
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnergy and charge transfer cascade in methylammonium lead bromide perovskite nanoparticle aggregates†
Marine E. F.
Bouduban
,
Andrés
Burgos-Caminal
,
Rachele
Ossola‡
,
Joël
Teuscher
 and
Jacques-E.
Moser
*
and
Jacques-E.
Moser
*
Photochemical Dynamics Group, Institute of Chemical Sciences & Engineering and Lausanne Centre for Ultrafast Science (LACUS), École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland. E-mail: je.moser@epfl.ch
First published on 19th April 2017
Abstract
Highly photoluminescent hybrid lead halide perovskite nanoparticles have recently attracted wide interest in the context of high-stake applications, such as light emitting diodes (LEDs), light emitting transistors and lasers. In addition, they constitute ideal model systems to explore energy and charge transport phenomena occurring at the boundaries of nanocrystalline grains forming thin films in high-efficiency perovskite solar cells (PSCs). Here we report a complete photophysical study of CH3NH3PbBr3 perovskite nanoparticles suspended in chlorobenzene and highlight some important interaction properties. Colloidal suspensions under study were constituted of dispersed aggregates of quasi-2D platelets of a range of thicknesses, decorated with 3D-like spherical nanoparticles. These types of nanostructures possess different optical properties that afford a handle for probing them individually. The photophysics of the colloidal particles was studied by femtosecond pump-probe spectroscopy and time-correlated single-photon counting. We show here that a cascade of energy and exciton-mediated charge transfer occurs between nanostructures: upon photoexcitation, localized excitons within one nanostructure can either recombine on a ps timescale, yielding a short-lived emission, or form charge-transfer states (CTSs) across adjacent domains, resulting in longer-lived photoluminescence in the millisecond timescale. Furthermore, CTSs exhibit a clear signature in the form of a strong photoinduced electroabsorption evidenced in femtosecond transient absorption measurements. Charge transfer dynamics at the surface of the nanoparticles have been studied with various quenchers in solution. Efficient hole transfer to N,N,N′,N′-tetrakis(4-methoxyphenyl)benzidine (MeO-TPD) and 1,4-bis(diphenyl-amino)benzene (BDB) donors was attested by the quenching of the nanoparticles emission. The charge transfer rate was limited by the organic layer used to stabilize the nanoparticles, which acted as a wide spacer between reactants. The forward charge transfer was found to take place in the sub-microsecond time-scale in competition with slow carrier recombination, while back transfer was shown to occur with a time-constant τ = 25 ms.
Introduction
Organolead halide perovskites have emerged as a thrilling novel material for photovoltaic technology. Since their first demonstration a few years ago, perovskite solar cells (PSCs) have already reached power conversion efficiencies of 22%.1 While nanostructured perovskites were originally deposited within mesoscopic metal oxide scaffolds,2–4 architectures of efficient PSCs have now evolved towards the use of homogeneous thin films in planar multilayer configurations. Perovskite films and large single crystals have been so far the focus of experiments aiming at gaining a fundamental knowledge of the material bulk properties. Alternatively, methylammonium or cesium perovskite nanocrystals and colloidal nanoparticles dispersed in a solvent have been shown to be highly promising in the context of LEDs and lasing applications.5–11 The observation by photoluminescence nano-imaging and transient absorption microscopy of the surface of multigrain thin films in perovskite solar cells suggested that their efficiency is influenced by the nanoscopic structure of the active semiconducting layer.12–14 Recently, energy transfer between crystal grains and photon recycling effects were shown to contribute to a large extend to the photocarriers long path lengths.15 Controlled nanoparticle aggregates, thus, constitute ideal model systems for the characterization of interfacial non-radiative energy- and charge transfer processes at grain boundaries, which appear to be key to the exceptional photovoltaic performances of perovskite devices.The study of perovskite nanoparticles provides also an opportunity to focus on a different paradigm as for the photogenerated species. As it happens, it has been shown that above-bandgap photoexcitation of methylammonium lead trihalide perovskite thin-films mostly yields free carriers at room temperature, although an increasing proportion of excitons is observed as one moves up in the halogens group from iodine to chlorine.16–20 As a consequence, excitonic features in perovskites can hardly be studied for thin-film architectures. Synthesizing and characterizing their nanostructured counterpart that present much larger exciton binding energies,21 however, allows for direct observation of excitonic behavior.
A first synthesis of CH3NH3PbBr3 perovskite nanoparticles was proposed by Schmidt et al.,22 where the stability of the suspension in aprotic, moderately polar solvents is ensured by the presence of an organic capping layer, typically consisting of alkylammonium long-chain cations. Ulterior studies endeavored to characterize those colloidal suspensions and highlighted the presence of a broad distribution of nanostructures. On top of 3D perovskite nanoparticles, perovskite nanoplatelets of various thicknesses exhibiting 2D quantum confinement were identified.21,23 Further synthesis attempts, aiming at getting improved CH3NH3PbX3 (X = Br, I) suspensions in terms of stability, emission properties and monodispersity, were later reported, although with mitigated results.24,25
The presence of nanoplatelets in colloidal suspension was exploited in quantum confinement studies. It was shown in particular that the proportion and thickness of the quasi-2D nanoplatelets can be tuned by varying (i) the ratio between the long-chain alkylammonium capping ligand and the methylammonium halide precursor and (ii) the length of the capping alkyl chain with use of butylammonium and octadecylammonium instead of octylammonium, for example.21,26,27 Investigations on those species later demonstrated the presence of a strong quantum confinement, yielding a blue-shift of the emission and absorption as the thickness of the platelets decreases.26,28
Only few studies focused on the photophysical properties of methylammonium lead–bromide perovskite nanoparticles. Remarkably high values of the photoluminescence quantum yield of 17–20% were reported,28–30 which were generally attributed to an enhanced exciton binding energy compared to bulk perovskites, sustained by reported values around 320 meV.21
In this work, we focus on colloidal suspensions of methylammonium lead–bromide perovskite nanoparticles in chlorobenzene. Excitation wavelength-dependent ultrafast transient absorbance measurements were performed to characterize their optical properties. Two different reductive quenchers were then added to the solution, providing insights into charge-transfer dynamics and related interfacial phenomena that were probed with a combination of techniques, namely ultrafast transient absorbance spectroscopy, nanosecond flash photolysis and time-correlated single-photon counting.
Experimental
Samples
Colloidal CH3NH3PbBr3 nanoparticles were prepared following the method described by Schmidt et al.,20 yielding octylammonium-capped nanoparticles. Dried nanoparticles were stored in the dark and under dry air at all times.Thorough solvent tests were performed to maximize the stability of the colloidal suspensions. In this context, it appeared that colloidal suspensions in chlorobenzene exhibited desirable features, such as a high stability, in particular for highly concentrated sols, and favorable solvation interactions, limiting the adsorption of the nanoparticles on the walls of the vial/cuvette.
A stable stock colloidal suspension was prepared every month, and various dilutions were made and used for the different experiments. Transient-absorbance and steady-state absorbance data have been recorded at nanoparticle density of 100 μg mL−1, while TCSPC and steady-state fluorescence measurements have been recorded with 10 μg mL−1 samples. The stability of the stock solution was routinely checked through absorbance spectra measurements. In addition, every sample was carefully degassed by bubbling with argon for 30 minutes before optical measurements.
