
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisualising the membrane viscosity of porcine eye lens cells using molecular rotors†
Peter S.
Sherin
 *ab,
Ismael
López-Duarte
c,
Michael R.
Dent
c,
Markéta
Kubánková
c,
Aurimas
Vyšniauskas
c,
James A.
Bull
c,
Evdokiya S.
Reshetnikova
d,
Andrey S.
Klymchenko
e,
Yuri P.
Tsentalovich
ab and
Marina K.
Kuimova
*c
*ab,
Ismael
López-Duarte
c,
Michael R.
Dent
c,
Markéta
Kubánková
c,
Aurimas
Vyšniauskas
c,
James A.
Bull
c,
Evdokiya S.
Reshetnikova
d,
Andrey S.
Klymchenko
e,
Yuri P.
Tsentalovich
ab and
Marina K.
Kuimova
*c
aInternational Tomography Center SB RAS, Institutskaya Street 3A, 630090, Novosibirsk, Russia. E-mail: petr.sherin@tomo.nsc.ru
bDepartment of Natural Sciences, Novosibirsk State University, Pirogova Street 2, 630090, Novosibirsk, Russia
cDepartment of Chemistry, Imperial College London, Exhibition Road, London, SW7 2AZ, UK. E-mail: m.kuimova@imperial.ac.uk
dInstitute of Molecular and Cellular Biology SB RAS, 8/2 Lavrentiev Avenue, 630090, Novosibirsk, Russia
eLaboratoire de Biophotonique et Pharmacologie, UMR 7213 CNRS, Faculté de Pharmacie, Université de Strasbourg, 74 Route du Rhin, 67401 ILLKIRCH Cedex, France
First published on 14th February 2017
Abstract
The plasma membranes of cells within the eye lens play an important role in metabolite transport within the avascular tissue of the lens, maintaining its transparency over the entire lifespan of an individual. Here we use viscosity-sensitive ‘molecular rotors’ to map the microscopic viscosity within these unusual cell membranes, establishing that they are characterised by an unprecedentedly high degree of lipid organisation.
Introduction
In an eye, the lens functions to focus incident light onto the retina. Nature has adopted a specific design for the lens tissue to make it transparent and flexible. Except for a thin layer of epithelial cells, the lens mostly consists of organelle-free fiber cells densely packed in an onion-like structure.1,2 The transparency of the lens is ensured through the ordered distribution of proteins within the cells, which have thin cell membranes and a small intercellular spacing compared to the wavelengths of visible light.1,2 A characteristic feature of lens cell membranes is a high proportion of gap junctions,3 which provide direct and extensive cell-to-cell transfer of cellular components that does not require energy consumption. Membrane defects that lead to the interruption of metabolite transport can induce many disorders of the lens, including some types of cataract.2Proteins and lipids of mammalian lenses have no turnover and during normal aging they undergo numerous modifications that change the properties of the tissue, ultimately resulting in lens coloration, an increase in stiffness and impairments of metabolite transport.4 These age-related changes may cause defects in the cell membrane structure. The formation of an age-related barrier that prevents the diffusion of metabolites between the nucleus and the cortex has also been reported in humans.5 The retarding of metabolite exchange predisposes the lens nucleus to oxidative stress – a key factor in the formation of cataracts.6 Therefore, a study of the normal functioning of the lens, as well as age- and cataract-related changes in metabolite transport through the lens cells may shed light on the general mechanism of cataractogenesis.
One of the most important parameters governing the dynamic properties of a cell membrane is its viscosity, which determines the diffusion and self-organisation of the proteins and lipids within. Existing reports on the viscosity of the whole lens have, however, provided contradicting values ranging from several cP7 to several hundred cP,8 perhaps reflecting the fact that these measurements were not specific to individual cell organelles. To the best of our knowledge no direct measurements of viscosity have been performed within eye lens cell membranes either in vivo or ex vivo.
