
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The development and mechanistic investigation of a palladium-catalyzed 1,3-arylfluorination of chromenes†
Richard T.
Thornbury‡
a,
Vaneet
Saini‡
b,
Talita de A.
Fernandes
ac,
Celine B.
Santiago
d,
Eric P. A.
Talbot
ab,
Matthew S.
Sigman
 d,
Jeffrey M.
McKenna
b and
F. Dean
Toste
*a
d,
Jeffrey M.
McKenna
b and
F. Dean
Toste
*a
aDepartment of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: fdtoste@berkeley.edu
bNovartis Institutes for Biomedical Research, Cambridge, Massachusetts 02139, USA
cInstituto de Química, Universidade de Brasília, Campus Universitário Darcy Ribeiro, Caixa Postal: 04478, 70904-970, Brasília, DF, Brazil
dDepartment of Chemistry, University of Utah, Salt Lake City, Utah 84112, USA
First published on 9th February 2017
Abstract
A mild palladium-catalyzed ligand-controlled regioselective 1,3-arylfluorination of 2[H]-chromenes has been developed. The products with a syn-1,3 substitution pattern were obtained with high enantiomeric excess using a PyrOx ligand, wherein the utility of these pyranyl-fluorides was further demonstrated through their participation in a diastereoselective C–C bond forming reaction. Ligand dependent divergent formation of both the 1,3- and 1,2- alkene difunctionalization products was observed. The nature of this bifurcation was investigated through experimental studies in combination with computational and statistical analysis tools. Ultimately, the site selectivity was found to rely on ligand denticity and metal electrophilicity, the electronics of the boronic acid, and the donor ability of the directing group in the substrate.
Introduction
Oxidative difunctionalization of alkenes via multi-component reactions is an attractive strategy to rapidly introduce complexity and diversity.1 As such, intercepting Mizoroki–Heck intermediates has recently garnered attention as a means to achieve this type of transformation.2 In this approach, the σ-alkyl palladium intermediate formed via insertion of an olefin into a [Pd]-aryl intermediate is functionalized rather than undergoing β-hydride elimination and alkene dissociation typical of the Heck reaction (Scheme 1). Although some of the earliest reports on the Heck reaction described the formation of alkene difunctionalization products, presumably formed via a similar mechanistic scenario, the outlined strategy had been applied sparingly to the development of new synthetic methods.3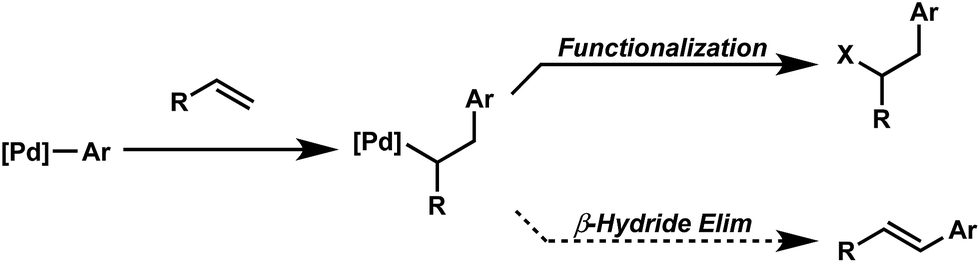 | ||
| Scheme 1 Palladium-catalyzed difunctionalization via Interrupted Mizoroki–Heck reaction. | ||
In particular, this strategy was recently revived in the development of palladium-catalyzed protocols for the 1,2- and 1,1-arylchlorination and bromination of unactivated α-olefins.2a,c In addition, one of our laboratories applied this strategy to the palladium catalyzed 1,2-diarylation of alkenes utilizing arylstannanes as the coupling partner and oxygen as the terminal oxidant.2b This technology was further extended to mixed diarylation reactions utilizing aryldiazonium salts and arylboronic acids for 1,1-diarylation of terminal alkenes and the 1,2-diarylations of dienes.2d,f The 1,4-divinylation of isoprene was achieved using vinyl triflates and boronic acids.2i The variety of coupling partners used to generate and intercept Mizoroki–Heck intermediates in these reports encouraged us to continue to explore the generality of this 3-component coupling platform.