Methods
Ultrafast transient absorbance (TA) spectra of methylammonium lead bromide perovskite nanoparticles were acquired using femtosecond pump-probe spectroscopy at two different excitation wavelengths. The pump beam at λex = 480 nm or 390 nm was obtained either by pumping a two-stage non-colinear optical parametric amplifier (NOPA) with the output of a chirped pulse amplified Ti:Sapphire laser (CPA-2001, Clark-MXR, 778 nm fundamental central wavelength, 120 fs pulse duration, 1 kHz repetition rate) or by frequency doubling in a BBO crystal, respectively. In the first case, pulses were compressed to ∼43 fs FWHM duration in a pair of SF10 prisms. The probe beam was generated by directing a portion of the 778 nm fundamental output of the laser into a CaF2 crystal, yielding a white light continuum spanning over a 400–780 nm wavelength domain. The probe fluence at the sample was much lower than that of the pump. Similarly, its diameter was smaller to ensure homogeneous excitation of the probed area. The dynamics of the photoinduced signals were obtained with a computer-controlled delay-line on the pump path. The probe beam was splitted before the sample into a beam going through the sample (signal beam) and a reference beam. Both signal and reference beams were directed to respective spectrographs (Princeton Instruments, Spectra Pro 2500i) and detected pulse-to-pulse with 512 × 58 pixels back-thinned CCD detectors (Hamamatsu S07030-0906). The pump beam was chopped at half of the laser frequency (500 Hz) and satisfying signal-to-noise ratio was obtained by typically averaging 3000 spectra. The time resolution of the experiment depended on the excitation wavelength and was 200 fs for λex = 390 nm (second harmonic) and 90 fs for λex = 480 nm.Similarly, long-time behavior of the sample was characterized using nanosecond flash photolysis. The pump beam (λex = 480 nm) was generated by a frequency-tripled Q-switched Nd:YAG laser (Ekspla NT-342, 355 nm, 20 Hz repetition rate) pumping an optical parametric oscillator (OPO), yielding pulses of ca. 5 ns duration (FWHM). The probe beam was obtained from a cw halogen lamp and was passed through a monochromator to select the detection wavelength and avoid undesired illumination before being focused onto the sample. The transmitted probe light was then passed through a second monochromator and detected either by a fast photomultiplier tube (R9910, Hamamatsu) in the visible or by an InGaAs photodiode (SM05PD5A, Thorlabs) for near-infrared measurements. The resulting signals were recorded and digitalized with a broad bandwidth digital oscilloscope (Tektronix, DPO 7254), with a typical averaging over 2000 shots.
In turn, the long-time time-dependence of the nanoparticles emission was unraveled via time-correlated single-photon counting (TCSPC) measurements. TCSPC dynamics were recorded by a Horiba Fluorolog-3 spectrofluorometer, using a 405 nm NanoLed laser diode as an excitation source (<200 ps pulse duration, 11 pJ per pulse). In order to avoid pile-up phenomena, the portion of photons reaching the detector per cycle was kept under 2% by adjusting the width of various slits within the setup.
Finally, steady-state characterization of the nanoparticles was performed. UV-vis absorbance spectra were recorded on a PerkinElmer Lambda 950 UV/vis/NIR spectrophotometer equipped with an integrating sphere to account for light-scattering by the sample. Steady-state photoluminescence spectra were measured using a PerkinElmer LS 50B spectrofluorometer.
Results and discussion
Evidence for cascade transfer: steady-state properties, excitation-dependent transient absorbance measurements and luminescence lifetimes
We prepared suspensions of CH3NH3PbBr3 nanoparticles in chlorobenzene. As presented in Fig. 1A, these suspensions exhibit several optical features, which were already reported and demonstrated to arise from a broad distribution of nanostructures:23 specifically, bulk-like tri-dimensional perovskite nanoparticles and various-sized nanoplatelets with formula (C6H13NH3)2(CH3NH3)m−1PbmBr3m+1.21,23,25,27,28 These are denominated “quasi-2D” (q-2D) perovskites, as they exhibit an almost two-dimensional structure comprising m layers of perovskite unit cells, surrounded by the octylammonium cations of the capping layer. | ||
| Fig. 1 Steady-state optical properties of a suspension of CH3NH3PbBr3 nanoparticles in chlorobenzene. (A) Absorption (blue) and photoluminescence (green) spectra. The emission spectrum was obtained with an excitation wavelength λex = 390 nm. (B) Photoluminescence excitation spectra measured for four emission wavelengths corresponding to 3D and q-2D perovskite nanoparticles, with m = 5, 4 and 3, respectively. Red: λobs = 520 nm, blue: λobs = 490 nm, green: λobs = 480 nm, and yellow: λobs = 455 nm. | ||
We observe an absorption spectrum similar to the one reported by Tyagi et al. From the abovementioned papers, the various absorption peaks in Fig. 1A were assigned as follows: the peak with λmax = 409 nm corresponds to 2D or q-2D perovskite with m = 1. The absorbance maxima at λmax = 453 nm, λmax = 472 nm and λmax = 489 nm are respectively attributed to q-2D perovskite with m = 3, q-2D perovskite with m = 4 and q-2D perovskite with m = 5. Finally, bulk 3D perovskite nanoparticles absorb at λmax = 520 nm, as expected for pristine CH3NH3PbBr3.21
Similarly, we observed that the five different perovskite nanostructures could also be distinguished in the emission spectrum, which displays peaks displaced by a Stokes shift of about 8 nm compared to the absorbance features. Interestingly, the relative intensities between the absorbance peaks and their corresponding emission features do not follow the same trend. In particular, the maximum of the emission spectrum at λobs = 530 nm corresponds to the lower edge of the absorbance spectrum and therefore to the 3D perovskite, with an absorbance maximum at 520 nm. Such a discrepancy can be rationalized in two ways: either a cascade of energy and/or charge transfer processes occurs between different nanostructures, with preferential radiative recombination from the 3D sites, or those nanostructures simply exhibit very different emission quantum yields. The latter is a reasonable postulate, as one expects the emission quantum yield in a semiconductor to increase with the exciton binding energy and decrease as the surface area increases, because of the higher density of trap state yielding non-radiative decay pathways. In this context, it was shown that the exciton binding energy diminishes as the thickness of the platelets increases, yielding a minimum for the bulk perovskite material.31 Thus, a trade-off emerges: the thinner platelets exhibit at the same time a larger binding energy, and more trap states and one, hence, expects an optimum quantum yield for the type of platelets exhibiting the best compromise between those two parameters. As a matter of fact, quantum yields for various-sized nanoplatelets up to m = 5, as well as for bulk bromide perovskite, were recently measured and this effect is perfectly illustrated. The quantum yield is indeed maximized for platelets with m = 4 and m = 5 (20–30%), and then decreases to reach 2% for single-layer platelets and 4% for bulk perovskite.26 In this light, the hypothesis of cascade energy and/or charge transfer within our suspensions appears very probable. As an evidence of the occurrence of cascade energy transfer, Fig. 1B displays a series of excitation spectra corresponding to four different nanostructures (3D and q-2D perovskites with m = 3, 4 and 5). It emerges that the shape of the excitation spectra is completely independent on the excitation wavelength, which indeed reflects excitation energy transfer between the different domains in presence.
Fig. 2 displays the transient absorbance change following λ = 390 nm pulsed excitation. We observe a strong bleaching feature peaking at 525 nm and strong oscillations on the blue side of the spectrum. Data for λex = 480 nm excitation are provided in ESI (Fig. S1†). These two excitation wavelengths have been chosen to selectively excite a given subset of the nanostructures in presence: while all nanostructures are excited at 390 nm, only three of them are activated upon λ = 480 nm excitation, 3D and q-2D perovskites with m = 4 and m = 5. To further support the occurrence of a cascade of energy and possibly charge transfer among nanostructures, a global fitting procedure of the time-resolved spectral data to a kinetic equation was performed. With such a process, we can express a time-series of transient absorbance spectra as a linear combination of the spectra of the various populations constituting the system, with their own dynamics (decay associated spectra, DAS). Population kinetics, as well as their individual spectra are obtained this way, allowing to shed light on interacting populations, and the possibility of charge and/or energy transfer processes. Details of the multiexponential fitting procedure are provided in the ESI† along with sample results. In addition, Fig. S2† assesses the fitting quality by displaying it on top of experimental points.
