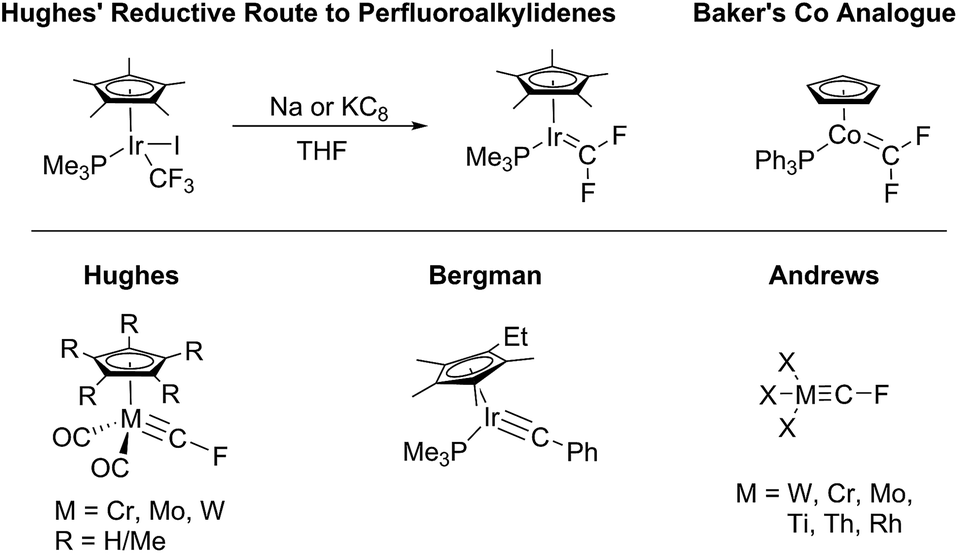 | ||
| Fig. 1 Perfluoroalkylidenes from Hughes and Baker. Isolated fluorocarbynes by Hughes, the Ir carbyne by Bergman, and matrix-trapped fluorocarbynes by Andrews. | ||
The only family of isolable terminal fluoromethylidyne complexes known to date are the Cp*M(CO)2(CF) compounds (M = Cr, Mo, W) reported by Hughes and co-workers (Fig. 1, bottom).10 The Andrews group has reported a number of fluoromethylidyne complexes of the general formula X3M(CF) (Fig. 1, bottom; X = halogen) via trapping laser ablated metal atoms in argon/halocarbon matrices at ca. 10 K.11 Most of the isolable terminal carbyne complexes are complexes of metals of groups 6,12,13 7,13,14 and 8.15,16 A few examples are known for group 5.17 In group 9, one 18-electron complex has been fully characterized for Ir by Bergman et al. (Fig. 1, bottom),18 and one square planar 16-electron complex was mentioned in passing for Rh by Werner et al.,19 as a component of a reaction mixture. The “concentration” of metal carbyne complexes in the middle of the transition metal series can be compared with similar trends for other metal-element multiple bonds.20,21 In this report, we describe the synthesis, characterization, and analysis of electronic structure of a rare cationic fluoromethylidyne complex of Rh, as well as related Rh perfluoroalkylidene complexes.
Results and discussion
Synthesis of CF2, C2F4, and CFCF3 complexes
We recently reported reactions of the (PNP)Rh fragment with aryl carboxylates, including aryl-oxygen oxidative addition.22 The (PNP)Rh acyl-oxygen oxidative addition product of phenyl trifluoroacetate, (PNP)Rh(COCF3)(OPh), could be thermolysed to produce (PNP)Rh(CO) and (PNP)Rh(CF3)(CO)(OPh) as major products. In that report, we noted that some other unidentified products were evident in trace amounts. We continued to be intrigued by one apparent trace product in particular that was consistently observed in 2–5% yield. For it, we observed a doublet of triplets both in the 31P{1H} NMR and 19F NMR spectra (coupling constants: 1JRh–P = 146 Hz, 2JRh–F = 49 Hz, 3JP–F = 30 Hz). These multiplicities implied a P2RhF2 NMR spin system – rather unexpected given the three fluorines in the CF3 group of the starting material. We noted that the 19F NMR chemical shift was itself uncommon (95.6 ppm) and in the range reported for various difluorocarbene complexes (i.e., MCF2).5,10a,23,24 The observed 2JRh–F = 49 Hz was also similar to that of Grushin's trans-(Ph3P)2(F)RhCF2, which possessed a 2JRh–F of 33 Hz.23
We hypothesized that this minor side product might be (PNP)RhCF2 and attempted an independent synthesis of it based on the procedure of Grushin et al. that yielded trans-(F)(PPh3)2RhCF2.23 Indeed, treatment of (PNP)Rh(TBE) (TBE = tert-butylethylene) with CsF/Me3SiCF3 (Ruppert's reagent) resulted in complete consumption of (PNP)Rh(TBE) and the formation of (PNP)RhCF2 and (PNP)Rh(C2F4) in about 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 ratio (NMR evidence).25 We were able to isolate (PNP)RhCF2 in 52% yield and of >98% purity by recrystallization. The 31P{1H} NMR and 19F NMR spectra of (PNP)RhCF2 obtained in this fashion were identical to that of the impurity we observed in the reaction in Scheme 1.
:15 ratio (NMR evidence).25 We were able to isolate (PNP)RhCF2 in 52% yield and of >98% purity by recrystallization. The 31P{1H} NMR and 19F NMR spectra of (PNP)RhCF2 obtained in this fashion were identical to that of the impurity we observed in the reaction in Scheme 1.
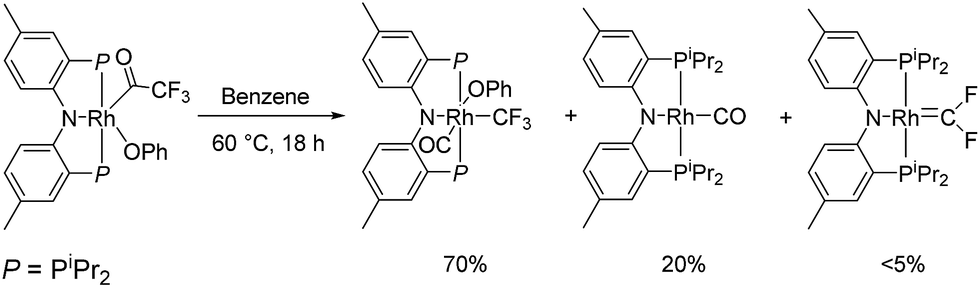 | ||
| Scheme 1 Initial observation of (PNP)RhCF2. | ||
(PNP)Rh(TBE) showed no reaction with Me3SiCF3 alone. Similar to the other cases of use of CsF/Me3SiCF3, we propose that these reagents generate a CF3 anion equivalent that displaces TBE and then loses fluoride, resulting in the formal transfer of CF2 to Rh. Alternatively, CsF/Me3SiCF3 could be generating free CF2 which then binds to Rh. Using the CsF/Me3SiCF3 protocol, we could not avoid the formation of (PNP)Rh(C2F4) due to the generation of free C2F4 from the CsF/Me3SiCF3 mixture. C2F4 was observed by 19F NMR spectroscopy in control experiments where CsF and Me3SiCF3 were mixed in C6D6 and heated at 80 °C. No reaction was observed when (PNP)RhCF2 was treated with another equivalent of CsF/Me3SiCF3. This contrasts the reactivity of Baker's difluorocarbene cobalt(I) complexes6 which undergo a [2 + 1] cycloaddition with free CF2 to form cobalt tetrafluoroethylene complexes.
To date, we have not been able to formulate a reasonable proposal for how (PNP)RhCF2 could be formed from (PNP)Rh(COCF3)(OPh) (Scheme 1). The formation of MCF2 by fluoride migration from M–CF3 is well precedented26 and is likely the key step in forming (PNP)RhCF2; the difficulty is with conceiving of a plausible fate of the other atoms of the original phenyl trifluoroacetate molecule.