The question we initially considered was that of oxidative fluorination of the σ-alkyl palladium intermediate on the basis of multiple studies describing sp3-C–F reductive elimination from high-valent metal species, including palladium.4,5 Specifically in this regard, we successfully integrated alkene difunctionalization reactions initiated via a Heck reaction to the enantioselective construction of sp3-C–F bonds,6 in our reported Pd-catalyzed directed enantioselective 1,2-arylfluorination of styrenes.2e This initial report was followed by the development of methods for enantioselective 1,1-arylfluorination of protected allylamines2h and β,β-arylfluorination of α,β-unsaturated carbonyls.2j
Although these reactions apply similar conceptual strategies, the regiochemical outcome of the transformations is dependent on substrate and conditions. The ultimate outcome of these reactions is determined by both the regioselectivity of the initial migratory insertion event, and the propensity of the palladium to migrate via successive β-hydride elimination and insertion events prior to oxidative functionalization. The latter suggests that oxidative alkene difunctionalization beyond the reported 1,1- and 1,2- regioselectivity should be accessible though pathways analogous to those proposed for the redox-relay Heck reaction reported by one of our laboratories.7 Herein, we present the development of the first Pd-catalyzed 1,3-arylfluorination reaction, including a catalytic enantioselective variant, as well as an integrated experimental, computational, and statistical analysis of the site selectivity as a function of substrate and ligand. The results of these studies shed light on the factors that govern site selectivity in the migratory insertion step, which should inform future applications of the strategies described.8
Discovery and development of 1,3-arylfluorination
In our previous studies, we noted that the alkene was arylated at a single site resulting in a 2,1-fluoroarylation of a trans-di-substituted styrene (Scheme 2a). In light of this result, the degree of substitution and geometry of alkenes that were competent to participate in this reaction was further examined. During these studies, our attention turned to the ortho-carboxamide of [2H]-chromene (Scheme 2b, R = CONHPh) as a substrate. Under the previously optimal conditions to achieve a directed 2,1-arylfluorination process, the resulting product was observed only in a trace amount. Surprisingly, the 1,3-arylfluorination product was the major product (Scheme 2b), thereby overcoming the expected directing group bias.2e,9 Given this unprecedented site selectivity of the initial migratory insertion event and the impact of ligand on such selectivity (vide infra), we posited that this transformation would be an ideal platform to investigate the subtle factors that contribute to regioselectivity in migratory insertion step of interrupted Mizoroki–Heck reactions.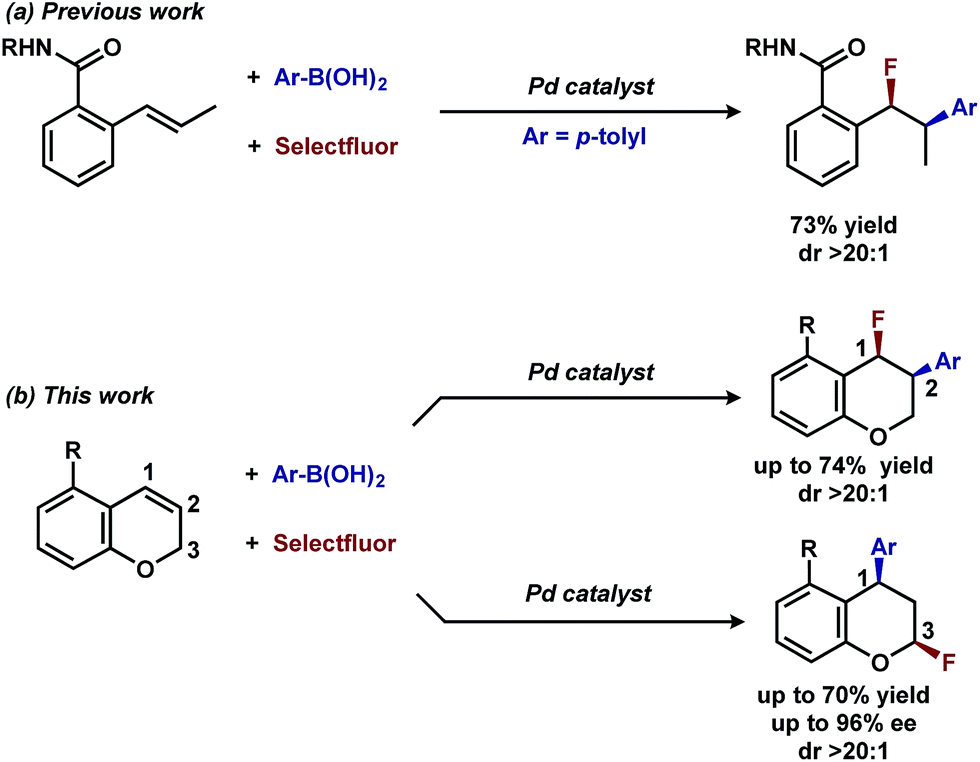 | ||
| Scheme 2 Palladium-catalyzed fluoroarylation reactions. | ||
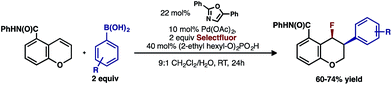
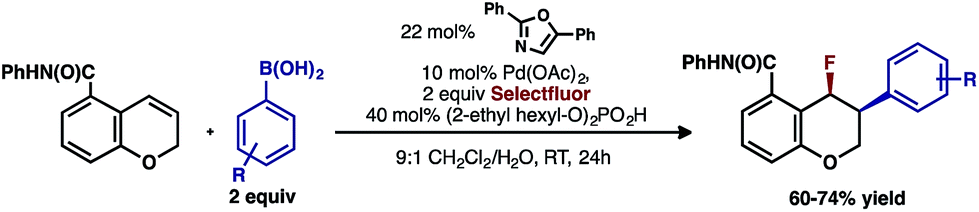
Intrigued by the 1,3-disposition of the introduced substituents, we sought to initially probe the scope of this transformation (Table 1). Using 4,4′-di-tert-butyl-2,2′-bipyridine L1 as the ligand, a wide variety of boronic acids (bearing electron withdrawing and donating groups) were evaluated. Under these conditions, the 1,3 display of the introduced functional groups was conserved with a range of boronic acids (2a–h). Interestingly, [2H]-chromenes bearing different substitution patterns were either less efficient (3–5a) or ineffective (5b–5d) under these reaction conditions.
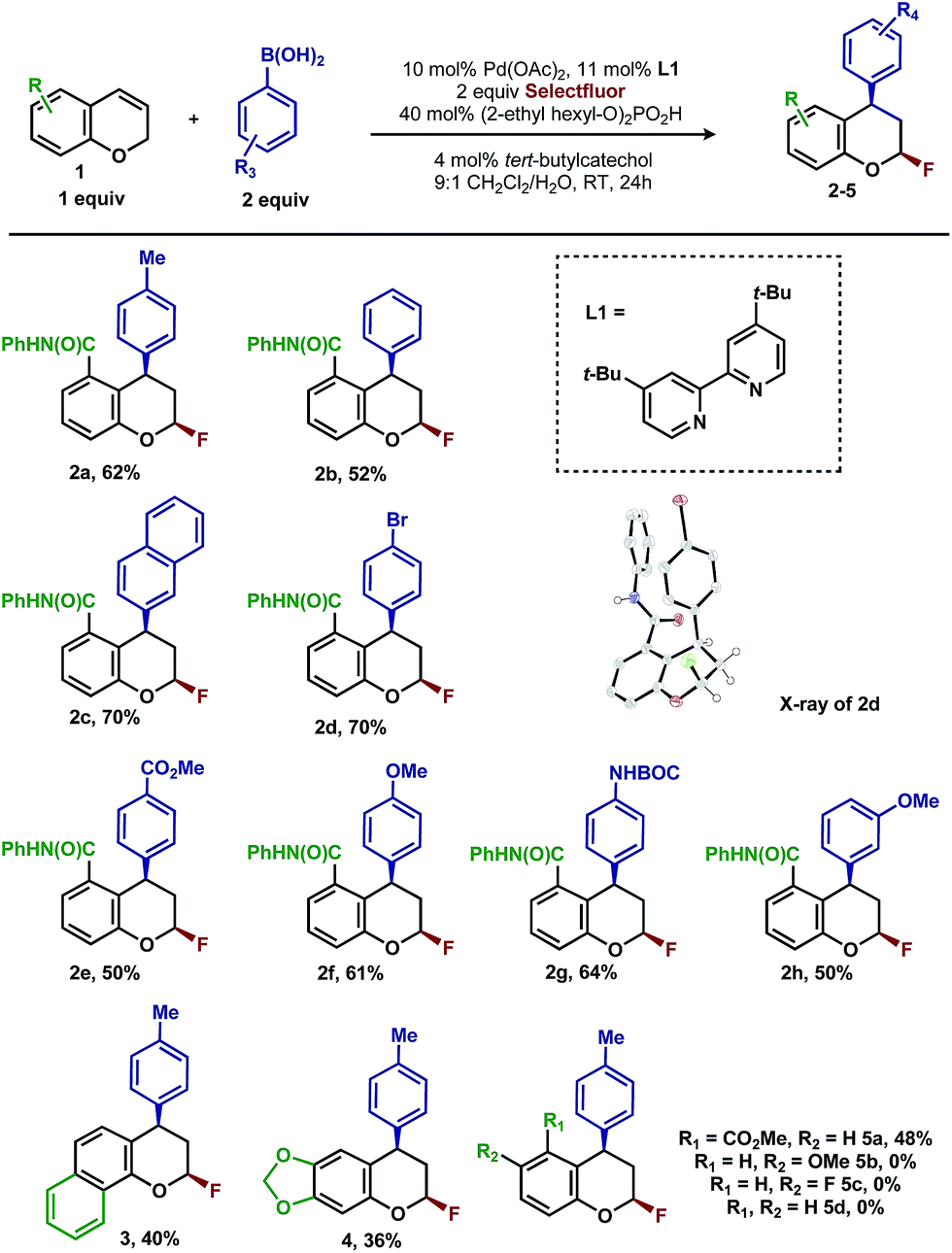