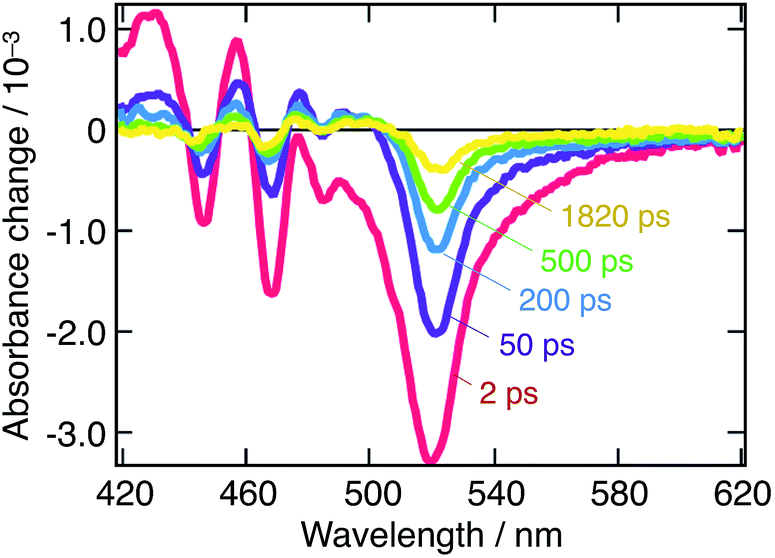 | ||
| Fig. 2 Transient absorption spectra of a suspension of CH3NH3PbBr3 nanoparticles in chlorobenzene at different time delays following λex = 390 nm pulsed excitation. Red: t = 2 ps, purple: t = 50 ps, blue: t = 200 ps, green: t = 500 ps, and yellow: t = 1820 ps. | ||
A biexponential decay for excitation at 480 nm and a triexponential decay for excitation at 390 nm were extracted from the fitted dynamical data. These were obtained in conditions, where the excitation energy fluence was kept sufficiently low as to remain below the non-geminate recombination regime. The corresponding decay associated spectra are reported in Fig. 3A and B, along with the dynamics recorded at 520 nm (Fig. 3C and D). Those dynamics are attributed to a combination of ground-state bleaching (GSB) and stimulated emission (SE).
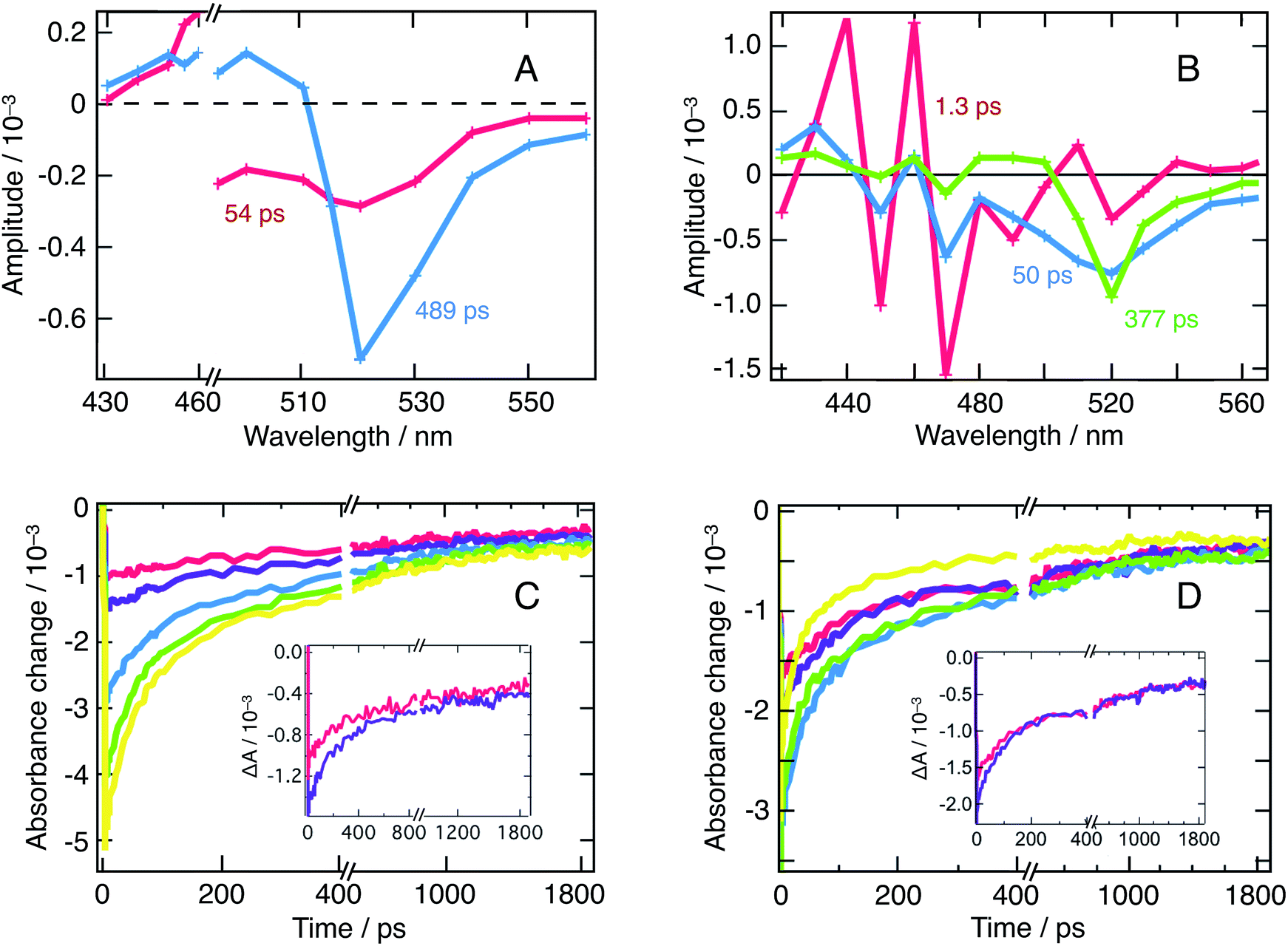 | ||
| Fig. 3 (A) Result of a global fitting procedure of transient absorbance data obtained for a suspension of CH3NH3PbBr3 nanoparticles in chlorobenzene for the excitation wavelength λex = 390 nm at an energy fluence of 10.5 μJ cm−2. Red: τ = 54 ps, blue: τ = 489 ps. (B) Result of the global fitting procedure of transient absorbance data obtained in the same conditions for λex = 390 nm and an energy fluence of 14 μJ cm−2. Red: τ = 1.3 ps, blue: τ = 50 ps, and green: τ = 377 ps. (C) Combination of ground-state bleaching (GSB) and stimulated emission (SE) peak (520 nm), intensity-dependent dynamics for λex = 480 nm. Red: 7 μJ cm−2, purple: 10.5 μJ cm−2, blue: 21 μJ cm−2 green: 35 μJ cm−2 and yellow: 52 μJ cm−2. Inset: zoom on the two transient signals obtained at lower fluences. The appearance of a third exponential emerges clearly with fluences ≥21 μJ cm−2. (D) GSB + SE peak intensity-dependent dynamics for λex = 390 nm. Three exponentials are needed at all fluences. In this case, red: 9 μJ cm−2, purple: 14 μJ cm−2, blue: 27 μJ cm−2, green: 45.5 μJ cm−2, and yellow: 68 μJ cm−2. Inset: zoom on the two transient signals obtained at lower fluences. | ||
On Fig. 3A (λex = 480 nm), one distinguishes two decay associated spectra with time constants of 54 ps and 489 ps respectively. The former contribution covers the spectral region of the q-2D perovskites with m = 5 and exhibits a negative peak centered at 515 nm. The second contribution in turn mostly features a negative amplitude centered at 520 nm aside a weaker positive signal extending into the blue region. This second contribution reflects the long-time transient absorption spectra reported in Fig. S1,† and represents the decay kinetics of the bulk nanoparticles population. Alternatively, the first contribution is assigned to the transfer of population from q-2D perovskites with m = 5 towards the 3D sites. Those two decay associated spectra confirm that at least two nanostructures contribute to the dynamics of the 520 nm peak (GSB + SE). To further support the fact that those two contributions indeed arise from interacting nanostructures and do not reflect an excessively high carrier density, we refer to Fig. 3C that displays fluence-dependent dynamics at 520 nm. At short times, and as excitation energy fluence increases, it clearly appears that one moves from a monomolecular process (exponential decay) to a higher-molecularity kinetics attributed to non-geminate carrier recombination (fluence > 21 μJ cm−2). We can then conclude that the DAS obtained from global fitting our data with λex = 480 nm (Fig. 3A, recorded at an energy fluence of 10.5 μJ cm−2) describes the interaction of two nanostructures and is not due to an excitation density artifact.