Goldman et al. documented formation of (PCP)IrCF2 in a reaction of a (PCP)Ir source with HCF3.24 This reaction proceeded via C–H oxidative addition of HCF3 to Ir followed by loss of HF. In a similar vein, we found that (PNP)Rh(TBE) reacted with HCF3 at 80 °C to provide a mixture of compounds containing (PNP)RhCF2 as a major product (>80%) with (PNP)Rh(CO) and [(PNP)Rh]2(μ-N2) as minor products (Scheme 2). Commercial HCF3 contains dinitrogen as an impurity. Hydrolysis of a difluorocarbene complex to a carbonyl complex has precedent,27 but attempts to purposefully hydrolyze (PNP)RhCF2 proved to be unsuccessful, reminiscent of Baker's cobalt fluorocarbene complexes.5 It is possible that hydrolysis of (PNP)RhCF2 only takes place in the presence of HF (a by-product of (PNP)RhCF2 generation). We observed no intermediates28 in the reaction of (PNP)Rh(TBE) with HCF3, which may indicate that dissociation of TBE29 is the rate-limiting step.
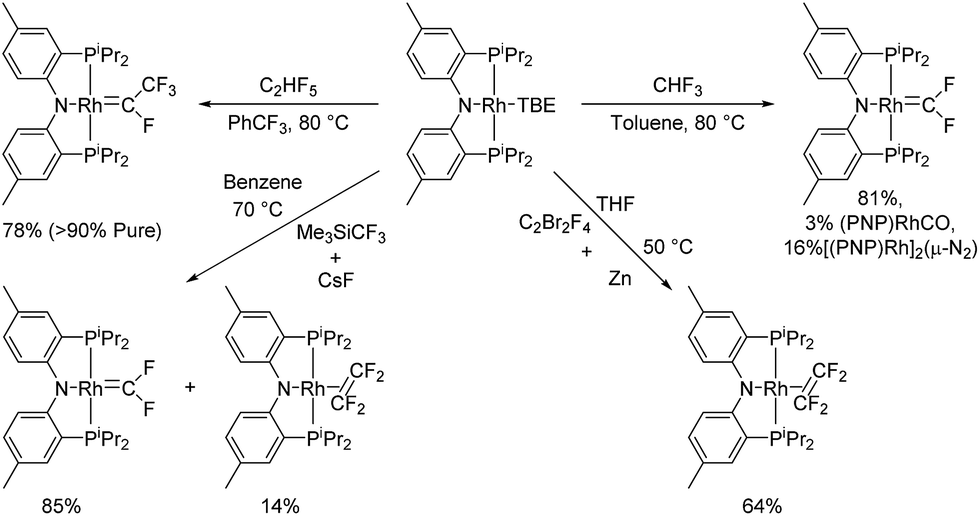 | ||
| Scheme 2 Synthesis of rhodium fluorocarbenes and tetrafluoroethylene complexes. | ||
An analogous reaction of (PNP)Rh(TBE) with C2HF5 was attempted as a potential means to access (PNP)Rh(C2F4). However, the major product of this reaction turned out to be a tetrafluoroethylidene complex (PNP)RhC(F)(CF3) (Scheme 2). Dinitrogen impurity in C2HF5 led to the known29 [(PNP)Rh]2(μ-N2) as a major side product, whose content could be reduced by degassing C2HF5 using the “freeze–pump–thaw” technique. (PNP)RhCF2 was also observed as a side product composing 13% of the reaction mixture when (PNP)Rh(TBE) was treated with 2 atm of C2HF5 and heated overnight at 80 °C. (PNP)RhC(F)(CF3) could be isolated in >90% purity with (PNP)Rh(CO) composing the rest of the mixture. The synthesis of (PNP)Rh(C2F4) was instead accomplished by thermolysis of (PNP)Rh(TBE) in a solution containing C2F4 which was generated in situ by reducing C2F4Br2 with 1.5 eq. of Zn powder at 50 °C in THF (Scheme 2). (PNP)Rh(C2F4) was isolated in 64% yield as a pure solid.
The presence of multiple NMR-active nuclei provided for information-rich NMR spectra of (PNP)RhCF2, (PNP)RhC(F)(CF3), and (PNP)Rh(C2F4). All three complexes displayed C2v-symmetric NMR spectra in solution at ambient temperature. The carbene complexes (PNP)RhCF2 and (PNP)RhC(F)(CF3) displayed characteristic 13C NMR resonances at 206.3 and 225.0 ppm. In the CF2 complex the observation that the two fluorines couple identically to both phosphorus nuclei, and vice versa, is consistent with rapid rotation about the RhCF2 bond on the NMR timescale at room temperature. Likewise the observation of identical coupling of both P-nuclei to all four fluorines in the C2F4 complex is consistent with rapid rotation about the Rh–alkene bond axis. Small energy barriers to these rotations are calculated by DFT (see below).
The identity of (PNP)RhCF2 and (PNP)Rh(C2F4) was confirmed by X-ray diffraction studies on suitable single crystals (Fig. 2). Treating the CF2 or C2F4 ligands as occupying a sole coordination site, the coordination environment about Rh is approximately square planar in both molecules. The CF2 unit in (PNP)RhCF2 lies approximately in that plane, while the C–C vector of the C2F4 ligand in (PNP)Rh(C2F4) is approximately perpendicular to it. The CF2 and C2F4 ligands evidently exert similar trans-influence as the Rh–N distances in (PNP)RhCF2 and (PNP)Rh(C2F4) are only different by ca. 0.01 Å. In general, the metrics of the RhCF2 unit in (PNP)RhCF2 are very similar to the RhCF2 unit in trans-(Ph3P)2(F)RhCF2. The structures of (PNP)RhCF2 and (PNP)Rh(C2F4) contain some close C–F⋯H contacts (C–F⋯H distances of 2.33–2.45) (F⋯C distances of 3.1–3.3 Å). While they are probably unavoidable in these molecules, these distances are short enough to be considered weak F⋯H interactions.30 C–F⋯H interactions have been observed in pincer-ligated zirconium complexes bearing a trifluoromethyl as a pendant group, which have also exhibited through-space H–F coupling visible in their 1H NMR spectra.31 However (PNP)Rh(C2F4) and (PNP)RhCF2 showed no through-space 19F–1H coupling to the isopropyl arms.
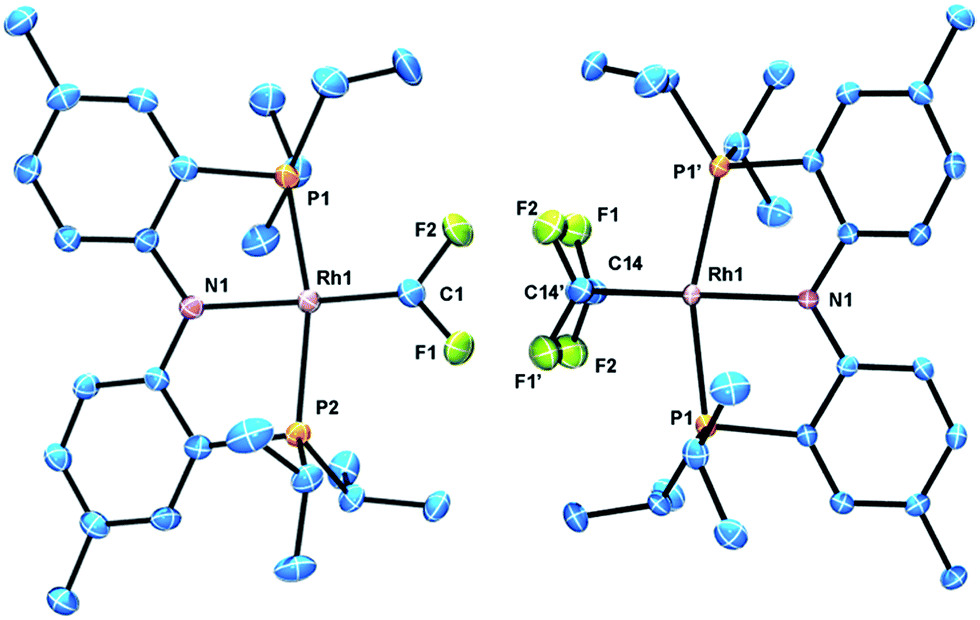 | ||
| Fig. 2 ORTEPs of (PNP)RhCF2 (left) and (PNP)Rh(C2F4) (right). The ellipsoids are set at the 50% probability level, and hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°) for (PNP)RhCF2: Rh1–C1, 1.821(4); Rh1–N1, 2.043(3); C1–F1, 1.335(4); C1–F2, 1.348(5); N1–Rh–C1, 171.39(15); F2–C1–F1, 100.8(3); Rh1–C1–F2, 130.1(3); Rh1–C1–F1, 128.6. (PNP)Rh(C2F4): Rh1–C14, 2.006(3); Rh1–N1, 2.054(3); C14–F1, 1.378(3); C14–F2, 1.361(3); C14–C14′, 1.354(7); C14–Rh–C14′, 39.4(2); C14–Rh–N1, 160.28(10). | ||
Synthesis of cationic fluoromethylidyne
With compounds (PNP)RhCF2, (PNP)Rh(CFCF3), and (PNP)Rh(C2F4) in hand, we contemplated whether one of the fluorides could be removed to yield cationic CxFy complexes. Hughes et al. previously demonstrated proton-induced loss of fluoride from α-positions of Ir perfluoroalkyls,3a and Baker recently demonstrated a Lewis-acid abstraction of a fluoride from N-heterocyclic fluoroalkenes to yield polyfluoroalkenyl imidazolium salts.32 There is significant precedent for electrophilic abstraction of an anionic heteroatom substituent from late-metal carbene complexes by a Lewis acid.18,33 Trialkylsilylium cations, in the form of their salts with halogenated carborane anions, are powerful Lewis acids with high affinity for fluoride.34 We and others have exploited them in catalytic C–F activation reactions35 and thus a [R3Si]+ reagent appeared perfect for fluoride abstraction.