Having already achieved an enantioselective 2,1-arylfluorination utilizing styrene as a substrate,2e we anticipated that the novel 1,3-arylfluorination manifold could also be rendered enantioselective. Several commercially available and readily accessible chiral N,N ligands were evaluated (see ESI†); however, only trace amounts of the target product were observed in nearly all cases. In a similar fashion to our previous study, the most promising class of ligand for this transformation were the PyrOx class, of which (S)-4-tert-butyl-2-(2-pyridyl)oxazoline led to the formation of 2a with the highest enantioselectivity (see ESI†). After extensive optimization (see ESI†), we identified that the addition of 1.5 equivalents of sodium fluoride10 and a DCE/H2O solvent mixture rendered the reaction selective for the 1,3-product (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 relative to the 2,1-arylfluorination product), while also maintaining a high enantioselectivity. Using these modified reaction conditions, the scope of the enantioselective reaction was explored. It should be noted that the preformed palladium(II) complex 6 was utilized in this investigation as this aided the reproducibility.11 The 1,3-difunctionalized products were obtained in moderate yields and high enantiomeric excess (Table 2, 2a–2h), when chromenes with an ortho-amide substituent were employed. When the corresponding ester substituent was employed in the ortho position, the product 5a was formed in both lower yield and reduced enantiomeric excess. The synthetic utility of these pyranyl fluorides was further demonstrated by treating 2a with potassium trifluoroborate salt 7 in the presence of BF3·etherate to form alkyne 8 in good yield and high diastereoselectivity, without erosion of the enantiomeric purity.12
:1 relative to the 2,1-arylfluorination product), while also maintaining a high enantioselectivity. Using these modified reaction conditions, the scope of the enantioselective reaction was explored. It should be noted that the preformed palladium(II) complex 6 was utilized in this investigation as this aided the reproducibility.11 The 1,3-difunctionalized products were obtained in moderate yields and high enantiomeric excess (Table 2, 2a–2h), when chromenes with an ortho-amide substituent were employed. When the corresponding ester substituent was employed in the ortho position, the product 5a was formed in both lower yield and reduced enantiomeric excess. The synthetic utility of these pyranyl fluorides was further demonstrated by treating 2a with potassium trifluoroborate salt 7 in the presence of BF3·etherate to form alkyne 8 in good yield and high diastereoselectivity, without erosion of the enantiomeric purity.12

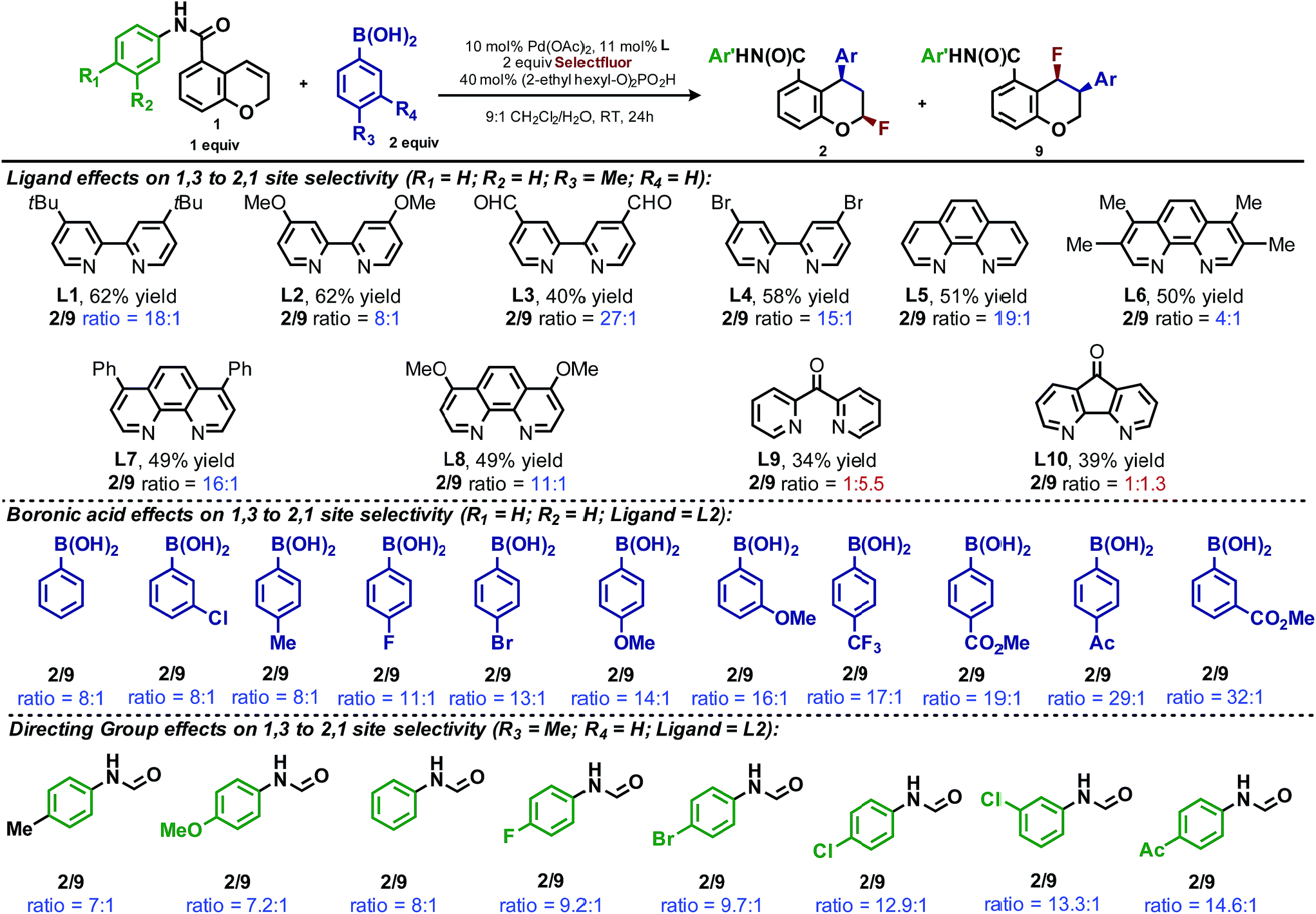
During the development of the enantioselective variant of this reaction, we observed the formation of the 2,1-arylfluorination product 9 (Table 3) in significant quantities. Given the unique nature of the 1,3-fluoroarylation reaction and the apparent ligand effect on regioselectivity, we sought to better understand the origin of this divergence. We set out to determine what experimental parameters affect the site selectivity of migratory insertion, the results of which are summarized in Table 3. We have identified three variables that significantly influence the observed ratio of products 2 and 9: choice of ligand, aryl boronic acid coupling partner, and directing group. The data was utilized in the following computational and statistical analysis to develop our mechanistic hypothesis and better our understanding the factors governing reaction performance and site selectivity.