Alternatively, following λex = 390 nm excitation, one obtains three decay associated spectra with time constants of 1.3 ps, 50 ps and 377 ps, displayed on Fig. 3B. The first contribution mainly consists in strong oscillations between 420 nm and 490 nm and represents the rise of the oscillatory signal already described in Fig. 2. The two other contributions feature diminishing oscillations and a growing and sharpening negative amplitude centered at 520 nm. This is assigned, again, to population transfer from the blue side of the spectrum to the 3D sites. Fig. 3D (fluence-dependent dynamics at 520 nm following λex = 390 nm excitation) unambiguously demonstrates that the decay rate is triphasic at all fluences. This confirms that the three decay associated spectra described above result from the excitation of all the nanostructures in presence and their ensuing interactions, and, again, are not an excitation density artifact.
As an additional supporting evidence of cascade transfer processes, we characterized the photoemission of the nanoparticles suspension, as it is particularly sensitive to those phenomena. In this respect, we resorted to the TCSPC technique to consider the long-time behavior, at the microsecond time-scale, of emission at λobs = 455, 475, and 523 nm, following λex = 405 nm excitation. The signals measured at these three wavelengths are dominated by the emission of the m = 3 and m = 4 platelets, as well as of the 3D bulk-like nanoparticles.
Similarly, we addressed the short-time luminescence behavior with transient absorbance (TA) following 390 nm excitation and extracting the dynamics at wavelengths involving a pure or a major fraction of emission: λobs = 476, 491, and 555 nm. The small discrepancy between the TCSPC and TA selected wavelengths arises from the high complexity of the TA spectrum, demanding a selection outside of the expected emission maxima.
Each of the traces on Fig. 4A and B can be fitted with a triexponential model, the latter moreover involving a convolution with the Gaussian response function of the instrument. We present the resulting time constants in Table 1. Note that the shortest TA time constant reflects the rise of the signal, while the two others correspond to its decay. On the contrary, TCSPC dynamics effectively feature a triexponential decay. In both cases, we attribute these multiexponential decays to the entanglement of various nanostructures, as already discussed here above.
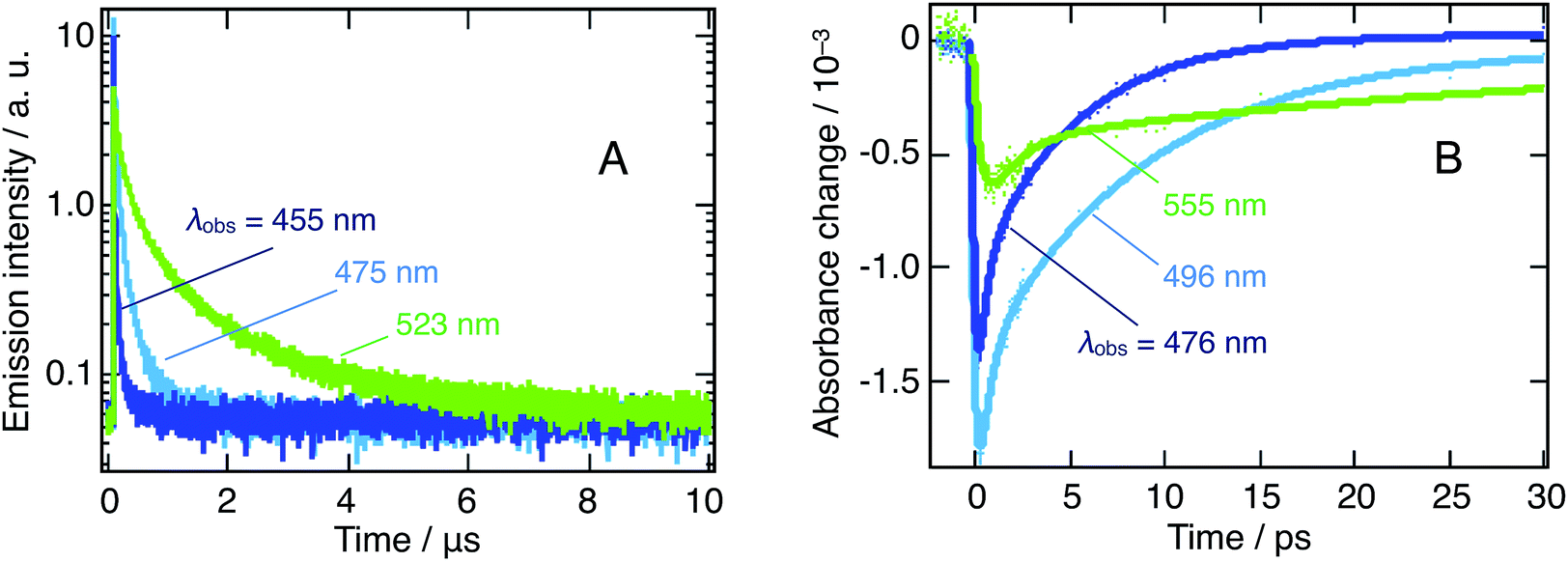 | ||
| Fig. 4 Dynamics of the emission of a suspension of CH3NH3PbBr3 nanoparticles in chlorobenzene on two different timescales. (A) Microsecond timescale from TCSPC following 405 nm excitation. Dark blue: λobs = 455 nm, light blue: 475 nm and green: 523 nm. (B) Picosecond timescale, extracted from the λex = 390 nm TA spectrum in regions where emission could be isolated. Dark blue: λobs = 476 nm; light blue: 496 nm and green: 555 nm. Note that we chose to display here dynamics recorded at a higher fluence (52 μJ cm−2), to further highlight the considered trends. Excitation energy fluences as low as 10 μJ cm−2 have been applied, yielding identical behaviors. | ||
| (A) | |||
|---|---|---|---|
| λ obs | τ 1 (ns) | τ 2 (ns) | τ 3 (ns) |
| 455 nm | 2.05 ± 0.02 | 21 ± 1 | 145 ± 10 |
| 475 nm | 6.82 ± 0.16 | 51 ± 1 | 262 ± 3 |
| 523 nm | 29 ± 1 | 203 ± 5 | 1200 ± 25 |
| (B) | |||
|---|---|---|---|
| λ obs | τ 1 (ps) | τ 2 (ps) | τ 3 (ps) |
| 476 nm | 0.153 ± 0.04 | 0.381 ± 0.09 | 5.14 ± 0.25 |
| 496 nm | 0.186 ± 0.03 | 0.507 ± 0.09 | 8.67 ± 0.36 |
| 555 nm | 0.61 ± 0.4 | 1.2 ± 0.8 | 32 ± 5 |
At all wavelengths under study, the photoluminescence appears to decay over two very different timescales (ps vs. μs) and the photoluminescence lifetime increases significantly as one moves to longer wavelengths for both timescales. In addition, we stress the notably slower rise time for 3D nanoparticles.