Reactions of (PNP)RhCF2, (PNP)Rh(C2F4), and (PNP)RhC(F)(CF3) with [Et3Si–H–SiEt3][HCB11Cl11]36 or [(Et3Si)2OTf][HCB11Cl11]37 all generated the Et3SiF by-product, indicating that fluoride abstraction took place in all three cases. However, reactions of (PNP)Rh(C2F4) and (PNP)RhC(F)(CF3) resulted in mixtures of several products as seen by 19F NMR spectroscopy and typically broad or no signals were observed by 31P{1H} NMR spectroscopy. The reaction mixtures produced from the reaction of (PNP)Rh(C2F4) or of (PNP)RhC(F)(CF3) with [(Et3Si)2OTf][HCB11Cl11] did regenerate the corresponding starting material when treated with CsF. This indicates that fluoride abstraction from these two isomeric complexes generates isomers of [(PNP)Rh(C2F3)]+ that do not interconvert on the experimental time scale. Although we were not able to identify these compounds experimentally, DFT computational studies were used to investigated possible structures of the [(PNP)Rh(C2F3)]+ isomers (vide infra).
On the other hand, reaction of (PNP)RhCF2 with [Et3Si–H–SiEt3][HCB11Cl11] cleanly and reproducibly generated a new Rh complex that displayed a P2RhF NMR spin system (Scheme 3). The key NMR spectroscopic features of this compound were the unusual 19F NMR chemical shift (66.2 ppm), the very high 1JC–F coupling constant of 470 Hz,38 and the rather substantial 2JRh–F = 136 Hz.
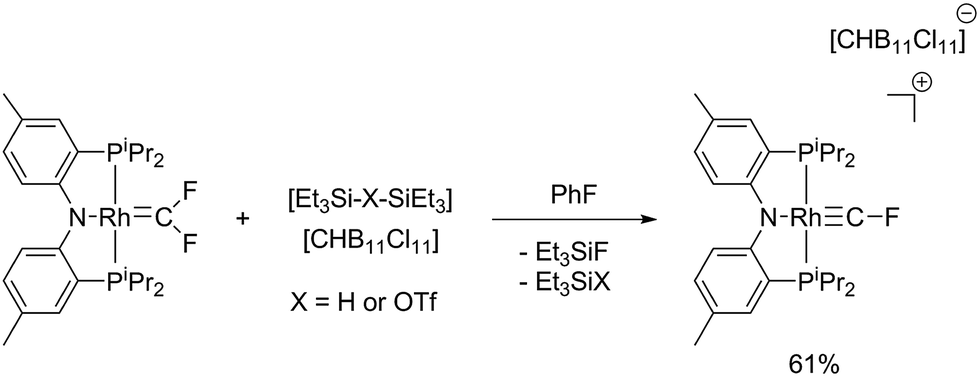 | ||
| Scheme 3 Synthesis of [(PNP)RhCF][CHB11Cl11] via fluoride abstraction from (PNP)RhCF2. | ||
These spectroscopic data are similar to those exhibited by Cp*(CO)2MoCF, whose 19F NMR spectrum contained a singlet at 78.15 ppm, with a large 1JC–F coupling constant of 556 Hz evident by 13C NMR spectroscopy.10a Hughes's other Cp(CO)2MCF (M = Cr, W) complexes also exhibited 19F NMR chemical shifts in this region with high JC–F coupling constants.10b Due to limited solubility in non-interactive solvents and the extensive coupling inherent to the fluoromethylidyne 13C NMR resonance in [(PNP)RhCF]+, it was not observed by 13C{31P}, 13C{1H}, nor 13C{19F} NMR spectroscopy.
X-ray quality crystals of [(PNP)RhCF][CHB11Cl11] were studied using X-ray diffraction to yield a structure fully supportive of a fluorocarbyne formulation (Fig. 3). The structural and NMR spectroscopic features of [(PNP)RhCF]+ are best reviewed in comparison with Cl3RhCF and a few other relevant compounds. Andrews et al. observed IR spectroscopic evidence for Cl3RhCF in reactions of laser-ablated rhodium atoms with CFCl3. A DFT calculation of this product predicted a Rh–C bond length of 1.740 Å and a Rh–C–F bond angle of 143.4°.11d This compares with our observed Rh–C bond length of 1.702(7) Å and a Rh–C–F bond angle of 173.46°. Although both [(PNP)RhCF]+ and Cl3RhCF are four-coordinate, they contain different numbers of valence electrons: from a hypothetical point of view of a [CF]+ ligand, it is attached to a d8 Rh center in [(PNP)RhCF]+, but to a d7 [Cl3Rh]− fragment in Cl3RhCF. The geometry of the RhCF unit in [(PNP)RhCF]+ is similar to Bergman's iridium carbyne complex (Scheme 1), which possesses an Ir–C bond length of 1.734(6) Å and an Ir–C–C bond angle of 175.7(4).18 The Rh–C distance in [(PNP)RhCF]+ is ca. 0.12 Å shorter than that in (PNP)RhCF2, consistent with the increase in the Rh–C bond order. The Rh–C bond distance in [(PNP)RhCF]+ is also ca. 0.07 Å shorter than that in Werner's trans-(PiPr3)2ClRhCC(Me)(H) square planar vinylidene complex.39
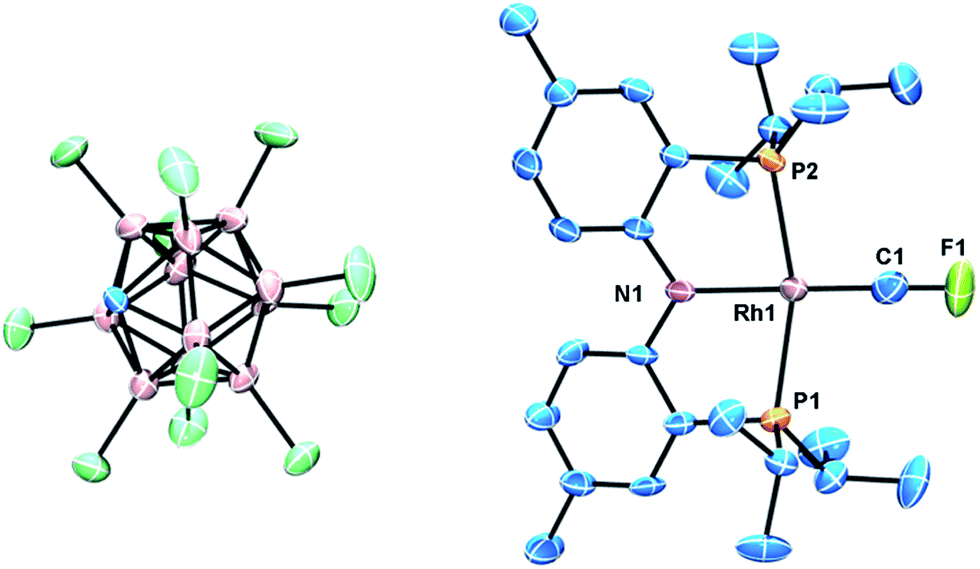 | ||
| Fig. 3 ORTEP of [(PNP)RhCF][HCB11Cl11]. The ellipsoids are set at the 50% probability level, and hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Rh1–C1, 1.702(7); Rh1–N1, 2.019(4); C1–F1, 1.257(8); C1–Rh1–N1, 174.1(3); F1–C1–Rh1, 173.4(7). | ||
Computational studies and discussion
DFT structural studies
Modern Density Functional Theory (DFT) is a powerful tool with which to examine electronic structures and bonding trends in organometallic compounds.40,41 In addition, application of Natural Bond Orbital (NBO)42–47 methods allows insight into some of the subtleties of metal–ligand bonding.42,44,46 The NBO analysis also generates Wiberg Bond Indices (WBI),48 determined within the natural atomic orbital basis, providing one means of estimating bond orders between atoms. Trends in WBI values are also useful in tracking variations in bond multiplicities. The newly synthesized family of fluorocarbon complexes (PNP)Rh(C2F4), (PNP)RhCF2, and [(PNP)RhCF]+ prompted a computational comparison with their (hypothetical) hydrocarbon analogues (PNP)Rh(C2H4), (PNP)RhCH2 and [(PNP)RhCH]+ in order to assess the effects of fluorination on the metal–carbon bonding, and, for the carbene and carbyne complexes, to probe the nature and extent of the multiple bonding between the metal and carbon. In addition, since the CF+ ligand is isoelectronic with the well-known ligands NO+ and CO, it was of interest to establish trends in metal ligand and intraligand bonding between [(PNP)RhCF]+, [(PNP)Rh(NO)]+, and (PNP)Rh(CO). Full molecule DFT studies were performed using the M06 functional49,50 and the triple-ζ LACV3P**++ basis set, which uses extended core potentials51–54 on heavy atoms and a 6-311G**++ basis55–58 for other atoms, as implemented in the Jaguar59,60 suite of programs. Full details are available as ESI.†
Selected bond lengths and computed WBI values for the calculated complexes are provided in Table 1, with metric comparisons to the available crystallographic structures reported here. The DFT calculated metrics are in good agreement with crystallographic numbers, giving confidence in the DFT metrics for the unknown complexes. One exception appears in (PNP)Rh(C2F4) in which the C–C distance for the coordinated alkene (1.354 Å) is only slightly longer than that in C2F4 itself (1.318 Å)61 and is much shorter than all other transition metal complexes of this perfluoroalkene in the Cambridge Structure Database.62 In contrast, the DFT calculated value (1.416 Å) is in good agreement with other crystallographically determined values62 and is much more sensible with respect to calculated WBI values (see below).