Ligand effects on product distribution
To elucidate the ligand features that influence the site selectivity in the arylfluorination reaction of chromenes, various ligands were evaluated in the palladium-catalyzed arylfluorination reaction of [2H]-chromene 1a and para-tolyl boronic acid (Table 3). The results illustrate a wide variation of reaction outputs from an undiscriminating (1:1.3; Table 3, L10) to a highly selective (27:1; Table 3, L3) 1,3-arylfluorination process. Following these experiments the ground state structure of various ligated palladium complexes, PdLCl2, were calculated using DFT.
Numerous parameters were gathered from the geometry optimized structures of the palladium complexes including Natural Bond Orbital (NBO) charges, N–Pd–N bite angle, Sterimol values, and IR frequencies and intensities.13 Through linear regression analysis, Pd NBO charge was found to correlate well with the difference in transition state energies of the two regioisomers (ΔΔG‡), which can be related to the log of the product ratios (Fig. 1b).14 As the Pd NBO charge becomes more positive, the ΔΔG‡ increases, which is directly correlated to greater selectivity for the 1,3-product. We conclude from this correlation that enhanced electrophilicity of the palladium center results in greater selectivity for the 1,3-arylfluorination product.
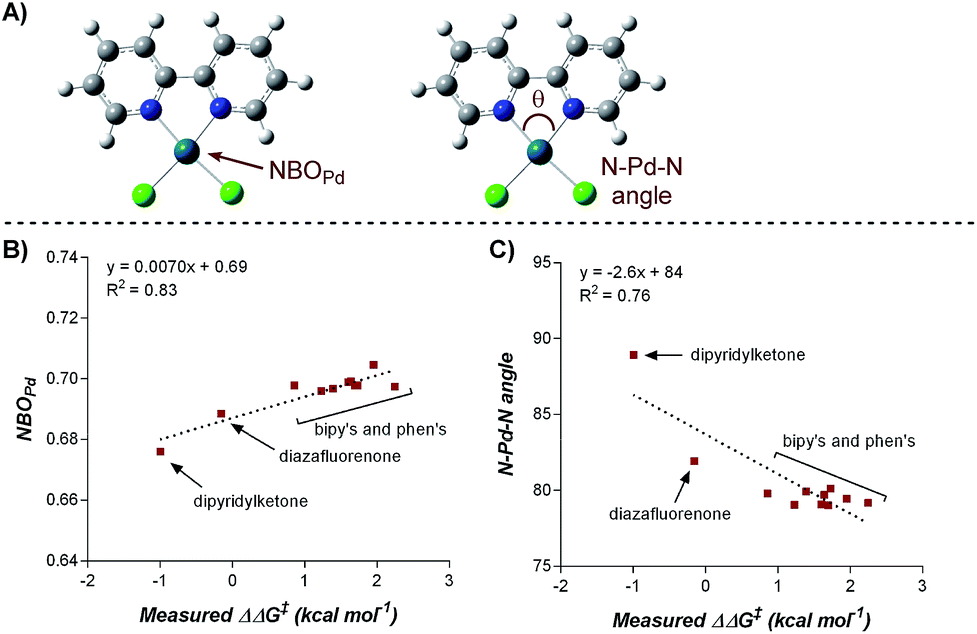 | ||
| Fig. 1 Correlation of Pd-NBO charge and ligand bite angle with regioselectivity. | ||
Additionally, the dipyridylketone and diazafluorenone were computed to have wider bite angles as compared to the other ligands tested, which correlates to the formation of more of the 2,1-product (Fig. 1c). This structural distortion has previously been shown to result in complex co-ordination chemistry for diazafluorenone with palladium(II) acetate.15a As a result, these ligands have hemi-labile behavior, suggesting they can act as both monodentate and bidentate ligands.15b We hypothesized that this ability to act in some cases as a monodentate ligand was critical for the preferred formation of the 2,1-product. This hypothesis was further supported by the fact that when monodentate ligands, such as oxazoles and pyridines, were employed with 1a and a variety of boronic acids exclusive formation of the 1,2-product was observed (see ESI†).
Effect of aryl coupling partner
The impact of the aryl boronic acid on site selectivity was explored in order to further understand the features contributing to the site of migratory insertion in these reactions. For this study, the standard reaction conditions were applied with substrate 1a in conjunction with a range of arylboronic acids; 4,4′-dimethoxy-2-2′-bipyridine L2 was chosen as the ligand since its use demonstrated moderate selectivity in the initial ligand screen (8:1; Table 3). Under the standard reaction conditions, the site selectivity observed in the arylfluorination was significantly impacted as a result of changes to the boronic acid coupling partner (Table 3). To understand what factors drive these changes, a similar correlation analysis was applied for this collection of arylboronic acids. Through the use of univariate linear regression analysis, it was found that the IR COH bending frequency of the corresponding benzoic acid correlated to the site selectivity in migratory insertion for this diverse set of boronic acids (Fig. 2).16 A more electron-withdrawing group (EWG) enhanced the formation of the 1,3-product. This correlation is consistent with our previous hypothesis that an increase in the cationic character of palladium complex favors formation of the 1,3-product.