We attribute the first of those features to the presence of different types of excitons with different lifetimes (vide infra). The second and third feature arises from interacting nanostructures and can be considered symptomatic of a cascade transfer process from the larger bandgap, quantum-confined, nanoplatelets all the way to the bulk 3D nanoparticles.
Evidence for CT excitons: analysis of the transient absorption spectra and of the different emission lifetimes
Looking back at the transient absorption data on Fig. 2, one observes strong oscillations on the blue side of the spectrum. This feature looks very much like the first or second derivative of the absorption spectrum of q-2D nanoplatelets within the 400–480 nm wavelength region. To shed light on the underlying process, the transient absorption spectrum at a time-delay of 2 ps is shown in Fig. 5, along with a simulated differential electroabsorption spectrum (see ESI† for details). The correspondence between the measured and simulated differential spectra allows to assign a significant part of the transient optical features observed below 500 nm to a photoinduced electroabsorption.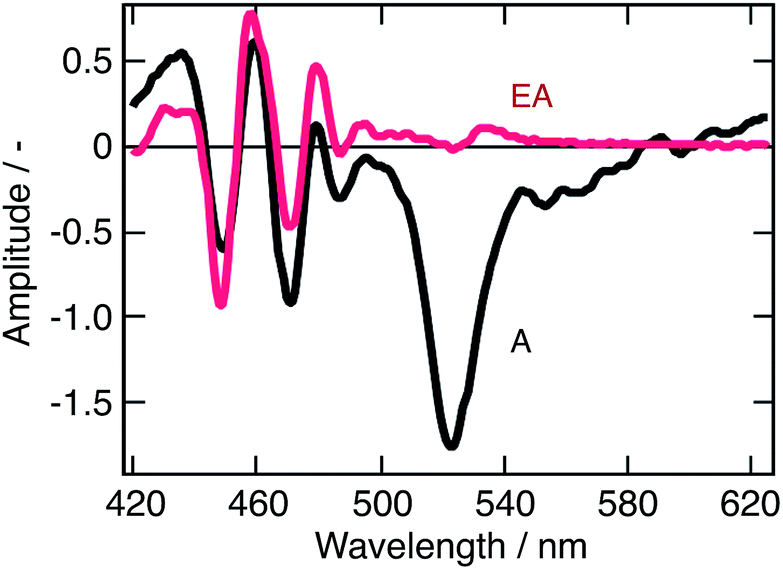 | ||
| Fig. 5 Modeled electroabsorption signal of a suspension of CH3NH3PbBr3 nanoparticles in chlorobenzene. The blue part of the observed transient absorption spectrum measured at a time delay of 2 ps (black line) fits well with a simulated electroabsorption spectrum (red) dominated by the second derivative component of the ground state absorption spectrum (see ESI† for details). | ||
The term “electroabsorption” designates the change in the absorption spectrum of a system subjected to an electric field, be it applied externally or originating from photogenerated carriers. For molecular and excitonic systems, the electroabsorption generally results from a quadratic Stark effect.32 Based on a perturbative approach, such electroabsorption signals can be shown to correspond to a linear combination of the first and second derivatives of the linear absorption spectrum (see ESI†). For anisotropic samples such as nanoparticles suspensions, the related amplitudes of these two components are then respectively given by the change in polarizability of the material and by the change in dipole moment upon the electronic transition of interest.
Fig. 5 shows that the observed differential electroabsorption signal appears to be dominated by the second derivative of the absorption spectrum (see ESI† for details), which amplitude is given by the change of a permanent dipole moment. Because Frenkel and Wannier-Mott bulk excitons do not display a permanent dipole moment, they are not likely to generate any significant electric field that could induce the observed electroabsorption.33 On the contrary, interfacial charge transfer (CT) states, where the electron and the hole occupy adjacent domains, exhibit a permanent dipole. These CT excitons are known to occur primarily in ionic solid materials,34 such as the lead halide perovskites, and are believed to be responsible for the observed photoinduced electroabsorption. Aside the photoinduced electroabsorption signal, the presence of CT excitons fits well with the observation underlined above of two different emission timescales (ps vs. μs). We indeed assigned this phenomenon to the recombination of two different exciton populations. From the long lifetime of the photoinduced electroabsorption signal (longer than 2 ns), we deduce that the CT states are long-lived. As a consequence, those two exciton populations would be localized excitons (within one nanostructure) that recombine on the ps timescale and those CT excitons.
Furthermore, the presence of CT excitons is of high interest as it evidences the occurrence of interstructure charge transfer in addition to the previously discussed energy transfer processes.
Interaction between nanostructures and global photophysical perspective
In the above sections, we gathered evidence towards cascade energy and CT exciton-mediated charge transfer, from the larger bandgap to the lower bandgap nanostructures. The cascade transfer is supported by steady-state optical measurements, global analysis of transient absorption data at two different wavelengths and emission lifetime characterization. Excitation energy transfer was evidenced by the wavelength independence of the excitation spectra (Fig. 1B). Similarly, the occurrence of charge transfer is sustained by the presence of interfacial charge transfer states, which involvement is deduced from the observation of a second derivative-dominated photoinduced electro-absorption signal in TA experiments and by the presence of two very different emission timescales (ps vs. μs), assigned to different excitons populations.We hence come up with a photophysical model where our colloidal suspensions involve two distinct excitons populations generated upon photoexcitation: localized exciton whose localized character increases as we move to thinner platelets and CT excitons, yielding the processes illustrated in Fig. 6.