An attractive approach for the investigation of the biophysical properties of biomembranes is the use of environmentally sensitive fluorescent probes, which change their color, fluorescence intensity and/or lifetime in response to their microenvironment.9 Firstly, solvatochromic probes operate through excited-state charge transfer with representative dyes such as Laurdan,10 styryl dyes,11 Nile Red12 and pyrene13 derivatives. They enable the monitoring of the polarity and hydration of cell membranes and visualisation of their lipid order.14 The other important family of membrane probes operates through intramolecular rotation, which includes molecular rotors15,16 and planarizable dyes (flippers).17
In this manuscript we report a methodology that enables the measurement of the spatially-resolved viscosity of lens membranes ex vivo, using the fluorescence lifetime imaging microscopy (FLIM) of molecular rotors.15,16 Molecular rotors are small synthetic fluorophores for which the fluorescence quantum yield and fluorescence lifetime (τf) show strong responses to the viscosity of their immediate environment. In order to perform quantitative and concentration-independent measurements of viscosity, τf of a molecular rotor can be calibrated as a function of viscosity.16,18,19 Following this calibration, the FLIM of molecular rotors allows the imaging of the distribution of microscopic viscosities across a variety of heterogeneous samples.19
Results and discussion
Thin slices of lenses (14–20 μm) from young animals were obtained using a cryotome and stained using solutions of membrane-soluble molecular rotors (1a–c and 2, see Chart 1), that have been used previously for measuring the viscosity within the plasma membranes of bacterial spores and bacterial cells, as well as in several types of eukaryotic cells.19–24 Surprisingly, none of the existing molecular rotors were deemed ideal for the imaging of the lens cells due to poor uptake leading to dim images (in the case of 1b and 2) or strong evidence for dye aggregation (in the case of 1a), see Fig. S4 of the ESI.†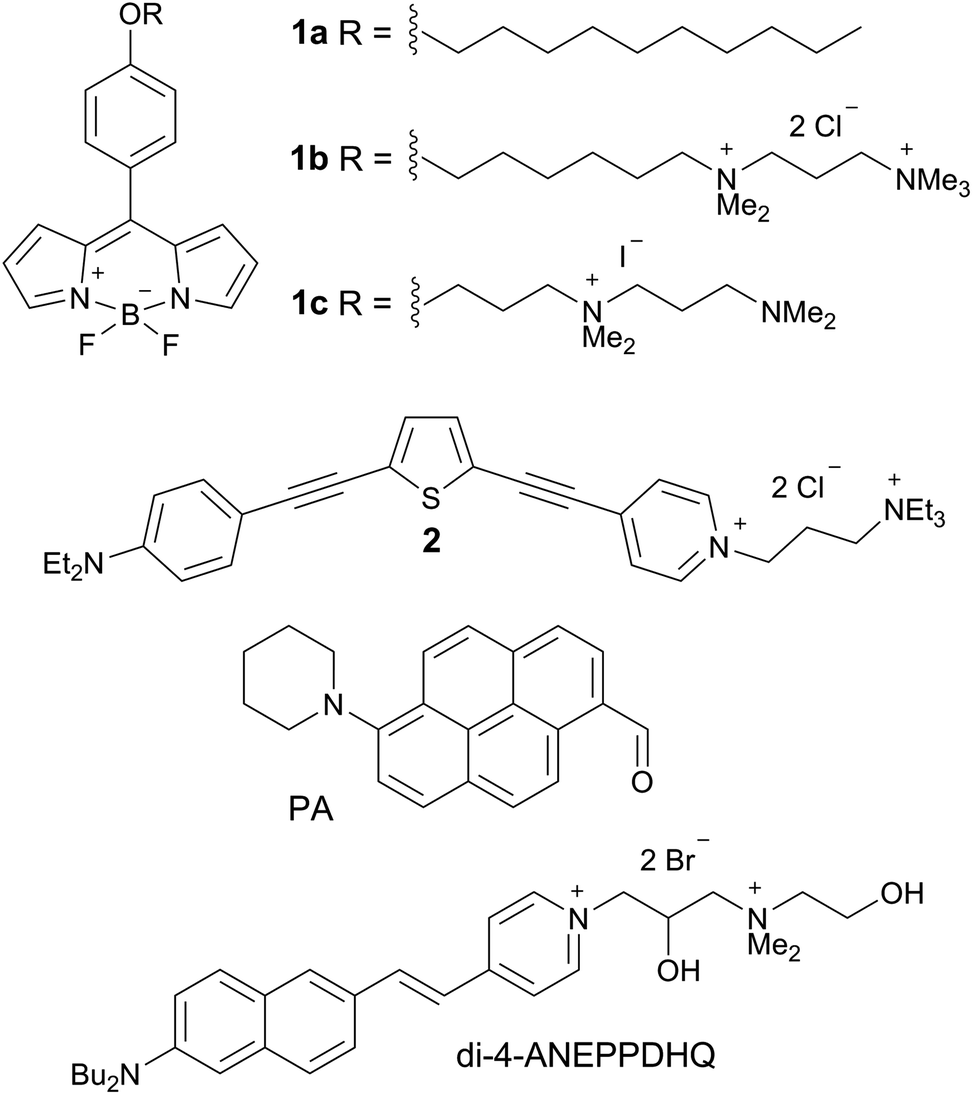 | ||
| Chart 1 Molecular structures of 1a–c, 2, PA and di-4-ANEPPDHQ. | ||
We hypothesised that the staining of the lens cell membranes might be improved through increasing the aqueous solubility of the molecular rotor, aiding its delivery into tissues while retaining its ability to localise in lipid membranes. Consequently, we have designed and synthesised the derivative 1c, which displays higher aqueous solubility compared to both 1a and 1b. The synthetic pathway to 1c, detailed synthetic procedure and characterisation are reported in the ESI.† We confirmed that 1c indeed acts as a molecular rotor and shows a strong increase in fluorescence lifetime with viscosity, and this lifetime calibration is independent of the solution temperature, Fig. 1A and B and S2.† The viscosity dependence of the τf value could be fitted using a variant of a Hill function,25,26 which is presented in Fig. S3† and enables the quantitative conversion of fluorescence lifetime values to viscosities.