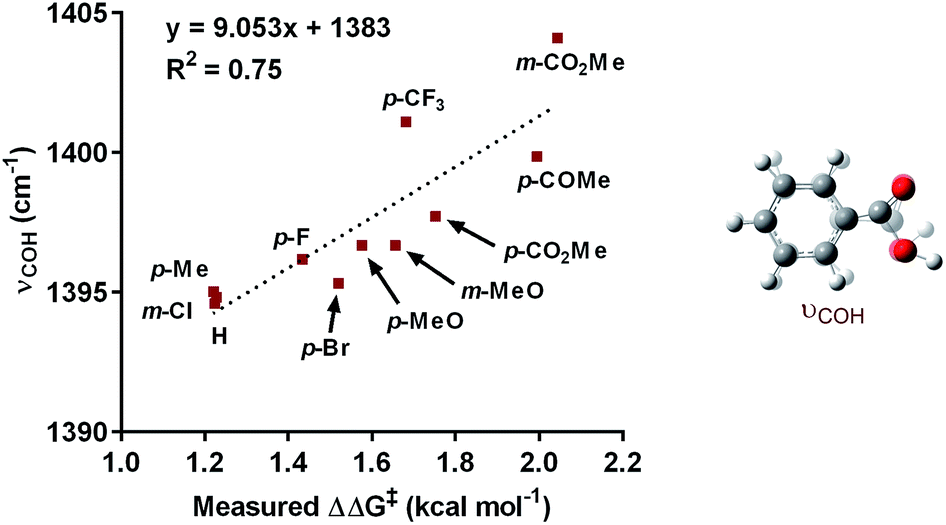 | ||
| Fig. 2 Correlation of IR stretch with regioselectivity. | ||
Directing group influence on regioselectivity
The final point for manipulation and analysis concerned the effect that the “directing group” has on the reaction outcome. To investigate this consequence, the standard reaction conditions were once again employed with 4,4′-dimethoxy-2-2′-bipyridine L2 as the ligand. The arylfluorination reaction site selectivity was impacted through alteration of the aryl-amide substituent, suggesting that the initial olefin insertion process is also controlled by the electronics on the arylamide of the chromene substrate (Table 3). In fact, a linear correlation was identified between the Hammett σ-values of various aryl substituents on the amide versus differential transition state energies for the formation of two constitutional isomers yielded in the reaction (Fig. 3).17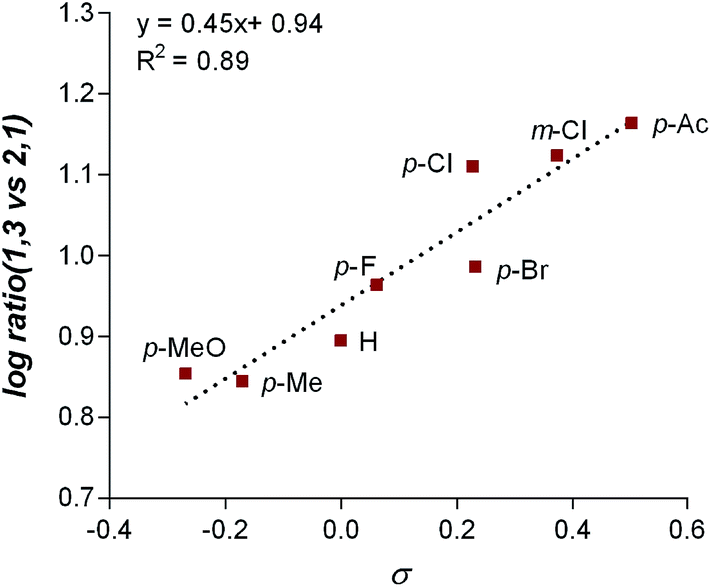 | ||
| Fig. 3 Plot of Hammett σ-values versus Measured Gibbs free energy change. | ||
A positive slope in the Hammett plot suggests that the electronics on the arylamide is impacting the orientation of [Pd]–Ar species via coordination to the metal center, thus influencing the migratory insertion pathway. In general, electron donating groups (EDG) on the arylamide decreases the selectivity for 1,3-products. We rationalized that with the use of EDG on the arylamide, coordination to the [Pd]–Ar species is more favorable, and thus the increased 2,1-product formation is a result of enhanced efficiency of coordination of the directing group.
To further probe the mechanistic divergence, we sought validation of our developing hypothesis that bifurcation occurs from the initial migratory insertion event. We assumed that the 2,1-arylfluorination product was formed in an analogous fashion to our previously reported reaction with styrenes, namely by a migratory insertion that places the palladium in the benzylic position, and the aryl-group in the homo-benzylic position, followed by C–F bond formation. For the 1,3-arylfluorination reaction, we anticipated an oxidative Heck-type mechanism would also be operative, although the possibility of alternative mechanisms, including allylic C–H palladation,18 was considered. We hypothesized that the 1,3-product was formed by a migratory insertion with the opposite sense of selectivity, followed by palladium migration, and C–F bond formation.
Deuterium labeling experiments
To gather further support for this latter hypothesis, we performed a deuterium labeling experiment with chromene d2-1, (Scheme 3a). The resulting product d2-2a, in which one deuterium migrated to the adjacent carbon, was the exclusive arylfluorination product. This is possible if a [Pd]-alkyl intermediate undergoes β-hydride elimination and reinsertion events on the carbon α to oxygen, which is suggestive of an oxidative Heck-type mechanism. In a second experiment designed to understand the palladium chain walking and probe the stereochemistry of the C–F bond-forming step, a cross-over experiment with chromene 1 and deuterated substrate d2-1 was performed (Scheme 3b). The observation of a 1:1 mixture of product 2a with no deuterium and product d2-2a with two deuterium atoms indicated that dissociation of an intermediate olefin from a palladium hydride species and subsequent isotopic scrambling likely did not occur,19 thereby supporting an inner-sphere C–F bond forming reductive elimination.4a,20
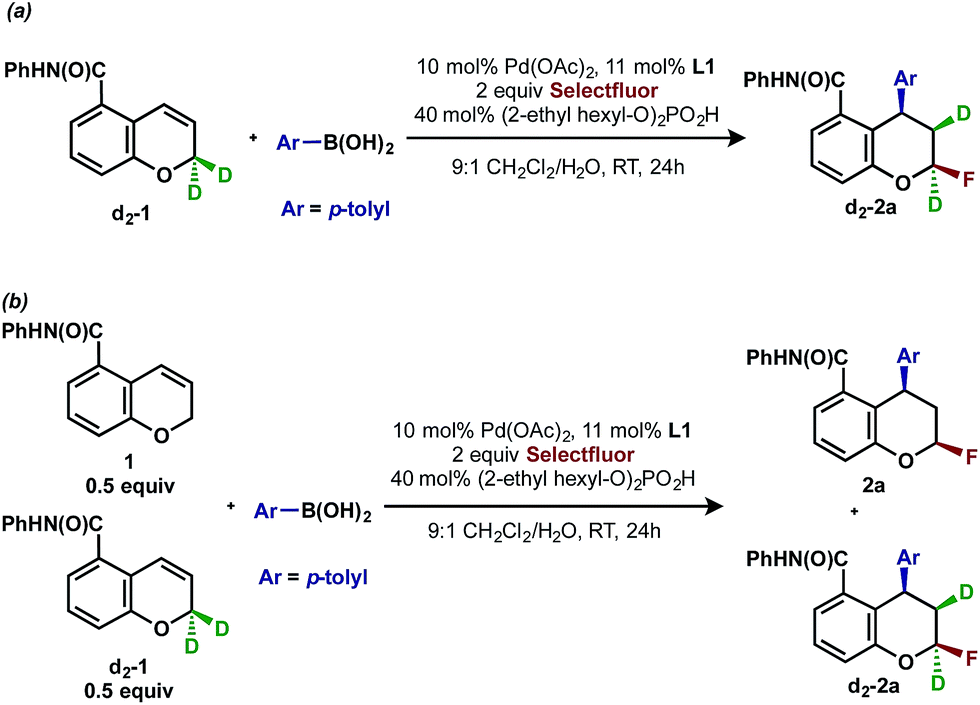 | ||
| Scheme 3 Deuterium labeling experiment with chromene d2-1. | ||
Mechanistic proposal