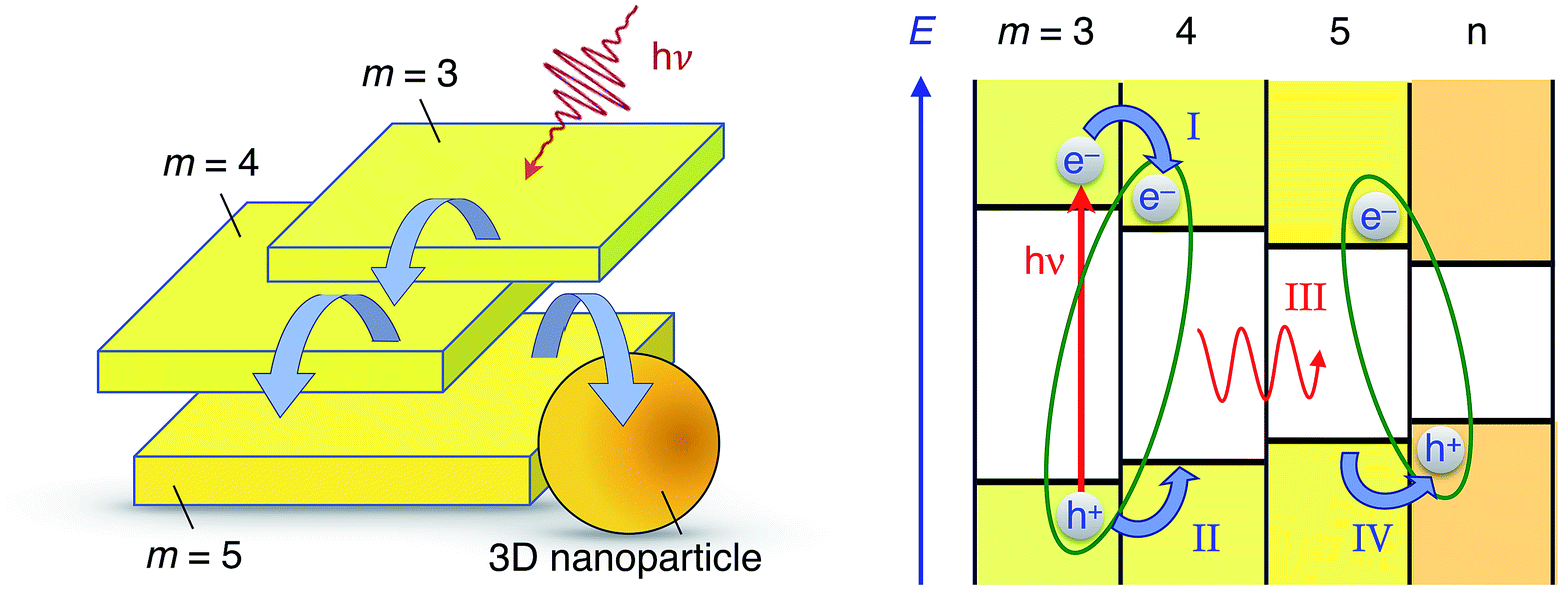 | ||
| Fig. 6 Cartoon illustrating the energy- and charge transfer processes occurring between the various nanostructures constituting CH3NH3PbBr3 perovskite colloidal aggregates. Left: Energy- and/or charge transfer cascade (curved blue arrows) between q-2D nanoplatelets of increasing thicknesses and eventually a 3D bulk-like nanoparticle. Right: Energetic scheme of some examples of photophysical processes taking place in a nanoparticle aggregate: upon photoexcitation of a thin q-2D (m = 3) nanoplatelet, interfacial electron transfer can take place to the adjacent particle (process I). Electrostatic interaction of the electron with a hole remaining on the other side of the interface yields a CT exciton (green ellipse). Subsequent hole transfer (process II) leads to the excitation of the m = 4 q-2D nanoplatelet. Energy transfer to a neighbouring nanostructure characterized by a narrower bandgap is then possible (process III). Interfacial hole transfer (process IV) finally enables the formation of a new interfacial CT excitonic species. | ||
First, the photoexcitation of two or more nanostructures forms localized excitons yielding a bleaching of the excitonic transition in the transient absorbance measurements. Then, localized exciton emission for all excited nanostructures, with a picosecond lifetime, competes with the formation of CT excitons with neighboring, smaller band-gap nanostructures. Finally, dissociation of the CT excitons on the nanosecond timescale allows for the radiative recombination of relocalized electron–hole pairs and results in the observed long-lived emission.
The cascade of charge transfer processes reported here have been recently highlighted in solution-processed, highly efficient, LEDs devices.9 They are thus likely to occur in the same way between different perovskite domains that could coexist within a thin-film photovoltaic device and, therefore, take place within the charge transport process following light absorption. Hence, this picture could occur in parallel and similarly to the recently highlighted photon recycling (long distance radiative energy transfer), playing a crucial role in the remarkable efficiency of PSCs.13
Interaction with donor and acceptors: dynamics of charge transfer at the solution interface
Charge-transfer processes occurring at the particle/solution interface were scrutinize to provide a comprehensive characterization of the nanoparticles interfacial properties. Two organic electron donor molecules N,N,N,N-tetrakis(4-methoxyphenyl)benzidine (MeO-TPD) and 1,4-bis(diphenyl-amino)benzene (BDB) (molecular structures provided in ESI, Fig. S4†) were selected based on three different criteria. The first criterion concerns the thermodynamics: the energetics of the quencher must match that of the nanoparticles for an electron transfer to occur. The second emerges as a discrimination criterion, as the absorption of the quencher must not overlap with that of the nanoparticles to allow optical observation of the reaction. The third and last criterion also aims at optimizing the monitoring of the reaction: the oxidized quencher must exhibit an optical signature in an accessible spectral range, typically in the vis-NIR domains, and the transient species should ideally not exhibit any overlap with the optical features of the nanoparticles.Fig. 7 shows that MeO-TPD and BDB absorption spectra and that of the corresponding oxidized species fulfil the abovementioned criteria.35–37 Besides their favorable thermodynamics, both molecules show an absorbance cut-off below 400 nm, fulfilling the discrimination criterion. In addition, their oxidized species have intense bands in the IR with maxima at 1400 and 900 nm, respectively, which do not overlap with the bare nanoparticles absorption spectrum.
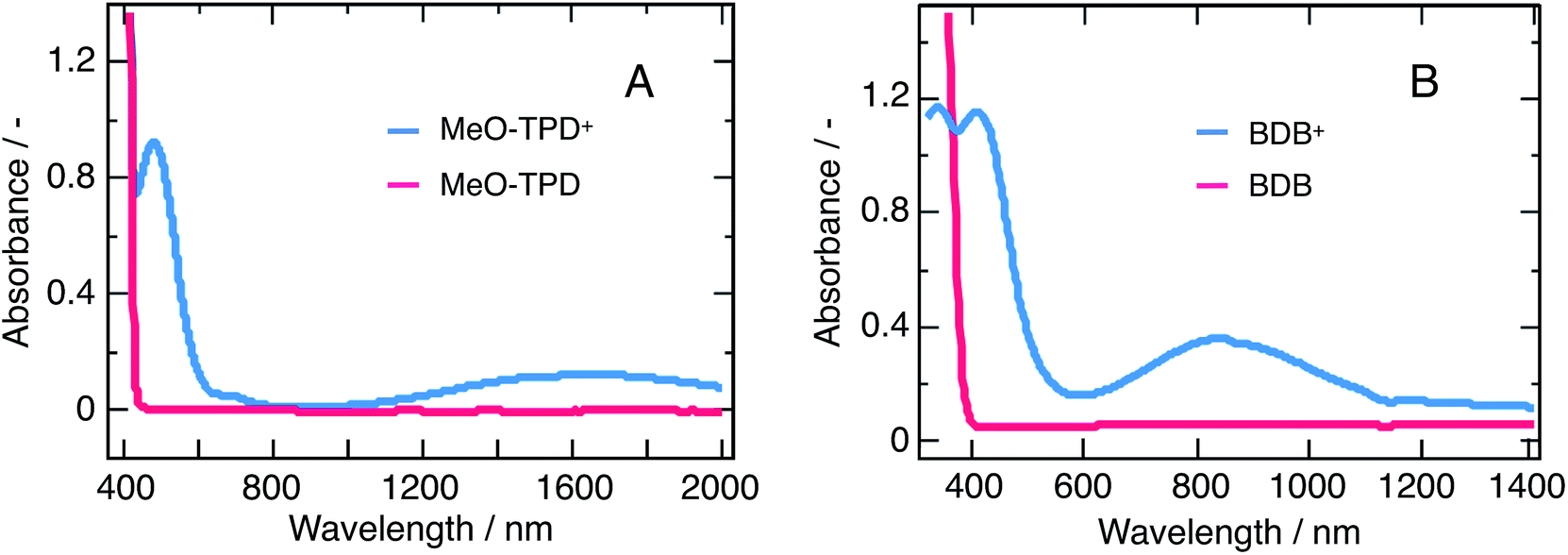 | ||
| Fig. 7 Absorption spectra of both quenchers in the neutral (red) and oxidized (blue) forms. (A) MeO-TPD and (B) 1,4-bis(diphenylamino)benzene (BDB). The spectra of the oxidized species have been recorded after quantitative reaction of the neutral compounds with the one-electron oxidizer NOBF4. | ||
Thorough Stern–Volmer analyses have demonstrated that the two selected electron donors efficiently quench the emission of a nanoparticle mixture as reported in the ESI.† The dynamics of those charge-transfer processes have been studied on two different timescales and with two different techniques, namely ultrafast transient absorbance (TA) to monitor the forward electron transfer and nanosecond flash photolysis to resolve the charge recombination reaction.
In the framework of time-resolved spectroscopy, a charge transfer should result in sizeable changes in the dynamics of bleaching or stimulated emission signals and the concomitant appearance of an optical signature corresponding to an oxidized quencher species. No such features could be resolved with ultrafast transient absorbance on a timescale up to 2 ns. Hence, we were not able to resolve the electron transfer dynamics from the quencher to the nanoparticles.
Fig. 8 shows flash photolysis transient dynamics of oxidized quenchers following λex = 480 nm excitation of the nanoparticles solution under inert Ar atmosphere up to 5 ms. The prompt appearance of transient absorption signals corresponding to the oxidized state of MeO-TPD at λobs = 1500 nm and of BDB at 950 nm was observed. The rise of the signal lies within the laser pulse, and we can then assume that photoinduced interfacial charge separation occurs with a time constant comprised between 5 ns (TA timescale) and 1 μs (flash photolysis time resolution in the near infrared). The analysis of Stern–Volmer plots (see ESI†) shows that electron transfer to the valence band of perovskite involves donor molecules associated within the organic capping layer. Time constants for forward electron transfer of τ = 170 ns and 60 ns were estimated for MeO-TPD and BDB, respectively.
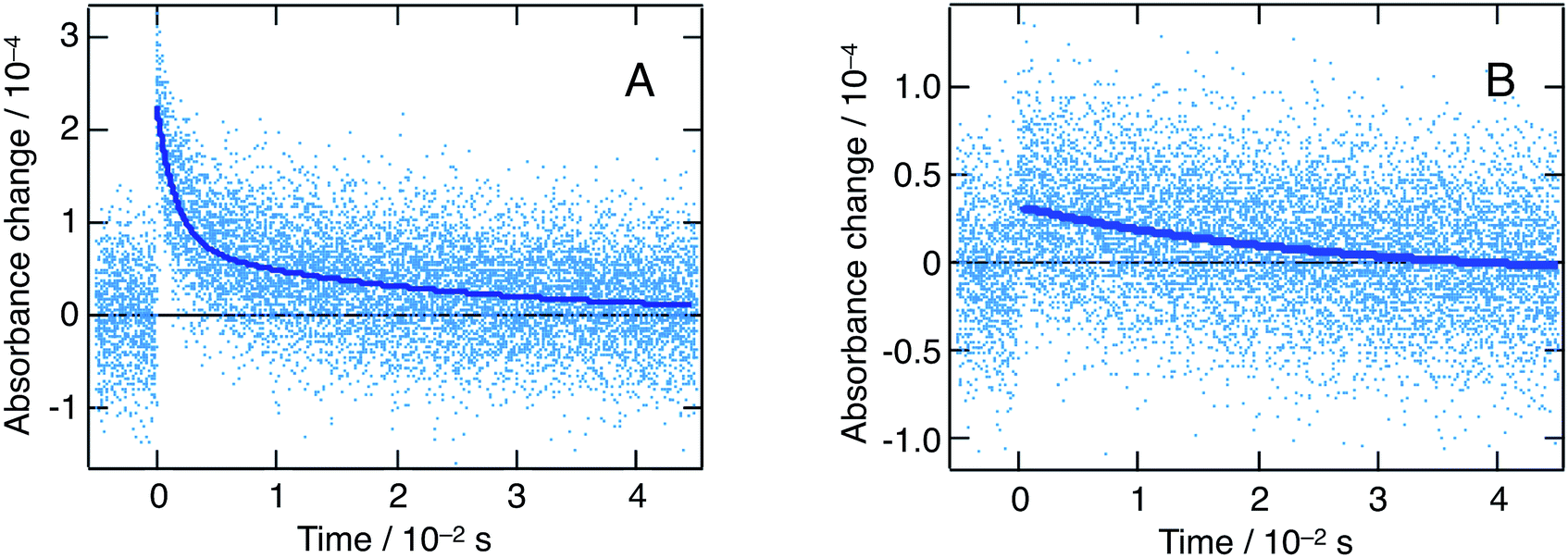 | ||
| Fig. 8 Nanosecond flash photolysis dynamics (λex = 480 nm, Ar atmosphere) for a mixture of CH3NH3PbBr3 nanoparticles with two electron donors: MeO-TPD (A) and 1,4-bis(diphenylamino)benzene (B). A different detection wavelength was selected for each quencher, in order to match their respective absorption maxima: λobs = 1500 nm (MeO-TPD) and 950 nm (1,4-bis(diphenylamino)benzene). Dark blue lines represent biexponential fits of the experimental points. | ||
The recombination of conduction band electrons with the oxidized MeO-TPD+ and BDB+ was followed by monitoring the time evolution of the transient absorption of the latter species (Fig. 8). The decay of the transient absorption of MeO-TPD+ at λobs = 1500 nm was fitted with a biexponential rate law, yielding time constants τ1 = 1.6 ms and τ2 = 22 ms. In turn, the decay of the oxidized BDB appeared mainly monoexponential with τ1 = 26 ms. Although, great care was taken to minimize the excitation energy fluence to the actual limits of the instrument detection (hence the large noise marring the kinetic traces reported in Fig. 8), it was not possible to ensure that at most one conduction band electron-oxidized donor cation pair is produced per particle and per laser pulse. Therefore, it is not expected that the recombination dynamics can be fitted by a single exponential and the initial fast component of the kinetics is likely to be due to a bimolecular process.
A complex kinetics for forward electron transfer and recombination could also result from a distribution of the distances separating the perovskite surface from the donor molecules that could penetrate to some degree the organic capping layer of the particle or rest at its external boundary. Due to the large difference in timescale, the photophysical processes intrinsic to the nanoparticles are not affected by the addition of quencher molecules. When electron transfer occurs, most of the inter-nanostructures cascade processes must, then, already be over, implying that the interfacial electron transfer predominantly takes place from the 3D bulk-like nanostructures. Depending on the morphology of the nanoparticle aggregates, the latter 3D structures might be more or less exposed to the solution and the donor molecules, constituting an additional source of dispersion for the kinetics of interfacial charge transfer.
Conclusion
We synthesized CH3NH3PbBr3 perovskite nanoparticles and studied their suspension in chlorobenzene. Such suspensions contain various nanostructures: quasi-2D nanoplatelets of variable thickness and 3D bulk-like nanoparticles. These structures exhibit several optical signatures that were previously reported and assigned. The nanoplatelets are blue-shifted compared to the bulk perovskite, due to a significant confinement regime. Using a combination of steady-state, excitation-dependent ultrafast transient absorbance and TCSPC measurements, we unraveled the presence of significant inter-structures interactions in the form of a cascade of energy and charge transfer, the latter being mediated by the formation of interparticle charge transfer states. Upon photoexcitation, localized excitons are formed within one nanostructure. They either rapidly recombine, yielding a short-lived emission on the picosecond timescale, or turn into CT states following injection of one type of carrier into a narrower band-gap, neighboring nanostructure. These CT excitons posses a permanent dipole and submit the material at close proximity to an electric field, which produces a significant photoinduced electroabsorption contribution to the transient absorption spectra. Carrier pairs contained in CT excitons eventually recombine, resulting in the long-lived, microsecond emission observed with TCSPC. Similar cascade charge transfer processes in highly efficient LEDs and photovoltaic devices is likely to occur in materials characterized by a multigrain morphology. As much as long-distance radiative energy transfer within the active film of a perovskite solar cell (photon recycling), non-radiative energy transfer and inter-domain charge transfer mediated by interfacial CT states could play an important role in slowing down the recombination of photocarriers and increasing their diffusion length.The dynamics of interfacial charge transfer from donor molecules in solution to the CH3NH3PbBr3 nanoparticles was studied. Efficient electron transfer from N,N,N′,N′-tetrakis(4-methoxy-phenyl)benzidine (MeO-TPD) and 1,4-bis(diphenylamino) benzene (BDB) resulted in an efficient quenching by up to respectively 90% and 96% of the photoemission of the semiconductor. The time constants of forward electron transfer were estimated at τ = 170 ns and 60 ns, while a remarkably long photoinduced charge separation could be sustained for more than 20 ms in the presence of both donors.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support by the Swiss National Science Foundation (SNSF) and NCCR MUST, a research instrument of the SNSF, is gratefully acknowledged.Notes and references
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 1–7 Search PubMed.