| Compound | Rh–Pave | Rh–N | Rh–Cave | C–C | C–Xave (ligand) |
|---|---|---|---|---|---|
| a DFT calculated (M06/LACV3P**++) values are in plain text; X-ray crystallographic values are in bold. b WBI values are in italics. c This crystallographic value is questionable. See discussion in the text. | |||||
| (PNP)Rh(C2H4) | 2.332 | 2.098 | 2.165 | 1.394 | 1.088 |
| 0.462 | 0.378 | 0.453 | 1.474 | 0.930 | |
| (PNP)Rh(C2F4) | 2.378 | 2.100 | 2.048 | 1.416 | 1.346 |
| 2.3309(11) | 2.054(3) | 2.006(3) | 1.354(7) | 1.369(3) | |
| 0.436 | 0.352 | 0.594 | 1.186 | 0.858 | |
| (PNP)RhCH2 |
2.338 | 2.216 | 1.850 | — | 1.104 |
| 0.476 | 0.225 | 1.250 | 0.955 | ||
| (PNP)RhCF2 |
2.331 | 2.156 | 1.864 | — | 1.325 |
| 2.302(12) | 2.043(3) | 1.821(4) | 1.341(5) | ||
| 0.462 | 0.297 | 1.168 | 0.911 | ||
| [(PNP)RhCH]+ |
2.378 | 2.075 | 1.728 | — | 1.110 |
| 0.440 | 0.332 | 1.714 | 0.942 | ||
| [(PNP)RhCF]+ |
2.384 | 2.061 | 1.740 | — | 1.247 |
| 2.337(16) | 2.019(4) | 1.702(7) | 1.257(8) | ||
| 0.432 | 0.341 | 1.587 | 1.065 | ||
| [(PNP)RhC–CF3]+ |
2.384 | 2.054 | 1.734 | — | 1.499 |
| 0.453 | 0.330 | 1.770 | 0.960 | ||
| (PNP)Rh(CO) | 2.336 | 2.117 | 1.855 | — | 1.152 |
| 0.462 | 0.291 | 1.041 | 2.031 | ||
| [(PNP)Rh(NO)]+ | 2.393 | 2.019 | 1.778 (Rh–N) | — | 1.141 (NO) |
| 0.439 | 0.445 | 1.151 | 1.917 | ||
All the complexes examined here can be formally viewed as square planar d8 compounds, i.e., as complexes of a d8, three-coordinate fragment (PNP)Rh with neutral or cationic ligands. The NBO perspective of bonding interactions in such compounds42,46 requires 4-electron/3-center bonds between the pair of trans ligands such that the alkene, carbene, and carbyne ligands of interest are always involved in a shared bonding interaction with the N of the PNP pincer. Clearly contributions to this shared interaction may be weighted differently in each case, and the WBI values should reflect this.
Comparison of C2H4 and C2F4 ligands
The bonding between alkenes and transition metal fragments is well understood,63 but a comparison between C2H4 and C2F4 coordinated to identical metal–ligand fragments is rare. A classic intramolecular example involves CpRh(C2H4)(C2F4)64 but the hydrocarbon and fluorocarbon alkenes are necessarily bound to different fragments in this molecule. Fig. 4 illustrates the key Natural Localized Molecular Orbitals (NLMOs)42,45,46 arising from NBO calculations of interactions between C2H4 and C2F4 and the truncated65 (PNP)Rh fragment. The bonding orbitals (σ and π) are essentially localized on Rh and the alkene ligand, while the corresponding antibonding NLMOs show significant “tailing” involving the σ and π orbitals on the trans-N of the pincer ligand. This “tailing” is indicative of delocalization of these N electrons into the corresponding σ* and π* components of the Rh–alkene interaction; it is significantly greater for the σ* component and corresponds to the 3-center/4-electron bonding expected between trans-ligands in a d8 Rh(I) complex. The WBI values indicate significantly greater reduction in C–C bonding and increase in Rh–C bonding in coordinated C2F4 than in C2H4. This is consistent with the shorter Rh–C distances and with the idea of a more metallacyclopropane structure and stronger Rh–C bonding for the fluorinated alkene complex. Not surprisingly, stronger bonding to the fluorinated alkene results in weaker bonding to the trans ligand, with correspondingly lower WBI values for the Rh–N bonds (Table 1). In the π*-perp NMLO for the C2F4 there is also evidence for delocalization from F-lone pairs (see Fig. 4).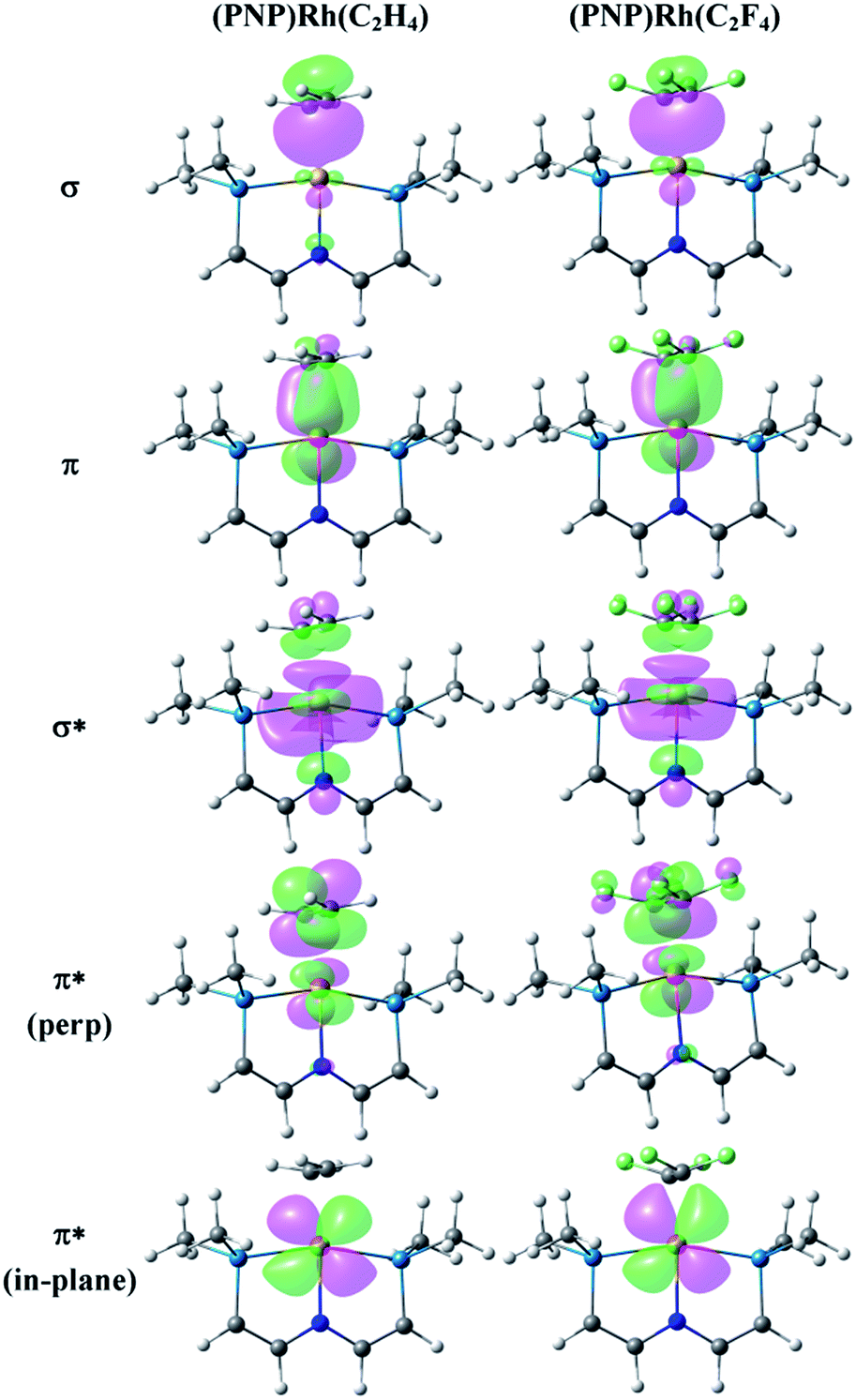 | ||
| Fig. 4 NLMOs for the bonding interactions between C2H4 (column 1) and C2F4 (column 2) and the (PNP)Rh fragment. For clarity the PiPr2 groups have been replaced by PMe2 groups and the aryl part of the pincer truncated to P–CHCH–N linkers. | ||
There are two Rh→alkene backbonding options involving π*-perp or π*-in-plane. Clearly the ground state conformation of the C2F4 complex utilizes the former, but the latter is available for an in-plane C2F4 conformation, leading to a low barrier for C2F4 rotation. Similar arguments for facile rotation of C2F4 ligands in Ru(II) complexes have been put forth elsewhere.66 The free energy profile for C2F4 rotation was calculated using a truncated65 version of the PNP ligand (identical to that shown in Fig. 4), and is unusual. Relative to the perpendicular conformation observed in the ground state, two transition states were located. The first, lying 7.1 kcal mol−1 above the ground state, corresponds to a 45 degree rotation about the Rh–alkene bond axis, and the second, lying 8.7 kcal mol−1 above the ground state, is the conformation in which the fluoroalkene lies in the coordination plane. These barriers are low enough in energy that rotation should be fast on the NMR timescale, consistent with the observed NMR data. The barriers contrast with those for the corresponding C2H4 analogue, for which the in-plane conformation is a minimum, lying only 0.4 kcal mol−1 above the ground state, and the 45 degree conformation is a transition state lying 2.5 kcal mol−1 above the ground state.
Comparison of CH2 and CF2 ligands
Fig. 5 presents the corresponding NLMOs for the CF2 complex with a truncated65 PNP ligand. Those for the CH2 analogue are very similar, except for the fluorine delocalizations into the π*-NLMO, and are not illustrated here but can be found in the ESI (Fig. S17†). The σ and π NLMOs are consistent with a formal double bond between Rh and the CF2 (or CH2) ligands, with the p-orbital on C and d-orbital on Rh providing the π-component. These NLMOs look essentially identical to those in Fig. 4, except for a more significant delocalization of the fluorine lone pairs in the π*-perp NLMO. But now there is competition between the rhodium d-orbital and the fluorine lone pair p-orbitals for π-bonding with the carbene carbon, as expected; in (PNP)RhCH2 only the metal can provide this π-bonding. Consequently π-bonding with fluorines diminishes π-bonding with Rh and, relative to the CH2 complex, the Rh–C WBI decreases significantly and the Rh–C distance increases; notably the C–F WBI is greater than in the previously discussed C2F4 complex. In contrast to the alkene ligands discussed above there is overall weaker metal–carbon bonding to CF2 than CH2 and the corresponding trans-Rh–N WBI value is larger for (PNP)RhCF2.
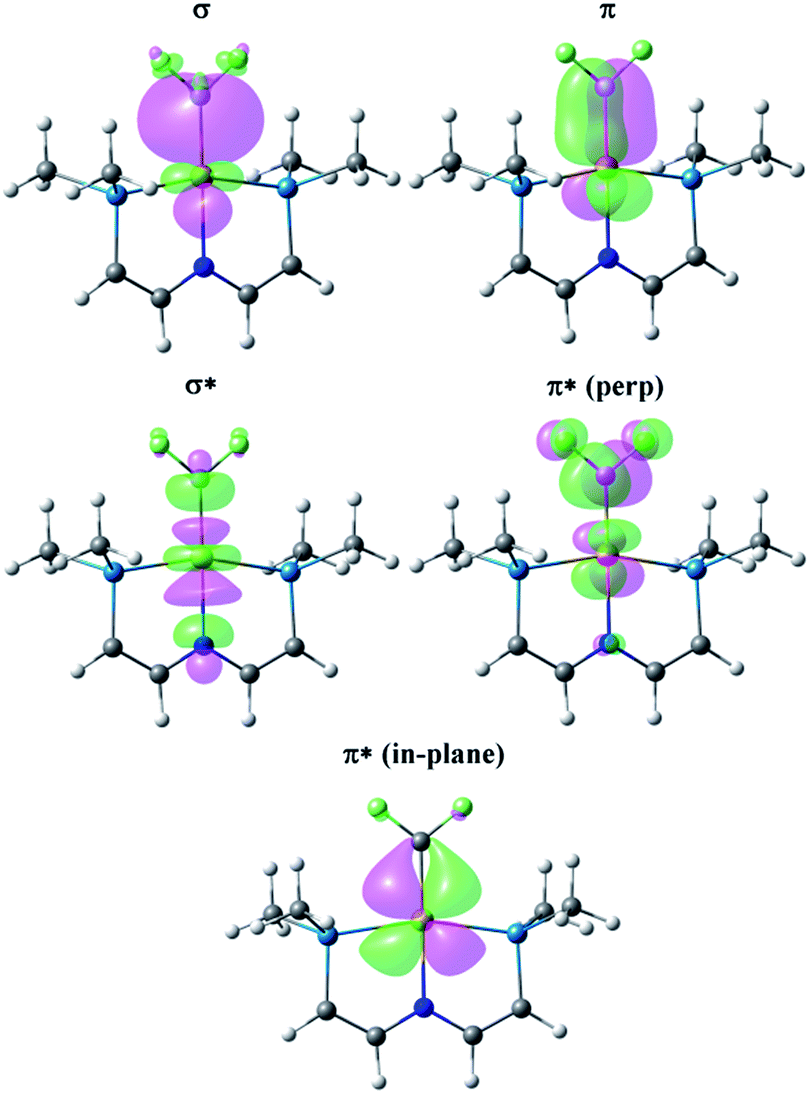 | ||
| Fig. 5 NLMOs for the bonding interactions between the CF2 ligand and the (PNP)Rh fragment. For clarity the PiPr2 groups have been replaced by PMe2 groups and the aryl part of the pincer truncated to P–CHCH–N linkers. | ||
Facile CF2 ligand rotation is expected due to the availability of the π*-perp and π*-in-plane interactions. Calculations on the truncated65 ligand analogue of the CF2 complex reveal the same trend in conformational energetics observed for the C2F4 complex (vide supra). The in-plane CF2 conformation, with the π*-perp interaction, is the ground state, with two transition states at 45 degree and 90 degree (perpendicular to the coordination plane) lying 2.0 and 4.8 kcal mol−1 higher in energy, respectively. The barriers are consistent with the experimental observation of fast rotation on the NMR timescale. In contrast, the rotation of the corresponding CH2 ligand is even more facile, with the perpendicular, 45 degree and in-plane conformations lying at essentially equal energies.
Comparison of CH+, CF+ and CCF3+ ligands
Fig. 6 presents the corresponding NLMOs for [(PNP)RhCF]+. As with the carbene complexes, those for [(PNP)RhCH]+ are similar except for enhanced “tailing” in the antibonding NLMOs for the CF+ complex. In contrast to the alkene and carbene complexes (vide supra) there is now a second fully engaged π-component for the Rh–C bond involving the in-plane d-orbital and a second p-orbital on the CF+ (or CH+) ligand. In the antibonding NLMOs we see the expected σ-donation from the trans-N in σ*, a small π-donation from trans-N in π*(perp), and a small donation from the Rh–P bonds in π*(in-plane). But once again the largest delocalizations in the π* NLMOs comes from the F lone pairs, interactions that cannot occur in [(PNP)RhCH]+. Consequently the Rh–C WBI for [(PNP)RhCH]+ is substantially larger than that for the CF+ analogue, consistent with the shorter Rh–C distance in the former; as before, a smaller Rh–C WBI in [(PNP)RhCF]+ leads to a larger WBI for the trans-Rh–N bond. In the CCF3 analogue, in which F lone pair participation with the carbyne carbon is removed, a larger Rh–C WBI is calculated, with a correspondingly smaller Rh–N WBI.
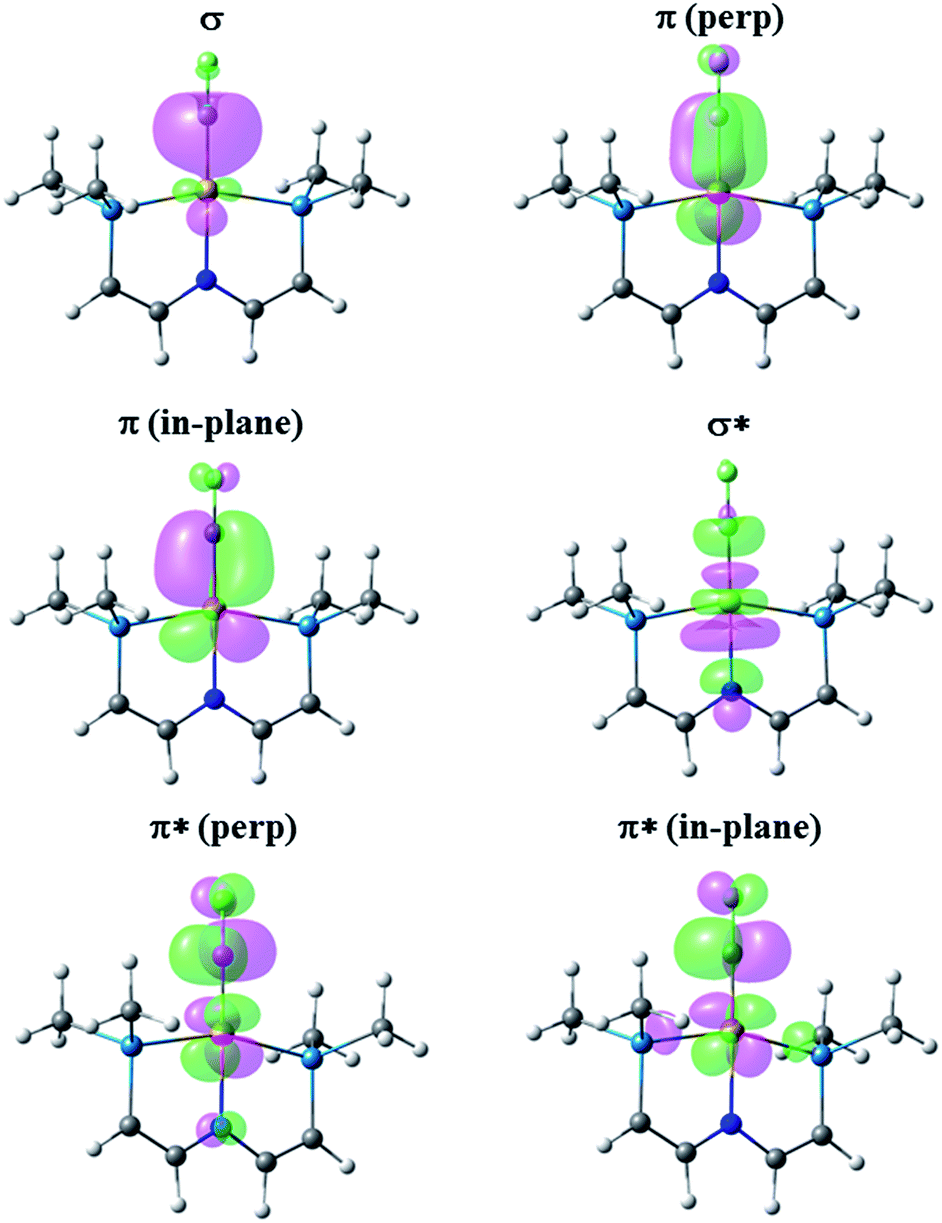 | ||
| Fig. 6 NLMOs for the bonding interactions between the CF+ ligand and the (PNP)Rh fragment. For clarity the PiPr2 groups have been replaced by PMe2 groups and the aryl part of the pincer truncated to P–CHCH–N linkers. | ||
Degree of Rh–C multiple bonding
The Rh–C bonding in these carbene and carbyne complexes is inextricably linked to interactions with the trans-N since there are necessarily shared bonding components between trans-ligands, with additional complexities introduced by the fluorine substituents on carbon. So we cannot expect the RhCX2 interaction to be a true double bond, or that in the cationic RhCX to be a triple bond, even though we may draw resonance structures that reflect these prejudices. However, the WBI values for both RhCX2 bonds (CH21.250; CF21.168) are significantly larger than unity, though not close to two, while those for the RhCX cations are significantly larger still (CH 1.714; CF 1.587; CCF31.770), though not close to the bond order of three. Clearly there is significant multiple bonding between Rh and these unsaturated carbon ligands with higher bond orders to these ligands being reflected in lower bond orders to the trans-N.
Comparison of CF+, NO+, and CO ligands
The NLMOs for the CO and NO+ complexes are similar to those of the CF+ compounds discussed previously and are not shown here. Considering this series of isoelectronic complexes as involving a linear Rh–X–Y array the three resonance forms (A, B, C) for the contiguous π-system are shown in Fig. 7, along with the WBI values for the appropriate bonds in the Rh–C–O, Rh–N–O and Rh–C–F complexes. The WBI values are consistent with progressively increased contributions of resonance forms C > B > A on changing the ligand from CO to NO+ to CF+, as expected from their relative π-acceptor abilities. Similar conclusions were reached for the fragments M(CO)2(XY) [M = Cr, Mo, W; XY = CO, NO+, CF+] in a previous study.10b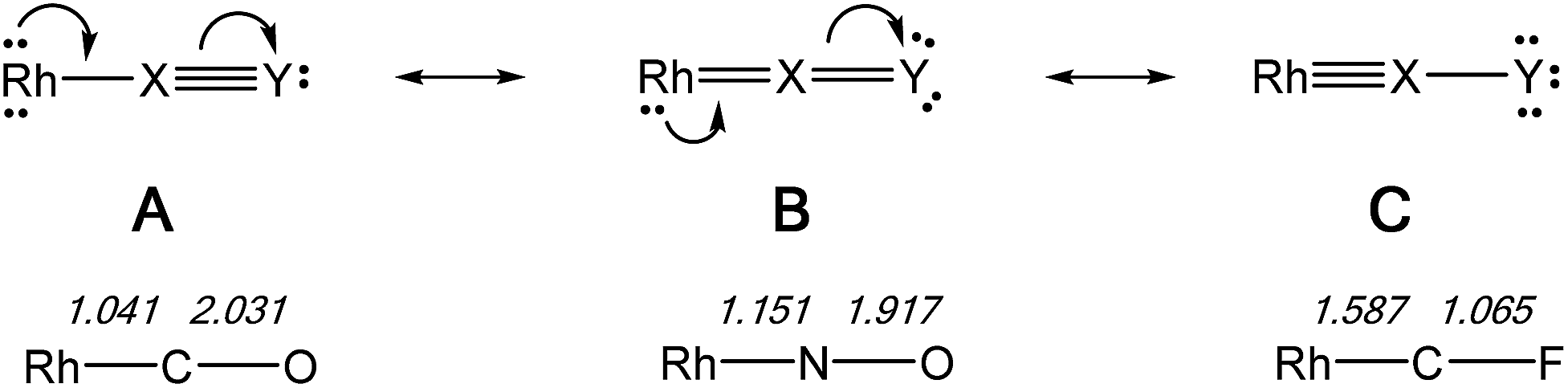 | ||
| Fig. 7 Resonance forms for the π-system in a linear Rh–X–Y ligand array, with WBI values for the bonds in Rh–C–O, Rh–N–O, and Rh–C–F complexes. All three complexes are isoelectronic and no formal charges are shown. | ||
Relative energies of isomeric fluorocarbon ligands
It was of interest to compare the relative energies of the (PNP)Rh(C2F4) complex with its carbene isomer (PNP)Rh(CFCF3). At the DFT/M06/LACV3P**++ level the free energy of the carbene isomer is found to be 2.4 kcal mol−1 uphill from its alkene analogue. Interestingly the carbene CFCF3 ligand lies perpendicular to the (PNP)Rh plane, in contrast to the CF2 analogue described above, presumably due to steric interactions between the CF3 and the cis-PR2 groups.Potential products arising from fluoride abstraction from these isomeric complexes were also subjected to DFT evaluation. Abstraction of fluoride from (PNP)Rh(CFCF3) could occur from the α-position to yield a carbyne complex (PNP)Rh(CCF3)+, analogous to the characterized CF+ complex described above, or from the β-position to afford the corresponding isomeric η1-perfluorovinyl cation (PNP)Rh(CFCF2)+. These are found to have almost identical free energies, with the carbyne complex lying only 0.4 kcal mol−1 higher than its perfluorovinyl isomer. An η2-perfluorovinyl isomer, the potential initial product of fluoride abstraction from the (PNP)Rh(C2F4) was found to lie 12.1 kcal mol−1 above its η1-perfluorovinyl analogue. Structures of all these compounds and their relative energies are provided in Fig. 8.
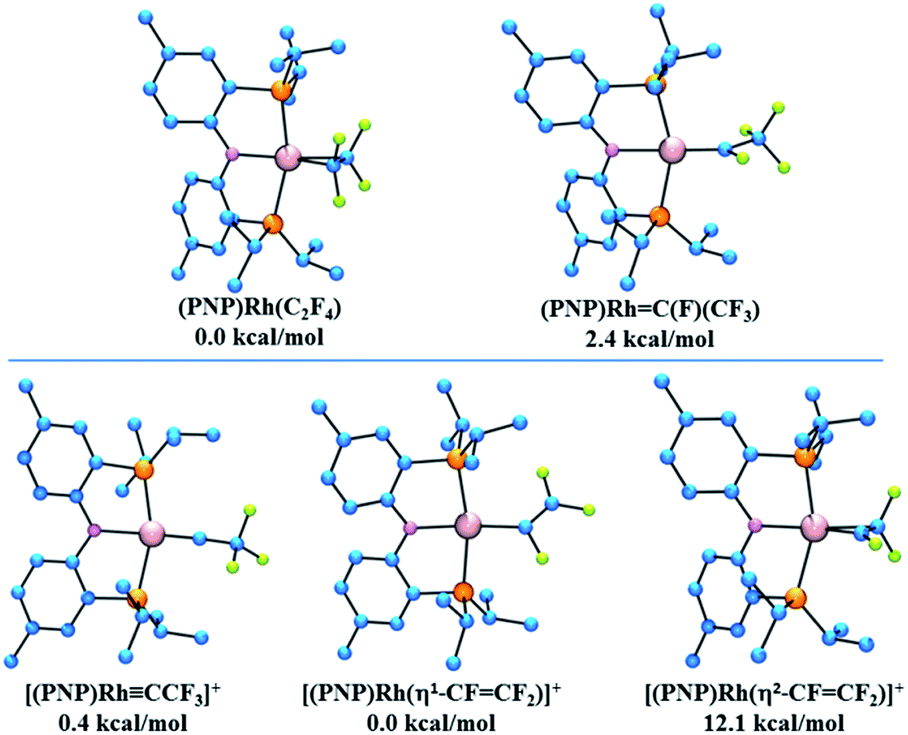 | ||
| Fig. 8 (Top) Calculated structures and relative energies for (PNP)Rh C2F4 isomers. (Bottom) Calculated structures and relative energies for proposed structures resulting from fluoride abstraction from (PNP)RhC(F)(CF3). | ||
Conclusions
In summary, we have shown that (PNP)Rh perfluorocarbene complexes can be synthesized by treating a Rh(I) precursor with Ruppert's reagent or a fluoroalkane containing a C–H bond. Using silylium reagents, a fluoride can be abstracted from (PNP)RhCF2 to form a cationic fluoromethylidyne. Thus the (PNP)Rh system conveniently allows synthesis and comparison of perfluoroolefin, perfluorocarbene, and perfluorocarbyne complexes. Using DFT calculations we were able to compare the natural localized molecular orbitals of these fluoroorganic complexes to their hypothetical hydrocarbon analogues, as well as to the CO and NO+ complexes. We established that the fluorine atoms on the carbene and carbyne ligands participate in π donation to the acceptor orbitals on carbon to compete with back donation from the metal. This resulted in a longer Rh–C bond in the fluorinated complexes compared to their hydrocarbon analogues. However, C2F4 was calculated to form a shorter Rh–C bond than the C2H4 complex. Calculated Wiberg bond indices also showed that although the unsaturated fluorocarbon ligands have bond orders greater than one to rhodium, the nitrogen trans to these ligands interacts with their antibonding orbital and decreases the bond order to less than a true double and triple bond.
Acknowledgements
We are grateful for the support of this research by the Welch Foundation (grant A-1717 to O. V. O.).Notes and references
- F. L. Taw, A. E. Clark, A. H. Mueller, M. T. Janicke, T. Cantat, B. L. Scott, P. J. Hay, R. P Hughes and J. L. Kiplinger, Organometallics, 2012, 31, 1484 CrossRef CAS.
- (a) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (b) W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Aceña, K. Izawa, H. Liu and V. A. Soloshonok, J. Fluorine Chem., 2014, 167, 37 CrossRef CAS; (c) M. Ohashi, H. Shirataki, K. Kikushima and S. Ogoshi, J. Am. Chem. Soc., 2015, 137, 6496 CrossRef CAS PubMed.
- (a) R. P. Hughes, R. B. Laritchev, J. Yuan, J. A. Golen, A. N. Rucker and A. L. Rheingold, J. Am. Chem. Soc., 2005, 127, 15020 CrossRef CAS PubMed; (b) C. J. Bourgeois, R. P. Hughes, J. Yuan, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2006, 25, 2908 CrossRef CAS; (c) R. P. Hughes, J. Fluorine Chem., 2010, 131, 1059 CrossRef CAS.
- J. Yuan, R. P. Hughes, J. A. Golen and A. L. Rheingold, Organometallics, 2010, 29, 1942 CrossRef CAS.
- D. J. Harrison, S. I. Gorelsky, G. M. Lee, I. Korobkov and R. T. Baker, Organometallics, 2013, 32, 12 CrossRef CAS.
- G. M. Lee, D. J. Harrison, I. Korobkov and R. T. Baker, Chem. Commun., 2014, 50, 1128 RSC.
- (a) D. J. Harrison, G. M. Lee, M. C. Leclerc, I. Korobkov and R. T. Baker, J. Am. Chem. Soc., 2013, 135, 18296 CrossRef CAS PubMed; (b) J. T. Fuller, D. J. Harrison, M. C. Leclerc, R. T. Baker, D. H. Ess and R. P. Hughes, Organometallics, 2015, 34, 5210 CrossRef CAS.
- D. J. Harrison, A. L. Daniels, I. Korobkov and R. T. Baker, Organometallics, 2015, 34, 5683 CrossRef CAS.
- M. C. Leclerc, J. M. Bayne, G. M. Lee, S. I. Gorelsky, M. Vasiliu, I. Korobkov, D. J. Harrison, D. A. Dixon and R. T. Baker, J. Am. Chem. Soc., 2015, 137, 16064 CrossRef CAS PubMed.
- (a) H. Huang, R. P. Hughes, C. R. Landis and A. L. Rheingold, J. Am. Chem. Soc., 2006, 128, 7454 CrossRef CAS PubMed; (b) H. Huang, R. P. Hughes and A. L. Rheingold, Dalton Trans., 2011, 40, 47 RSC.
- (a) J. T. Lyon and L. Andrews, Inorg. Chem., 2006, 45, 9858 CrossRef CAS PubMed; (b) J. T. Lyon, H.-G. Cho and L. Andrews, Organometallics, 2007, 26, 6373 CrossRef CAS; (c) J. T. Lyon and L. Andrews, Eur. J. Inorg. Chem., 2008, 1047 CrossRef CAS; (d) H.-G. Cho and L. Andrews, Organometallics, 2010, 29, 2211 CrossRef CAS.
- R. R. Schrock, Acc. Chem. Res., 1986, 19, 342 CrossRef CAS.
- R. R. Schrock, Chem. Rev., 2002, 102, 145 CrossRef CAS PubMed.
- O. V. Ozerov, L. A. Watson, M. Pink and K. G. Caulton, J. Am. Chem. Soc., 2007, 129, 6003 CrossRef CAS PubMed.
- G. Jia, Coord. Chem. Rev., 2007, 251, 2167 CrossRef CAS.
- (a) S. R. Caskey, M. H. Stewart, Y. J. Ahn, M. J. A. Johnson, J. L. C. Roswell and J. W. Kampf, Organometallics, 2007, 26, 1912 CrossRef CAS; (b) M. Shao, L. Zheng, W. Qiao, J. Wang and J. Wang, Adv. Synth. Catal., 2012, 354, 2743 CrossRef CAS.
- (a) R. Ramírez-Contreras, N. Bhuvanesh and O. V. Ozerov, Organometallics, 2015, 34, 1143 CrossRef and references within; (b) T. Kurogi, P. J. Carroll and D. J. Mindiola, J. Am. Chem. Soc., 2016, 138, 4306 CrossRef CAS PubMed.
- H. F. Luecke and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 11008 CrossRef CAS.
- T. Rappert, O. Nürnberg, N. Mahr, J. Wolf and H. Werner, Organometallics, 1992, 11, 4156 CrossRef CAS.
- W. A. Nugent and J. M. Mayer, Metal-Ligand Multiple Bonds, John Wiley & Sons, New York, 1988 Search PubMed.
- (a) R. H. Holm, Chem. Rev., 1987, 87, 1401 CrossRef CAS; (b) J. M. Mayer, Comments Inorg. Chem., 1988, 8, 125 CrossRef CAS; (c) T. A. Betley, Q. Wu, T. V. Voorhis and D. G. Nocera, Inorg. Chem., 2008, 47, 1849 CrossRef CAS PubMed.
- Y. Zhu, D. A. Smith, D. E. Herbert, S. Gatard and O. V. Ozerov, Chem. Commun., 2012, 48, 218 RSC.
- J. Goodman, V. V. Grushin, R. B. Larichev, S. A. Macgregor, W. J. Marshall and D. C. Roe, J. Am. Chem. Soc., 2009, 131, 4236 CrossRef CAS PubMed.
- J. Choi, D. Y. Wang, S. Kundu, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Science, 2011, 332, 1545 CrossRef CAS PubMed.
- (PNP)Rh(CO) accounted for approximately 1% of the reaction mixture.
- D. Huang, P. R. Koren, K. Folting, E. R. Davidson and K. G. Caulton, J. Am. Chem. Soc., 2000, 122, 8916 CrossRef CAS.
- (a) R. P. Hughes, Eur. J. Inorg. Chem., 2009, 4591 CrossRef CAS; (b) P. J. Brothers and W. R. Roper, Chem. Rev., 1988, 88, 1293 CrossRef CAS.
- We do not observe the HF by-product spectroscopically, but it is possible it is consumed by a reaction with borosilicate glass.
- S. Gatard, C. Guo, B. M. Foxman and O. V. Ozerov, Organometallics, 2007, 26, 6066 CrossRef CAS.
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22 RSC.
- S. C. F. Kui, N. Zhu and M. C. W. Chan, Angew. Chem., Int. Ed., 2003, 42, 1628 CrossRef CAS PubMed.
- M. C. Leclerc, S. I. Gorelsky, B. M. Gabidullin, I. Korobkov and R. T. Baker, Chem.–Eur. J., 2016, 22, 8063 CrossRef CAS PubMed.
- (a) E. O. Fischer, G. Kreis, C. G. Kreiter, J. Müller, G. Huttner and H. Lorenz, Angew. Chem., 1973, 85, 618 CrossRef CAS; (b) E. O. Fischer, U. Schubert, W. Kleine and H. Fischer, Inorg. Synth., 1979, 19, 172 CrossRef CAS.
- D. G. Gusev and O. V. Ozerov, Chem.–Eur. J., 2011, 17, 634 CrossRef CAS PubMed.
- C. Douvris and O. V. Ozerov, Science, 2008, 29, 1188 CrossRef PubMed.
- S. P. Hoffmann, T. Kato, F. S. Tham and C. A. Reed, Chem. Commun., 2006, 767 RSC.
- L. P. Press, B. J. McCulloch, W. Gu, C.-H. Chen, B. M Foxman and O. V. Ozerov, Chem. Commun., 2015, 51, 14034 RSC.
-
1
J
C–F coupling constant was determined in an experiment using [BArF20]− as a counter anion for [(PNP)RhCF]+.
- F. J. G. Alonso, A. Höhn, J. Wolf, H. Otto and H. Werner, Angew. Chem., Int. Ed. Engl., 1985, 24, 406 CrossRef.
- J. N. Harvey, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2006, 102, 203 RSC.
- T. R. Cundari, Compr. Organomet. Chem. III, 2007, 1, 639 CAS.
- C. R. Landis and F. Weinhold in The Chemical Bond: Fundamental Aspects of Chemical Bonding, ed. G. Frenking and S. Shaik, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2014, vol. 1, pp. 91–119 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. K. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013 Search PubMed.
- F. Weinhold and C. R. Landis, Discovering Chemistry with Natural Bond Orbitals, John Wiley & Sons, Hoboken, 2012 Search PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1 CrossRef CAS.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, Cambridge, 2005 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- T. H. Dunning and P. J. Hay, Modern Theoretical Chemistry, in Applications of Electronic Structure Theory, ed. H. F. Schaefer III, Plenum, 1977, vol. 4, p. 461 Search PubMed.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110 CrossRef CAS.
- Jaguar, versions 7.0-9.3, Schrödinger, LLC, New York, 2007–2016 Search PubMed.
- D. Lentz, A. Bach, J. Buschmann, P. Luger and M. Messerschmidt, Chem.–Eur. J., 2004, 10, 5059 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- R. H. Crabtree and M. Mingos, Comprehensive Organometallic Chemistry III, Elsevier, New York, 2006 Search PubMed.
- L. J. Guggenberger and R. Cramer, J. Am. Chem. Soc., 1972, 94, 3779 CrossRef CAS.
- We refer to a PNP ligand in which the PiPr2 groups have been replaced by PMe2 groups and the aryl part of the pincer replaced by P–CHCH–N linkers as “truncated.” It was used in calculations from which the NLMO Figures were derived and for calculations of rotational barriers in the C2H4/C2F4/CH2/CF2 complexes. All other calculations used the full version of the ligand. The truncated PNP ligands has been previously used by Wu and Hall ( H. Wu and M. B. Hall, Dalton Trans., 2009, 5933 RSC ).
- (a) O. J. Curnow, R. P. Hughes and A. L. Rheingold, J. Am. Chem. Soc., 1992, 114, 3153 CrossRef CAS; (b) O. J. Curnow, R. P. Hughes, E. N. Mairs and A. L. Rheingold, Organometallics, 1993, 12, 3102 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1518497–1518499. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05391b |
| ‡ These authors contributed equally. |
| § Present address: The Dow Chemical Company, Freeport, Texas 77541, USA. |
| ¶ Present address: Department of Chemistry, University of Manitoba, Winnipeg, MB, Canada. |
| This journal is © The Royal Society of Chemistry 2017 |