Having established that both products likely arise from oxidative Heck-like mechanisms, we propose the following two pathways to rationalize the divergence in site selectivity outlined in Fig. 4 and 5. In the presence of a strong bidentate ligand such as 4,4′-di-tert-butyl-2,2′-bipyridine L1 (Fig. 4), transmetallation with an arylboronic acid, followed by displacement of an anionic ligand with a chromene olefin, results in the formation of cationic palladium species, C. The site of migratory insertion is then controlled by the polarity of the alkene. When considering the chromene as a vinylogous enol-ether, insertion of the aryl group at the position α to the aromatic ring and the palladium at the β-position gives rise to the expected regiochemical outcome for an electron rich olefin and a cationic palladium species.21 Subsequent migration and oxidation results in the formation of the observed 1,3-product. In this case, the polarity bias of the olefin outcompetes the influence of the directing group. The correlations we found between the increased cationic character on palladium to greater selectivity for the 1,3-product corroborates this hypothesis. Additionally, when considering the pre-migratory insertion intermediate C, the lack of readily available coordination site in the square planar complex, the directing group would be expected to have little influence on the regioselectivity of the migratory insertion. In the presence of either hemi-labile bidentate or monodentate ligands,22 we alternatively propose the mechanism outlined in Fig. 5. After transmetallation of an arylboronic acid to form intermediate F, a labile ligand may allow for the formation of an intermediate such as G, in which the substrate is ligated by both the olefin and the ortho-carboxamide directing group. We propose that subsequently a directed migratory insertion occurs, placing the palladium α to the aromatic ring, and proximal to the ortho-amide directing group. Finally, oxidation and reductive elimination would afford the 2,1-arylfluorination product. In this case, the open coordination site for the directing group to occupy would be more accessible, thus enhancing the influence of the directing group. In addition, our studies also indicate that the electrophilicity of the [Pd]–Ar also has an influence on the location of the migratory insertion. The proposed intermediate G, is a neutral palladium species and as a result is less-electrophilic than the corresponding intermediate in the mechanism in Fig. 4; thus, the polarity of the olefin has significantly less influence in the selectivity determining step. It should be noted that similar results where significant shifts in the site of migratory insertion as a function of ligand structure in Heck reactions is precedented.23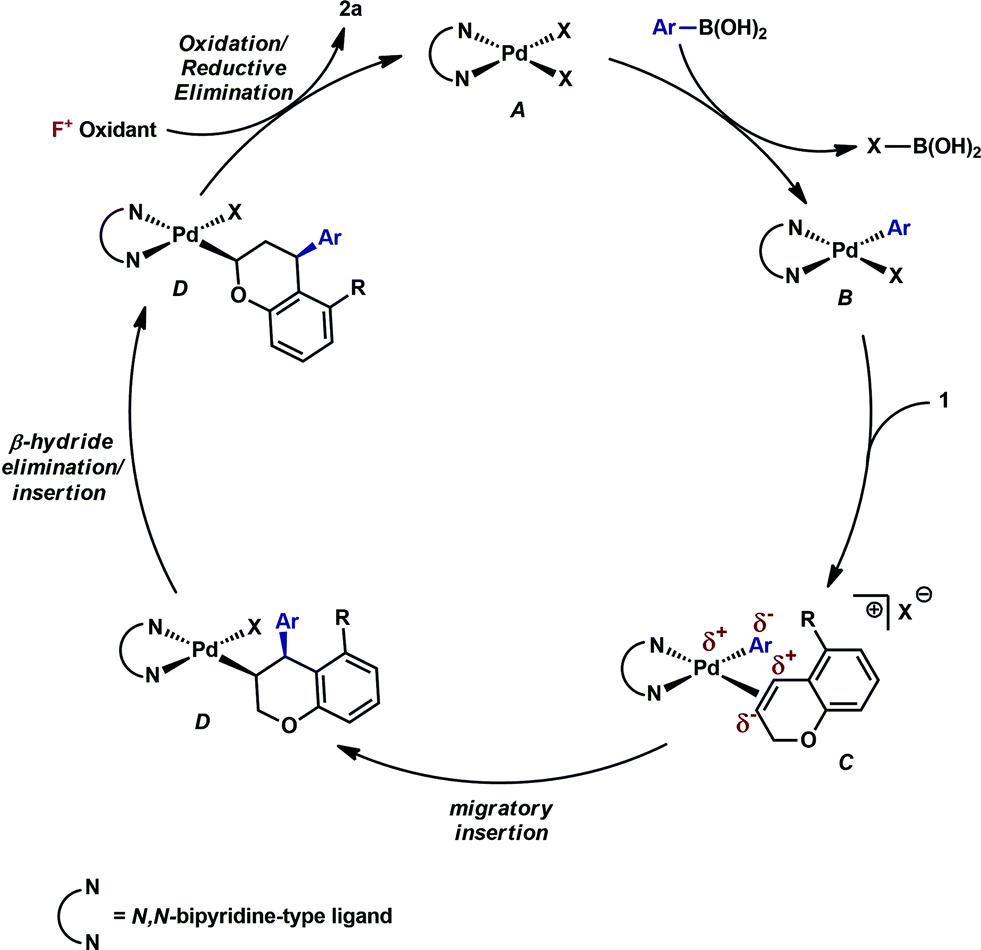 | ||
| Fig. 4 Proposed mechanisms for the formation of the 1,3-arylfluorination product. | ||
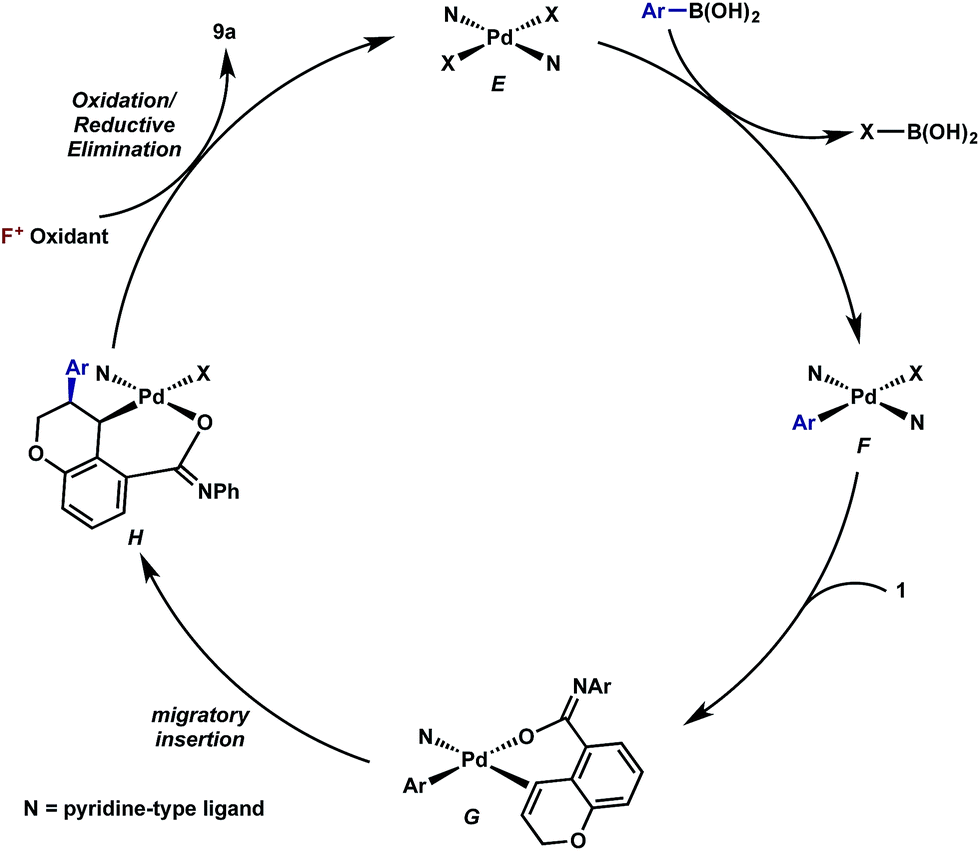 | ||
| Fig. 5 Proposed mechanisms for the formation of the 2,1-arylfluorination product. | ||
Finally, the low enantioselectivity observed (7% ee; Scheme 4) for 2,1-arylfluorination products when bidentate chiral ligands are employed may also be attributed to the partial dissociation of ligand allowing directing group ligation. In contrast, high enantioselectivity was observed with the chiral bidentate PyrOx ligand L* for the formation of 1,3-arylfluorination product (Table 2). This observation is consistent with a mechanistic hypothesis discussed above in which the 1,3-arylfluorination occurs through a palladium species wherein the chiral ligands maintain their bidentate coordination.24
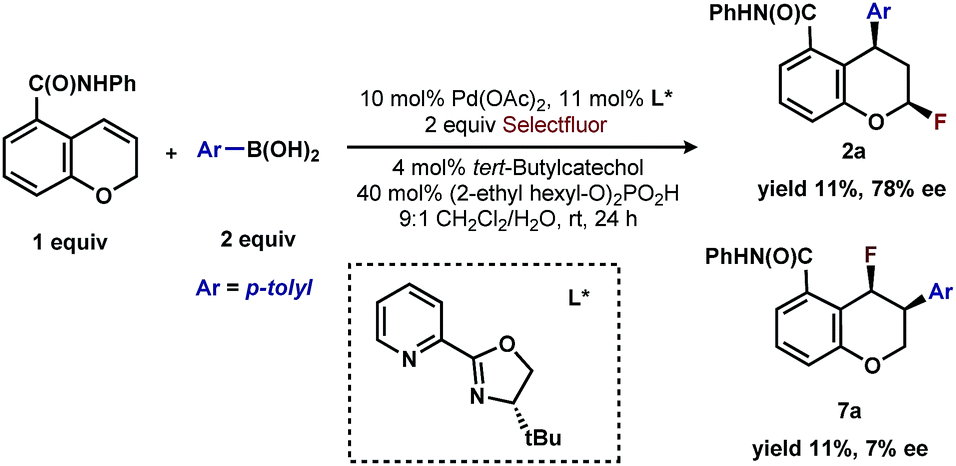 | ||
| Scheme 4 Comparison of enantioselective 1,3-arylfluorination to 2,1-arylfluorination. | ||
Conclusion