- J.-H. Im, C.-R. Lee, J.-W. Lee, S.-W. Park and N.-G. Park, Nanoscale, 2011, 3, 4088–4096 RSC.
- J. Xing, F. Yan, Y. Zhao, S. Chen, H. Yu, Q. Zhang, R. Zeng, H. V. Demir, X. Sun, A. Huan and Q. Xiong, ACS Nano, 2016, 10, 6623–6630 CrossRef CAS PubMed.
- S. Bai, Z. Yuan and F. Gao, J. Mater. Chem. C, 2016, 4, 3898–3904 RSC.
- S. Liu, W. Sun, J. Li, Z. Gu, K. Wang, S. Xiao and Q. Song, Opt. Eng., 2016, 55, 057102–057110 CrossRef.
- S. A. Veldhuis, P. P. Boix, N. Yantara, M. Li, T. C. Sum, N. Mathews and S. G. Mhaisalkar, Adv. Mater., 2016, 28, 6804–6834 CrossRef CAS PubMed.
- N. Wang, L. Cheng, R. Ge, S. Zhang, Y. Miao and W. Zou, Nat. Photonics, 2016, 10, 699–704 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- D. W. deQuilettes, S. M. Vorpahl, S. D. Stranks, H. Nagaoka, G. E. Eperon, M. E. Ziffer, H. J. Snaith and D. S. Ginger, Science, 2015, 348, 683–686 CrossRef CAS PubMed.
- A. R. Srimath Kandada and A. Petrozza, Acc. Chem. Res., 2016, 49, 536–544 CrossRef CAS PubMed.
- S. Y. Leblebici, L. Leppert, Y. Li, S. E. Reyes-Lillo, S. Wickenburg, E. Wong, J. Lee, M. Melli, D. Ziegler, D. K. Angell, D. F. Ogletree, P. D. Ashby, F. M. Toma, J. B. Neaton, I. D. Sharp and A. Weber-Bargioni, Nat. Energy, 2016, 1, 16093–16097 CrossRef CAS.
- L. M. Pazos-Outòn, M. Szumilo, R. Lamboll, J. M. Richter, M. Crespo-Quesada, M. Abdi-Jalebi, H. J. Beeson, M. Vrućinić, M. Alsari, H. J. Snaith, B. Ehrier, R. H. Friend and F. Deschler, Science, 2016, 351, 1430–1433 CrossRef PubMed.
- J. S. Manser and P. V. Kamat, Nat. Photonics, 2014, 8, 737–743 CrossRef CAS.
- C. Wehrenfennig, G. E. Eperon, M. B. Johnston, H. J. Snaith and L. M. Herz, Adv. Mater., 2013, 26, 1584–1589 CrossRef.
- D. A. Valverde-Chàvez, C. S. Ponseca, C. C. Stoumpos, A. Yartsev, M. G. Kanatzidis, D. G. Cooke and V. Sundström, Energy Environ. Sci., 2015, 8, 3700–3707 Search PubMed.
- A. Miyata, A. Mitioglu, P. Plochocka, O. Portugall, J. T.-W. Wang, S. D. Stranks, H. J. Snaith and R. J. Nicholas, Nat. Phys., 2015, 11, 582–587 CrossRef CAS.
- V. D'Innocenzo, G. Grancini, M. J. P. Alcocer, A. R. S. Kandada, S. D. Stranks, M. M. Lee, G. Lanzani, H. J. Snaith and A. Petrozza, Nat. Commun., 2014, 5, 3586 Search PubMed.
- K. Zheng, Q. Zhu, M. Abdellah, M. E. Messing, W. Zhang, A. Generalov, Y. Niu, L. Ribaud, S. E. Canton and T. Pullerits, J. Phys. Chem. Lett., 2015, 6, 2969–2975 CrossRef CAS PubMed.
- L. C. Schmidt, A. Pertegás, S. Gonzalez-Carrero, O. Malinkiewicz, S. Agouram, G. Mínguez Espallargas, H. J. Bolink, R. E. Galian and J. Pérez-Prieto, J. Am. Chem. Soc., 2014, 136, 850–853 CrossRef CAS PubMed.
- P. Tyagi, S. M. Arveson and W. A. Tisdale, J. Phys. Chem. Lett., 2015, 6, 1911–1916 CrossRef CAS PubMed.
- S. Gonzalez-Carrero, R. E. Galian and J. Pérez-Prieto, J. Mater. Chem. A, 2015, 3, 9187–9193 CAS.
- O. Vybornyi, S. Yakunin and M. V. Kovalenko, Nanoscale, 2016, 8, 6578–6583 RSC.
- J. A. Sichert, Y. Tong, N. Mutz, M. Vollmer, S. Fischer, K. Z. Milowska, R. García Cortadella, B. Nickel, C. Cardenas-Daw, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6521–6527 CrossRef CAS PubMed.
- Y. Hassan, Y. Song, R. D. Pensack, A. I. Abdelrahman, Y. Kobayashi, M. A. Winnik and G. D. Scholes, Adv. Mater., 2015, 28, 566–573 CrossRef PubMed.
- B. Luo, Y.-C. Pu, Y. Yang, S. A. Lindley, G. Abdelmageed, H. Ashry, Y. Li, X. Li and J. Z. Zhang, J. Phys. Chem. C, 2015, 119, 26672–26682 CAS.
- K. Zheng, Q. Zhu, M. Abdellah, M. E. Messing, W. Zhang, A. Generalov, Y. Niu, L. Ribaud, S. E. Canton and T. Pullerits, J. Phys. Chem. Lett., 2015, 6, 2969–2975 CrossRef CAS PubMed.
- F. Zhang, H. Zhong, C. Chen, X.-G. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- D. Sapori, M. Kepenekian, L. Pedesseau, C. Katan and J. Even, Nanoscale, 2016, 8, 6369–6378 RSC.
- G. Lanzani, The Photophysics Behind Photovoltaics and Photonics, Wiley-VCH, Weinheim, 2012 Search PubMed.
- M. T. Trinh, X. Wu, D. Niesner and X. Y. Zhu, J. Mater. Chem. A, 2015, 3, 9285–9290 CAS.
- P. Cudazzo, F. Sottile, A. Rubio and M. Gatti, J. Phys.: Condens. Matter, 2015, 27, 113204–113222 CrossRef PubMed.
- W. Z. Gao, X. G. Li and S. R. Wang, Appl. Mech. Mater., 2012, 161, 121–127 CrossRef CAS.
- H.-C. Cheng, K. Y. Chiu, S. H. Lu, C.-C. Chen, Y. W. Lee, T.-F. Yang, M. Y. Kuo, P. P.-Y. Chen and Y. O. Su, J. Phys. Chem. A, 2015, 119, 1933–1942 CrossRef CAS PubMed.
- H. D. Zhao, C. Tanjutco and S. Thayumanavan, Tetrahedron Lett., 2001, 42, 4421–4424 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Global fitting procedure, analysis of electroabsorption contribution to transient spectra, Stern–Volmer plots. See DOI: 10.1039/c6sc05211h |
| ‡ Present address: Environmental Chemistry Group, Department of Environmental Systems Science, ETH Zurich, CH-8092 Zurich, Switzerland. |
| This journal is © The Royal Society of Chemistry 2017 |