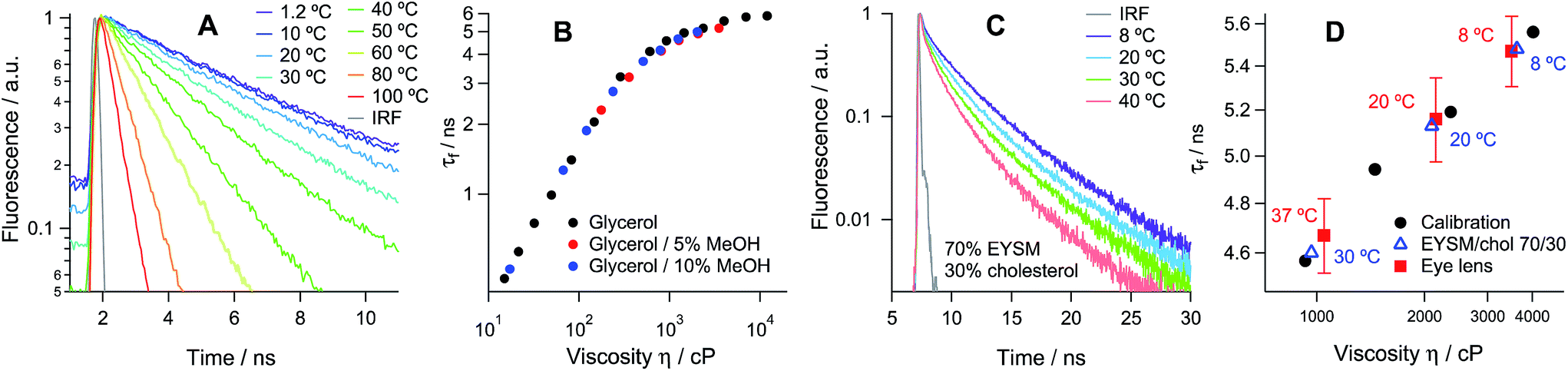 | ||
Fig. 1 (A) Time-resolved fluorescence decay of 1c obtained in glycerol and (B) fluorescence lifetime calibration vs. viscosity obtained in methanol/glycerol mixtures at various temperatures. (C) Time-resolved fluorescence decay of 1c in LUVs of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 EYSM:cholesterol recorded at 8–40 °C. (D) Determination of viscosity in porcine eye lens slices and in LUVs of 70:30 EYSM:cholesterol at 8–37 °C using the calibration curve in (B). The individual temperatures are shown next to the data. :30 EYSM:cholesterol recorded at 8–40 °C. (D) Determination of viscosity in porcine eye lens slices and in LUVs of 70:30 EYSM:cholesterol at 8–37 °C using the calibration curve in (B). The individual temperatures are shown next to the data. | ||
Hexagonal structures, typical of slices from the equatorial plane of the lens samples,2 were observed using transmission and confocal microscopy (Fig. S4 and S5†) and FLIM, stained with all available dyes. While all tested molecules displayed preferential membrane localisation in fiber cells, the best contrast in the signal between membranes and intracellular space was observed with the new rotor 1c, Fig. 2A and S5.†
 | ||
| Fig. 2 Fluorescence lifetime imaging of porcine eye lens slices stained with 1c. (A) Two-photon excited fluorescence image; (B and C) FLIM images of the τ1 and τ2 distributions recorded at 20 °C. Scale bars = 10 μm. (D) Time-resolved fluorescence decay obtained from whole images and (E) fluorescence lifetime histograms for τ1 and τ2 recorded at 8, 20 and 37 °C. | ||
The time-resolved fluorescence decay of 1c in lens cell membranes at 20 °C is best fitted with a biexponential model with characteristic time constants of τ1 = 1.06 ± 0.08 and τ2 = 5.16 ± 0.15 ns, Table S2 of the ESI.† It is well known that for BODIPY-based dyes the presence of biexponential decay can be indicative of dye aggregation,27 with aggregated species characterised by a weak emission band centred at 650–700 nm. Aggregate formation results in the quenching of the main emission band centred at 515 nm,27 which renders the lifetime-viscosity calibration curve unusable. While dye 1a showed strong evidence for aggregation in the lens cell membranes (Fig. S4†), the decays recorded for 1c in the two spectral windows for monomers and aggregates (480–560 and 600–700 nm, respectively) were identical and did not depend on the dye concentration in the range of 1 to 10 μM (Fig. S6†). This data confirms that the biexponential decay kinetics of 1c are not due to the presence of aggregates but rather indicates the fact that the rotor can probe the microscopic heterogeneities in the lens membranes, for example, a distribution of dye positions within the lipid bilayer (laterally or vertically) that is below the resolution of our diffraction-limited technique.
Previously, we have used experiments together with molecular dynamics simulations to demonstrate that in model membrane systems that display a high degree of lipid packing, such as liquid ordered (Lo) or gel phases, BODIPY rotors can occupy two positions, which results in biexponential fluorescence decay.28 The longer-lived component was determined to be representative of the high viscosity within the tail region of the lipid bilayer. Such biexponential decay was recently detected in the plasma membranes of living systems stained with 1a, e.g. in the inner membranes of live E. coli bacteria20 and B. subtilis spores,22 translating to a viscosity of ca. 1000 cP at 20 °C.
In order to examine the localisation and fluorescence decay properties of 1c in model membranes, we incorporated 1c into large and giant unilamellar vesicles (LUVs and GUVs) with various lipid compositions: pure Ld (1,2-dioleoyl-sn-glycero-3-phosphocholine, DOPC), pure Lo (egg yolk sphingomyelin, EYSM mixed with cholesterol 70:30), DOPC mixed with cholesterol and a ternary phase-separated system composed of DOPC:EYSM:cholesterol 20:55:25 (Fig. S7 and S8†). The analysis of this data revealed that in the pure Ld phase the rotor displays monoexponential decays, with lifetimes of 0.7–2.8 ns at 55–8 °C (corresponding to viscosities of 30–240 cP). Such decays indicate a single orientation of 1c in the Ld membranes. For all other LUV compositions, however, the decays were biexponential (Fig. S8†), consistent with at least two rotor orientations in these membranes. The fitting results are given in Table S1.†
Next we recorded FLIM images of the lens cells at three temperatures: 8, 20 and 37 °C. The time resolved fluorescence decays of 1c within the cells show an excellent agreement with its lifetimes within the model Lo membranes (EYSM:cholesterol 70:30%) at all temperatures, Fig. 1D and Tables S1–S3.† This indicates the similarity in their structures, which is supported by the fact that the membrane of the porcine eye lens is known to contain large quantities of sphingomyelin and cholesterol.29
We have used the lifetime/viscosity calibration graph for 1c, Fig. 1B, to assign viscosity values to the plasma membranes of the lens cells: 3500 cP at 8 °C, 2150 cP at 20 °C and 1050 cP at 37 °C (Fig. 1D). These are unprecedentedly high values that exceed the viscosities reported using the molecular rotor technique in the plasma membranes of all other types of cells studied so far, including E. coli live cells (ca. 1000 cP at room temperature and at 37 °C), B. subtilis spores (1000 cP) or germinated cells (ca. 400 cP) and eukaryotic cells (ca. 200 cP), all at room temperature.19–24 While E. coli cells exhibited a similar membrane viscosity at 37 °C, the values recorded at 20 °C were significantly lower than those in the lens, which might reflect the ability of the E. coli membrane lipid composition to adapt to temperature.20 We note that the short component of the decay of 1c corresponds to the viscosity of ca. 50 cP, assigned to 1c molecules localised in the lipid headgroup region of the membrane,28 which shows a smaller variation with temperature (Fig. 2E). The presence of a short component is consistent with the results previously reported for highly ordered membranes stained with rotors 1a–b.28
Next, we set out to test whether the high viscosities detected in the lens cell membranes using 1c were related to an abundance of Lo-like lipid domains. We used three previously reported fluorescence sensors of lipid organisation (Chart 1): a pyrene-based dye (PA),13 di-4-ANEPPDHQ30 and molecular rotor 2, which was previously shown to selectively stain the plasma membranes of eukaryotic cells24,31 and is characterised by equal partitioning of the Ld and Lo phases of model lipid bilayers. We recently used the phasor analysis32 of the fluorescence decays of 2 which indicated that 2 detects both Lo and Ld domains in the plasma membranes of live eukaryotic cells.24
Given the possible parameter uncertainties in fitting the biexponential decays of fluorophores it is useful to apply the phasor analysis,32 a model-free approach that uses the Fourier transforms of time-resolved fluorescence decays to produce a decay representation in a phasor space, on the universal circle. This approach is free from the uncertainties introduced by choosing a (multi)exponential decay model, and does not require multi-parameter fitting.
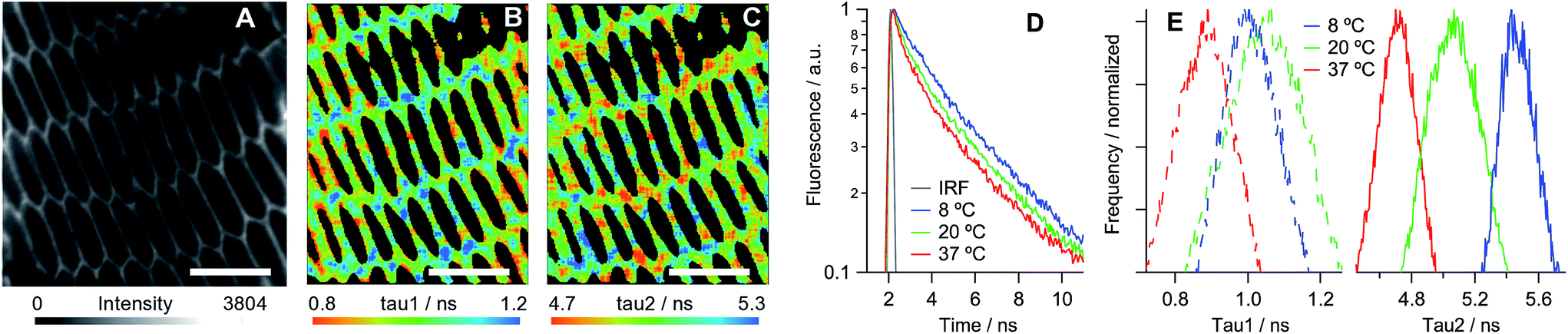
We have established that all three dyes selectively stain the plasma membrane of the eye lens slices (Fig. 3). While 2 is characterised by a low quantum yield, which in combination with biexponential fluorescence decays was prohibitive for quantitative viscosity determination in lens membranes, we were able to use phasor analysis of the FLIM images of 2 to confirm that the lens cell membranes are indeed present in the Lo phase, Fig. 3D. To establish this fact, we compared the fluorescence decays of 2 in lens cells within model Lo phases (DPPC/cholesterol LUVs) and model Ld phases (DOPC/cholesterol LUVs) that were reported previously.24 The decays recorded for 2 in lens cell membranes fell on the straight line between the pure Lo and Ld phases in the phasor space, with the cell phasors overlapping well with the phasors of the Lo model membranes.
 | ||
| Fig. 3 Fluorescence images of porcine eye lens slices stained with 10 μM solutions of (A) 2, (B) PA and (C) di-4-ANEPPDHQ in PBS. Scale bars = 10 μm. (D) Phasor analysis of the FLIM data for 2 in cells and in model lipid membranes containing 0–30% cholesterol24 (increase in % of cholesterol is shown with arrows). DPPC: 1,2-dipalmitoyl-sn-glycero-3-phosphocholine. (E) The emission spectra obtained from samples (B) (blue) and (C) (red). | ||
PA13 is a push–pull dye, operating through excited state charge transfer, and di-4-ANEPPDHQ30 is sensitive to the membrane potential. Both dyes are able to distinguish between the Lo and Ld phases based on a spectral shift in their emission: blue-shifted emission is expected in the case of an Lo phase compared to an Ld phase. We detected blue-shifted emission maxima at 510 and 580 nm in eye lens cell membranes for PA and di-4-ANEPPDHQ, respectively (Fig. 3E), corresponding to the Lo phase of model membranes.13,30 These results suggest that the Lo phase is abundant within the eye lens cell membranes, a conclusion that is supported by the phasor analysis of the FLIM of lens cells stained with di-4-ANEPPDHQ (Fig. S9†). The data for all three dyes indicate the presence of a higher proportion of Lo-like domains in lens cells compared to the reported literature values for eukaryotic cells studied to date.13,14,24,30
Conclusions
In summary, we have used fluorescent probes sensitive to lipid order and organisation to demonstrate that the plasma membrane of porcine eye lens fiber cells displays an unprecedentedly high degree of lipid ordering, very similar to the Lo phase in model lipid membranes. Furthermore, we have designed a fluorescent molecular rotor 1c that has allowed us to quantitatively determine the viscosity of these unusual membranes at various temperatures. We report the viscosity of the tail region of the porcine eye lens plasma membrane as 3500–1050 cP at 8–37 °C, a significantly larger variation than was reported for E. coli membrane viscosity at temperatures of 20–37 °C.19 These values represent the highest viscosities reported for the lipid tail regions of the plasma membrane of any live cell studied to date and appear to represent a pure ‘lipid raft’-like continuous phase. These results open up the opportunity to study the functioning of ‘lipid rafts’ in a controlled and easy-to-visualise live system that can be extended to studies in vivo with an appropriate FLIM setup. At the same time we believe that this data will be crucial for enhancing our understanding of the changes in viscosity and lipid organisation in eye lenses during normal aging and cataract progression that will be extremely useful for cataract research in human health.Experimental section
Materials
The synthesis of 1a,161b,23231 and PA13 has been reported previously, while di-4-ANEPPDHQ was obtained from Thermo Fisher Scientific. All solvents used were spectroscopic grade. Chloroform, methanol and DMSO were obtained from Sigma-Aldrich, and glycerol was obtained from Alfa-Aesar. The lipids 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), egg yolk sphingomyelin (EYSM) and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) were obtained from Avanti Polar lipids as chloroform solutions. Cholesterol was obtained from Avanti Polar lipids as a lyophilised solid and dissolved in chloroform at 1 mg ml−1.
Viscosity calibration of the fluorescence lifetime of compound 1c
A mode-locked femtosecond Ti:Sapphire laser (Chameleon Vision II, Coherent Inc., Germany) tunable over the 680–1080 nm range (140 fs pulse duration, 80 MHz) was used as an excitation source. The excitation light was frequency doubled using a second harmonic generation (SHG) crystal (Harmonic, Coherent Inc., Germany). The excitation was performed at 480 nm and fluorescence was detected at 520 nm after passing through a 500 nm long-pass filter. The detection system consisted of a DCC-100 detector control module (Becker&Hickl, Germany), PMC-100-1 photomultiplier tube (Hamamatsu, Japan), Omni-λ 150 monochromator (LOT-Quantum Design, Germany), and SPC-830 single-photon counting card (Becker&Hickl, Germany). The measurements were performed in quartz cuvettes with a 1 cm path length, measured in a qpod cuvette holder heated using a TC 125 Peltier thermostat (Quantum Northwest, USA). Measurements were performed in a temperature range of 1–100 °C. The temperature was checked before and after each decay acquisition with a thermocouple. The number of photons at the peak of all traces was 10000 counts. The viscosity values for glycerol/methanol solutions at various temperatures were taken from ref. 25, 27 and 33.
GUV/LUV preparation
LUVs were formed using an extrusion method. A solution of the appropriate lipid and a dye (1c or di-4-ANEPPDHQ) was prepared in chloroform, which was then evaporated off under nitrogen. Multilamellar vesicles (MLVs) were then prepared by hydrating the lipid film using enough water to give a 1 mM solution of lipid and vortexing for 1 minute above the gel transition temperature of the lipid. This was then extruded 10 times through a polycarbonate membrane with a pore diameter of 200 nm using a LIPEX extruder (Northern Lipids Inc., Canada), ensuring it was above the gel transition temperature of the lipid.GUVs were prepared using an electroformation method.34 A solution of 1 mg ml−1 lipid with 1c in chloroform was added dropwise (ca. 30 μl) onto a clean conductive indium tin oxide (ITO) coated glass slide and spread using a glass coverslip to give a thin film of lipid and 1c, before being lyophilised for 60 minutes. A polydimethyl siloxane spacer was placed over the lipid film and a second ITO coated slide was then placed on top of the spacer, creating a sealed chamber. The chamber was filled with 0.1 M aqueous sucrose solution using a syringe. An AC voltage of 1.0 V and 10 Hz was applied for 90 minutes, making sure the chamber was held above the gel transition temperature of the lipids being used. In order to be visible in phase contrast microscopy, the GUVs formed in this way were suspended in a 0.125 M aqueous solution of glucose.
Preparation of eye lens slices
Enucleated porcine eyes were obtained from a local abattoir (Novosibirsk, Russia) within 2–4 hours after the animal’s death and stored on ice for less than 2 hours. The extracted lenses were then frozen and stored at −80 °C until used in experiments 2–6 days after extraction. Thin slices of lenses in the range of 14–20 μm were obtained using a cryotome (OTF cryostat, Bright Instrument Company Ltd, UK) and mounted onto microscope slides (SuperFrost Plus, Thermo Fisher Scientific, Germany). Two animal lenses (ap. 100 slices in total) were used to optimise the sample preparation procedures and to get the preliminary data and three lenses (ap. 50 slices in total) were used to acquire the statistics. The age of the animals was 6–8 months.Incubation of eye lens slices with fluorescent dyes
Phosphate buffered saline (PBS) from Invitrogen was used in all experiments. Each fluorescent dye was dissolved in DMSO at a concentration of 1 mM and stored at −20 °C. PBS solutions of a dye in the concentration range of 1–10 μM were prepared by adding of 1 μl of DMSO stock solution in 1.0–0.1 ml of PBS; the resulting total concentration of DMSO was less than 1% by volume. Each eye lens slice was stained with a drop of 10–15 μl of a dye in PBS for 10 minutes and washed once with the same volume of PBS. After the removal of the excess of PBS using filter paper, the lens slices were covered by a cover slip and sealed with non-fluorescent lacquer to avoid the drying of the tissue slice during the data acquisition. In our experiments we used lenses from young animals that have no differences in lipid composition between the cortex and nuclear regions.35,36 Thus, frames for the acquisition of the fluorescent time resolved signal were chosen randomly in a slice.Fluorescence confocal imaging
Confocal fluorescence imaging of the eye lens slices stained with various dyes was performed using a confocal laser-scanning microscope Leica TCS SP5 II (Leica Microsystem Ltd, Germany). The room temperature imaging was carried out with a ×100 (N.A. 1.0) HCX PL APO CS oil immersion objective lens with a correction collar (11506279, Leica Microsystem Ltd, Germany), and the imaging at 8 and 37 °C was performed with a ×40 objective (N.A. 0.85) HCX PL APO (Leica Microsystem Ltd, Germany). The samples were excited at 476 nm with an internal argon ion laser and emission intensity was recorded at 490–700 nm.Fluorescence lifetime imaging
FLIM images of 256 × 256 pixels were obtained using a Leica TCS SP5 II inverted scanning confocal microscope coupled with a TCSPC module SPC830 (Becker&Hickl, Germany) and internal FLIM detector PMH-100 (Becker&Hickl, Germany). The module was synchronised to a Ti-Sapphire pulsed laser source (680–1080 nm, 80 MHz, 140 fs, Chameleon Vision II, Coherent Inc., Germany).Two-photon excitation was performed at 920 nm for compounds 1a–c and 2 and at 860 nm for PA and di-4-ANEPPDHQ, respectively. The emission was captured at the following wavelength ranges: 470–560 nm for 1a–c (monomers) and 600–680 nm (aggregates); 470–700 nm for 2; 440–590 nm for PA; 470–650 nm for di-4-ANEPPDHQ; corresponding to the maximum of the emission bands of each dye. The laser power was maintained at <150 mW before entering the microscope to avoid cell damage. The acquisition time was varied in the range of 100–800 s depending on the emission intensity from each dye.
FLIM data were analysed in SPCImage software (Becker&Hickl, Germany) using a two-exponential decay. Pixels were binned to maintain a minimum peak count in the decay maximum of 1000 counts per pixel and thresholding was used to remove the background noise. A pseudocolour scale was assigned to each fluorescence lifetime, amplitude and the goodness of fit (χ2) values (red for small values and blue for large values) to provide the corresponding lifetime maps.
Phasor analysis was performed using an in-house MATLAB R2012a code. In this technique the fluorescence decays collected are Fourier transformed and their imaginary parts are plotted against the real parts in a so-called phasor plot.32 All phasors were corrected using an instrument response function.
Acknowledgements
M. K. K. is grateful to the EPSRC for the Career Acceleration Fellowship (EP/I003983/1); A. V. thanks the EPSRC for the Prize Studentship. M. K. is thankful for the Imperial President’s Scholarship; P. S. S. acknowledges the RSC for the Researcher Mobility Fellowship. We thank Yosuke Niko for the synthesis of the PA probe. The animal eye lenses were harvested with the financial support from the Russian Scientific Foundation (project #14-14-00056).References
- A. van Leeuwenhoek, The Collected Letters of Antoni van Leeuwenhoek, letter no. 81, english, July 25, 1684, Swets Zeitlinger, Amsterdam, 1952, vol. IV, pp. 253–299 Search PubMed.
- Duane’s Ophthalmology, ed. W. Tasman and E. A. Jaeger, Williams & Wilkins, Philadelphia, USA, 2007, ch. 15 Search PubMed.
- (a) B. T. Philipson, L. Hanninen and E. A. Balazs, Exp. Eye Res., 1975, 21, 205 CrossRef CAS PubMed; (b) J. Kuszak, H. Maisel and C. V. Harding, Exp. Eye Res., 1978, 27, 495 CrossRef CAS PubMed.
- H. Bloemendal, W. de Jong, R. Jaenicke, N. H. Lubsen, C. Slingsby and A. Tardieu, Prog. Biophys. Mol. Biol., 2004, 86, 407 CrossRef CAS PubMed.
- (a) M. H. J. Sweeney and R. J. W. Truscott, Exp. Eye Res., 1998, 67, 587 CrossRef CAS PubMed; (b) B. A. Moffat, K. A. Landman, R. J. W. Truscott, M. H. J. Sweeney and J. M. Pope, Exp. Eye Res., 1999, 69, 663 CrossRef CAS PubMed.
- R. J. W. Truscott, Exp. Eye Res., 2005, 80, 709 CrossRef CAS PubMed.
- M. T. Thao, D. Perez, J. Dillon and E. R. Gaillard, Mol. Vision, 2014, 20, 125 CAS.
- (a) R. A. Schachar, R. W. Chan and M. Fu, Br. J. Ophthalmol., 2007, 91, 366 CrossRef PubMed; (b) R. A. Schachar, R. W. Chan and M. Fu, Br. J. Ophthalmol., 2011, 95, 1010 CrossRef PubMed.
- A. S. Klymchenko, Acc. Chem. Res., 2017, 50(2), 366–375 CrossRef CAS PubMed.
- L. A. Bagatolli, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1541 CrossRef CAS PubMed.
- L. Jin, A. C. Millard, J. P. Wuskell, X. M. Dong, D. Q. Wu, H. A. Clark and L. M. Loew, Biophys. J., 2006, 90, 2563 CrossRef CAS PubMed.
- O. Kucherak, S. Oncul, Z. Darwich, D. A. Yushchenko, Y. Arntz, P. Didier, Y. Mély and A. S. Klymchenko, J. Am. Chem. Soc., 2010, 132, 4907 CrossRef CAS PubMed.
- Y. Niko, P. Didier, Y. Mely, G. I. Konishi and A. S. Klymchenko, Sci. Rep., 2016, 6, 18870 CrossRef CAS PubMed.
- A. S. Klymchenko and R. Kreder, Chem. Biol., 2014, 21, 97–113 CrossRef CAS PubMed.
- M. A. Haidekker and E. A. Theodorakis, Org. Biomol. Chem., 2007, 5, 1669 CAS.
- M. K. Kuimova, Phys. Chem. Chem. Phys., 2012, 14, 12671 RSC.
- M. Dal Molin, Q. Verolet, A. Colom, R. Letrun, E. Derivery, M. Gonzalez-Gaitan, E. Vauthey, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2015, 137, 568 CrossRef CAS PubMed.
- T. Förster and G. Z. Hoffmann, Phys. Chem., 1971, 75, 63 Search PubMed.
- M. K. Kuimova, G. Yahioglu, J. A. Levitt and K. Suhling, J. Am. Chem. Soc., 2008, 130, 6672 CrossRef CAS PubMed.
- J. T. Mika, A. J. Thompson, M. R. Dent, N. J. Brooks, J. Michaels, J. Hofkens and M. K. Kuimova, Biophys. J., 2016, 111, 1528 CrossRef CAS PubMed.
- P. Loison, N. A. Hosny, P. Gervais, D. Champion, M. K. Kuimova and J.-M. Perrier-Cornet, Biochim. Biophys. Acta, 2013, 1828, 2436 CrossRef CAS PubMed.
- P. Loison, P. Gervais, J.-M. Perrier-Cornet and M. K. Kuimova, Biochim. Biophys. Acta, 2016, 1858, 2060 CrossRef CAS PubMed.
- I. López-Duarte, T. T. Vu, M. A. Izquierdo, J. A. Bull and M. K. Kuimova, Chem. Commun., 2014, 50, 5282 RSC.
- M. R. Dent, I. López-Duarte, C. J. Dickson, P. Chairatana, H. L. Anderson, I. R. Gould, D. Wylie, A. Vyšniauskas, N. J. Brooks and M. K. Kuimova, Chem. Commun., 2016, 52, 13269 RSC.
- A. Vyšniauskas, M. Qurashi, N. Gallop, M. Balaz, H. L. Anderson and M. K. Kuimova, Chem. Sci., 2015, 6, 5773 RSC.
- A. Vyšniauskas, M. Balaz, H. L. Anderson and M. K. Kuimova, Phys. Chem. Chem. Phys., 2015, 17, 7548 RSC.
- Y. Wu, M. Štefl, A. Olzyńska, M. Hof, G. Yahioglu, P. Yip, D. R. Casey, O. Ces, J. Humpolíčková and M. K. Kuimova, Phys. Chem. Chem. Phys., 2013, 15, 14986 RSC.
- M. R. Dent, I. López-Duarte, C. J. Dickson, N. D. Geoghegan, J. M. Cooper, I. R. Gould, R. Krams, J. A. Bull, N. J. Brooks and M. K. Kuimova, Phys. Chem. Chem. Phys., 2015, 17, 18393 RSC.
- J. M. Deeley, T. W. Mitchell, X. Wei, J. Korth, J. R. Nealon, S. J. Blanksby and R. J. W. Truscott, Biochim. Biophys. Acta, 2008, 1781, 288 CrossRef CAS PubMed.
- (a) D. M. Owen, C. Rentero, A. Magenau, A. Abu-Siniyeh and K. Gaus, Nat. Protoc., 2012, 7, 24–35 CrossRef CAS PubMed; (b) D. M. Owen, D. J. Williamson, A. Magenau and K. Gaus, Nat. Commun., 2012, 3, 1256 CrossRef PubMed.
- I. López-Duarte, P. Chairatana, Y. Wu, J. Pérez-Moreno, P. M. Bennett, J. M. Reeve, I. Boczarow, W. Kaluza, N. A. Hosny, S. D. Stranks, R. J. Nicholas, K. Clays, M. K. Kuimova and H. L. Anderson, Org. Biomol. Chem., 2015, 13, 3792 Search PubMed.
- M. A. Digman, V. R. Caiofla, M. Zamai and E. Gratton, Eur. Biophys. J., 2008, 94, L12 CrossRef.
- Glycerin Producers’ Association, Physical properties of glycerine and its solutions, Glycerine Producers’ Association, New York, 1963 Search PubMed.
- L. A. Bagatolli, T. Parasassi and E. Gratton, Chem. Phys. Lipids, 2000, 105, 135 CrossRef CAS PubMed.
- D. Borchman, N. A. Delamere, L. A. McCauley and C. A. Paterson, Lens Eye Toxic. Res., 1989, 6, 703 CAS.
- M. C. Yappert, M. Rujoi, D. Borchman, I. Vorobyov and R. Estrada, Exp. Eye Res., 2003, 76, 725 CrossRef CAS PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: Synthetic procedures, compound characterisation and additional spectroscopic and imaging data. See DOI: 10.1039/c6sc05369f |
| This journal is © The Royal Society of Chemistry 2017 |