We have developed a 1,3-arylfluorination of [2H]-chromenes; the first of the palladium-catalyzed arylhalogenation reactions that results in 1,3-relationship of the introduced substituents. In addition, we have developed an enantioselective variant of the 1,3-arylfluorination of [2H]-chromenes and demonstrated the utility of the enantioenriched pyranyl fluorides by further diastereoselective C–C bond formation. We have established that both the 1,3- and 2,1-products likely arise from oxidative Heck-type mechanisms that diverge at the initial migratory insertion event. Our integrated experimental, computational, and statistical analysis revealed that the identity of the ligand, the arylboronic acid coupling partner, and the directing group all affect the site of migratory insertion. The vinylogous enol ether selectivity leading to formation of the 1,3-product is enhanced by increased electrophilic character at palladium, either by the bipyridine/phenanthroline ligand or the electronics of the aryl coupling partner. Selectivity for the 1,2-product is enhanced by increased electron donating character of the amide directing group and decreased denticity of the supporting ligand. These results should help inform the design of future arylfluorination reactions, and more broadly shed light on the subtle factors, which influence the site of functionalization in interrupted Mizoroki–Heck reactions and the role of directing groups in high-valent palladium catalyzed reactions.Acknowledgements
F. D. T. thanks NIHGMS (R35 GM118190) for financial support and T. A. F. is grateful for a postdoctoral fellowship from CAPES. M. S. S. thanks NSF (CHE-1361296) for support. The authors thank the Novartis Education, Diversity and Inclusion (ED&I) office for a presidential postdoctoral fellowships to E. P. A. T. and V. S. We acknowledge Dr A. DiPasquale (UC Berkeley) for assistance with X-ray crystallography and support from NIH Shared Instrument Grant (S10-RR027172). We acknowledge Dr J. Coelho for assistance with DFT calculations.Notes and references
- For multicomponent reactions in organic synthesis see: J. Zhu and B. Hugues, Multicomponent Reactions, Wiley-VCH, Weinheim, 2005 Search PubMed.
- (a) D. Kalyani and M. S. Sanford, J. Am. Chem. Soc., 2008, 130, 2150 CrossRef CAS PubMed; (b) K. B. Urkalan and M. S. Sigman, Angew. Chem., Int. Ed., 2009, 48, 3146 CrossRef CAS PubMed; (c) D. Kalyani, A. D. Satterfield and M. S. Sanford, J. Am. Chem. Soc., 2010, 132, 8419 CrossRef CAS PubMed; (d) V. Saini, L. Liao, Q. Wang, R. Jana and M. S. Sigman, Org. Lett., 2013, 15, 5008 CrossRef CAS PubMed; (e) E. P. A. Talbot, T. de A. Fernandes, J. M. McKenna and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 4101 CrossRef CAS PubMed; (f) B. J. Stokes, L. Liao, A. Mendes, Q. Wang and M. S. Sigman, Org. Lett., 2014, 16, 4666 CrossRef CAS PubMed; (g) H. M. Nelson, B. D. Williams, J. Miro and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 3213 CrossRef CAS PubMed; (h) Y. He, Z. Yang, R. T. Thornbury and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 12207 CrossRef CAS PubMed; (i) M. S. McCammant and M. S. Sigman, Chem. Sci., 2015, 6, 1355 RSC; (j) J. Miro, C. del Pozo, F. D. Toste and S. Fustero, Angew. Chem., Int. Ed., 2016, 55, 9045 CrossRef CAS PubMed. For other palladium-catalyzed oxidative difunctionalizations see: (k) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413 CrossRef CAS PubMed.
- (a) R. F. Heck, J. Am. Chem. Soc., 1968, 90, 5538 CrossRef CAS; (b) Y. Tamaru, M. Hojo, H. Higashimura and Z. Yoshida, Angew. Chem., Int. Ed. Engl., 1986, 25, 735 CrossRef; (c) Y. Tamaru, M. Hojo, S. Kawamura and Z. Yoshida, J. Org. Chem., 1986, 51, 4089 CrossRef CAS. For more on the Heck reaction see: (d) M. Larhed and A. Hallberg, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi and A. De Meijere, Wiley, New York, 2002, pp. 1133–1178 Search PubMed; (e) M. Oestreich, The Mizoraki–Heck Reaction, Wiley, Hoboken, NJ, 2009 Search PubMed.
- From Pd(IV): (a) J. M. Racowski, J. B. Gary and M. S. Sanford, Angew. Chem., Int. Ed., 2012, 51, 3414 CrossRef CAS PubMed. From Pt(IV): (b) S.-B. Zhao, J. J. Becker and M. R. Gagne, Organometallics, 2011, 30, 3926 CrossRef CAS PubMed. From Au(III): (c) N. Mankad and F. D. Toste, Chem. Sci., 2012, 3, 72 RSC.
- For catalytic example involving Pt(IV) see: N. A. Chochrane, H. Nguyen and M. R. Gagne, J. Am. Chem. Soc., 2013, 135, 628 CrossRef PubMed.
- For alternative carbofluorination strategies see: (a) H. Wang, L.-N. Guo and X.-H. Duan, Chem. Commun., 2014, 50, 7382 RSC; (b) H. Peng and G. Liu, Org. Lett., 2011, 13, 772 CrossRef CAS PubMed; (c) M.-G. Braun, M. H. Katcher and A. B. Doyle, Chem. Sci., 2013, 4, 1216 RSC; (d) S. Kindt and M. R. Heinrich, Chem.–Eur. J., 2014, 20, 15344 CrossRef CAS PubMed. For reviews on enantioselective sp3-C–F reactions see: (e) S. Lectard, Y. Hamashima and M. Sodeoka, Adv. Synth. Catal., 2010, 352, 2708 CrossRef CAS; (f) D. Cahard, X. Xu, S. Couve-Bonnaire and X. Pannecoucke, Chem. Soc. Rev., 2010, 39, 558 RSC; (g) T. Liang, C. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (h) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (i) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073 CrossRef CAS PubMed.
- (a) H. H. Patel and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 3462 CrossRef CAS PubMed; (b) T.-S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef CAS PubMed; (c) Z.-M. Chen, M. J. Hilton and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 11461 CrossRef CAS PubMed.
- K. Subburaj, R. Katoch, M. G. Murugesh and G. K. Trivedi, Tetrahedron, 1997, 53, 12621 CrossRef CAS.
- For directed Mizoroki–Heck reaction see: N. Chatani and L. Ackermann, Directed Metallation, Springer Verlag, Berlin, Germany, 2007 Search PubMed.
- Although the role of the sodium fluoride is not certain, some possibilities include serving as a base for the phosphate or as a ligand on palladium to block coordination of the directing group.
- K. S. Yoo, C. P. Park, C. H. Yoon, S. Sakaguchi, J. O'Neill and K. W. Jung, Org. Lett., 2007, 9, 3933 CrossRef CAS PubMed.
- (a) J. Zeng, S. Vedachalam, S. Xiang and X. Liu, Org. Lett., 2011, 13, 42 CrossRef CAS PubMed; (b) For a related example utilizing a stannane nucleophile see: J. Li, P. Yang, M. Yao, J. Deng and A. Li, J. Am. Chem. Soc., 2014, 136, 16477 CrossRef CAS PubMed.
- M. S. Sigman, K. C. Harper, E. N. Bess and A. Milo, Acc. Chem. Res., 2016, 49, 1292 CrossRef CAS PubMed.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS; (b) K. C. Gross, P. G. Seybold and C. M. Hadad, Int. J. Quantum Chem., 2002, 90, 445 CrossRef CAS; (c) C. A. Hollingsworth, P. G. Seybold and C. M. Hadad, Int. J. Quantum Chem., 2002, 90, 1396 CrossRef CAS; (d) E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429 CrossRef CAS PubMed.
- (a) P. B. White, J. N. Jaworski, C. G. Fry, B. S. Dolinar, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2016, 138, 4869 CrossRef CAS PubMed; (b) P. B. White, J. N. Jaworski, G. H. Zhu and S. S. Stahl, ACS Catal., 2016, 6, 3340 CrossRef CAS PubMed.
- (a) R. N. Jones, W. F. Forbes and W. A. Mueller, Can. J. Chem., 1957, 35, 504 CrossRef CAS; (b) A. Milo, E. N. Bess and M. S. Sigman, Nature, 2014, 507, 210 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- For an example of palladium-catalyzed C–H fluorination see: (a) M. G. Braun and A. G. Doyle, J. Am. Chem. Soc., 2013, 135, 12990 CrossRef CAS PubMed. For reviews on palladium catalyzed allylic C–H activation see: (b) G. Liu and Y. Wu, Top. Curr. Chem., 2010, 292, 195 CrossRef CAS PubMed; (c) F. Liron, J. Oble, M. M. Lorion and G. Poli, Eur. J. Org. Chem., 2014, 27, 5663 Search PubMed.
- (a) T. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340 CrossRef CAS PubMed; (b) M. J. Hilton, L. Xu, P. Norrby, Y. Wu, O. Wiest and M. S. Sigman, J. Org. Chem., 2014, 79, 11841 CrossRef CAS PubMed.
- An alternative explanation for the observed 1,3-syn-stereodiad is reductive elimination of the α-oxy-palladium species to form the corresponding oxonium ion, which is trapped by fluoride under thermodynamic conditions. DFT calculations revealed that the syn-isomer is the thermodynamically more stable isomer by <0.5 kcal mol−1. The fact that we do not observe the trans-product together with the small calculated thermodynamic preference for the cis-isomer suggests that the product is formed via an inner sphere C–F reductive elimination. In either case, the C–F bond-forming step is independent of the regiodetermining step.
- (a) G. D. Daves Jr and A. Hallberg, Chem. Rev., 1989, 89, 1433 CrossRef; (b) W. Cabri, I. Candiani, A. Bedeschi and R. Santi, J. Org. Chem., 1992, 57, 3558 CrossRef CAS; (c) W. Cabri, I. Candiani, A. Bedeschi, S. Penco and R. Santi, J. Org. Chem., 1992, 57, 1481 CrossRef CAS; (d) W. Cabri and I. Candiani, Acc. Chem. Res., 1995, 28, 2 CrossRef CAS.
- In accord with this hypothesis, we identified an oxazole ligand as providing high selectivity for the 2,1-arylfluorination. See ESI† for additional details on screening of monodentate ligands and 2,1-arylfluorination substrate scope.
 .
. - (a) W. Cabri, I. Candiani, A. Bedeschi and R. Santi, J. Org. Chem., 1990, 55, 3654 CrossRef CAS; (b) J. Ruan and J. Xiao, Acc. Chem. Res., 2011, 44, 614 CrossRef CAS PubMed.
- (a) F. Ozawa, A. Kubo and T. Hayashi, J. Am. Chem. Soc., 1991, 113, 1417 CrossRef CAS; (b) E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef CAS PubMed; (c) J. P. Knowles and A. Whiting, Org. Biomol. Chem., 2007, 5, 31 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1518394. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05102b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |