
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcid/base triggered interconversion of μ-η2:η2-peroxido and bis(μ-oxido) dicopper intermediates capped by proton-responsive ligands†
V. E.
Goswami
a,
A.
Walli
a,
M.
Förster
b,
S.
Dechert
a,
S.
Demeshko
a,
M. C.
Holthausen
 *b and
F.
Meyer
*a
*b and
F.
Meyer
*a
aInstitut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstraße 4, 37077 Göttingen, Germany. E-mail: franc.meyer@chemie.uni-goettingen.de
bInstitut für Anorganische und Analytische Chemie, Goethe-Universität Frankfurt, Max-von-Laue-Straße 7, 60438 Frankfurt am Main, Germany. E-mail: max.holthausen@chemie.uni-frankfurt.de
First published on 17th February 2017
Abstract
CuII2(μ-η2:η2-peroxido) and CuIII2(μ-oxido)2 cores represent key intermediates in copper/dioxygen chemistry, and they are mechanistically important for biological hydroxylation and oxidation reactions mediated by dinuclear (type III) copper metalloenzymes. While the exact nature of the active species in different enzymes is still under debate, shifting equilibria between Cux/O2 species is increasingly recognized as a means of switching between distinct reactivity patterns of these intermediates. Herein we report comprehensive spectroscopic, crystallographic and computational analysis of a family of synthetic CuII2(μ-η2:η2-peroxido) and CuIII2(μ-oxido)2 dicopper complexes with a bis(oxazoline) (BOX) capping ligand. In particular, we demonstrate that a reversible peroxido/bis(μ-oxido) interconversion of the [Cu2O2] core can be triggered by peripheral (de)protonation events on the ligand backbone. As the copper ions in the enzymes are typically supported by histidine imidazoles that offer a backside N atom amenable to potential (de)protonation, it is well conceivable that the shifting of equilibria between the [Cu2O2] species in response to changes in local pH is biologically relevant.
Introduction
Dioxygen binding to copper in the active sites of metalloproteins has received much attention in past decades.1 These metalloenzymes serve as prototypes for the development of bioinspired catalysts that can mediate the selective oxidation and oxygenation of C–H bonds, which is increasingly relevant for viable fuel and chemical feedstock formation.2 Major research efforts in bioinorganic chemistry have been devoted to the development of small model complexes that mimic various aspects of the structure and function of the natural enzymes.3–8 Owing to their specific design, it is often possible to trap key intermediates in such systems and to characterize their intrinsic reactivities in much greater detail than for the metalloenzymes themselves. The ultimate challenge is to understand the origin of the remarkable selectivity of the biochemical role models and to exploit the same concepts for the development of efficient synthetic catalysts.2,9A variety of Cux/O2 intermediates with different dioxygen binding modes have been uncovered and their diagnostic spectroscopic features and distinct reactivities are reasonably well understood.1–3,6,7,10 Binuclear complexes with a μ-η2:η2 peroxido dicopper(II) core (P in Scheme 1), as found in oxy-haemocyanin or oxy-tyrosinase, are among the most prominent species, and synthetically useful biomimetic analogs capable of hydroxylating exogenous phenol substrates are starting to emerge.11–14 In model studies, it has been shown that dicopper complexes bearing a bis(μ-oxido) dicopper(III) core (O) are capable of C–H bond hydroxylation as well.15–18 In solution, the thermodynamic preference for the P or the O core depends on the subtle influence of the particular supporting ligand, the solvent and the nature of counterions, and in some cases both isomers have been reported to coexist in rapid equilibrium.15,19–22 These findings have spurred vivid discussions regarding the nature of the active species in hydroxylation reactions13,23–28 and the P/O core interconversion is increasingly recognized as being mechanistically relevant. Herein we report on an unprecedented pH-dependent P/O interconversion reaction, triggered by (de)protonation events in a bioinorganic model complex, which appears to be of particular relevance for such reactions in biological systems.
 | ||
| Scheme 1 P and O isomers found to be in equilibrium with each other (A). Bis-oxazoline(BOX) ligands used in this work (B). | ||
Results and discussion
We have recently shown that bidentate bis(oxazoline)s (BOXs), which are easily accessible and represent a privileged ligand class, are well-suited for supporting biomimetic Cu/O2 chemistry.29 By spectroscopic means we demonstrated that several ligands, R,HBOXs (Scheme 1B), reversibly bind to dioxygen to form μ-η2:η2-peroxidodicopper(II) complexes, both in solution and in the solid state; the thermodynamic and kinetic parameters of the P core formation have been determined.29 Furthermore, we have shown that certain free bis(oxazoline)s, R,HBOXs, exist as an equilibrium mixture between the diimine and iminoenamine tautomers.30 The latter are reminiscent of β-diketimines used extensively as anionic ligands after deprotonation, which led us to consider R,HBOXs as proton responsive ligands. We have now exploited this concept in bioinspired Cu/O2 chemistry to study an acid/base triggered interconversion between the P and O cores.Reactions of R,R′BOX with [Cu(MeCN)4]ClO4 in tetrahydrofuran (THF) gave the air sensitive copper(I) complexes [(R,R′BOX)Cu(MeCN)]ClO4, 1 (R = R′ = Me), 2 (R = Me, R′ = H), 3 (R = R′ = H)29 as colourless compounds (see ESI†). Oxygenation of 1, 2 and 3 in THF at 193 K led to deep purple coloured solutions of [{R,R′BOX(THF)Cu}2(μ-η2:η2-O2)](ClO4)2, P4 (R = R′ = Me), P5 (R = Me, R′ = H) and P6 (R = R′ = H) with intense optical features around 330 nm (ε ≈ 20 × 103 M−1 cm−1) and 500 nm (ε ≈ 1 × 103 M−1 cm−1). For a detailed assignment we performed time-dependent density functional theory (TD-DFT) calculations on the μ-η2:η2 peroxido complex P5, including a coordinating ClO4− anion in the simulations.31 In good agreement with experiment, the simulated spectrum for P5 shows an intense absorption at 340 nm and a shallow band around 535 nm (Fig. 1b, see ESI† for a detailed discussion). The former absorption originates from a σ → σ* intra-core charge transfer (i.e., from an occupied in-plane dxy(CuII)/π*(O22−)/dxy(CuII) bonding orbital combination into its antibonding counterpart), typically observed for μ-η2:η2 peroxido complexes.26,29,32 The shallow absorption band at 535 nm is assigned to an intra-core π* → σ* transition (i.e., from the out-of-plane π* of the O22− ligand into the in-plane dxy(CuII)/π*(O22−)/dxy(CuII) antibonding orbital combination).
 | ||
| Fig. 1 (a) UV-vis monitoring of the titration of P5 with DBU to give O7. The inset shows the decrease of the band at 333 nm (black triangles) and the rise of the new band at 395 nm (red circles) depending on the equivalents of DBU added. Simulated UV-vis spectra and simplified assignments of electronic transitions ignoring ligand contributions for (b) the dicationic peroxido complex P5 (FWHM = 40) and (c) the neutral bis(μ-oxido) complex O7, calculated at the BLYP-D3/def2-TZVP(SDD)/COSMO(THF) level (FWHM = 13). | ||
Single crystals of P4 and P5 were grown by Et2O diffusion into THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetone (1:1) solutions at 193 K. X-ray diffraction analysis revealed the centrosymmetric molecular structures of the dications (P4 and P5) as shown in Fig. S11† and Fig. 2. Each copper ion is found in a slightly distorted square pyramidal (SP-5) coordination environment (τ = 0.16 in P4, 0.14 in P5) consisting of the respective BOX capping ligand, the bridging side-on μ-η2:η2 peroxide (dCu–O = 1.92–1.93 Å) and a THF solvent molecule bound in the apical position (dTHFCu–O = 2.32/2.33 Å). While the Cu⋯Cu separations of 3.52 Å (P4) or 3.50 av. Å (P5) are typical for P cores, the bridging peroxido ligands exhibit the largest bond lengths (P4: 1.56 Å, P5: 1.58 Å) reported so far for synthetic and biological Cu(μ-η2:η2-O2)Cu systems (Table S3† presents a compilation of geometric and spectroscopic features of all literature-known μ-η2:η2-peroxidodicopper(II) species).33–38 Most of the metric parameters of P4 and P5 are in excellent agreement with those derived from Cu K-edge extended X-ray absorption fine structure (EXAFS) data, for the corresponding P cores obtained with the capping ligands tBu,HBOX (tBu,HP) and H,HBOX (H,HP).29
:acetone (1:1) solutions at 193 K. X-ray diffraction analysis revealed the centrosymmetric molecular structures of the dications (P4 and P5) as shown in Fig. S11† and Fig. 2. Each copper ion is found in a slightly distorted square pyramidal (SP-5) coordination environment (τ = 0.16 in P4, 0.14 in P5) consisting of the respective BOX capping ligand, the bridging side-on μ-η2:η2 peroxide (dCu–O = 1.92–1.93 Å) and a THF solvent molecule bound in the apical position (dTHFCu–O = 2.32/2.33 Å). While the Cu⋯Cu separations of 3.52 Å (P4) or 3.50 av. Å (P5) are typical for P cores, the bridging peroxido ligands exhibit the largest bond lengths (P4: 1.56 Å, P5: 1.58 Å) reported so far for synthetic and biological Cu(μ-η2:η2-O2)Cu systems (Table S3† presents a compilation of geometric and spectroscopic features of all literature-known μ-η2:η2-peroxidodicopper(II) species).33–38 Most of the metric parameters of P4 and P5 are in excellent agreement with those derived from Cu K-edge extended X-ray absorption fine structure (EXAFS) data, for the corresponding P cores obtained with the capping ligands tBu,HBOX (tBu,HP) and H,HBOX (H,HP).29
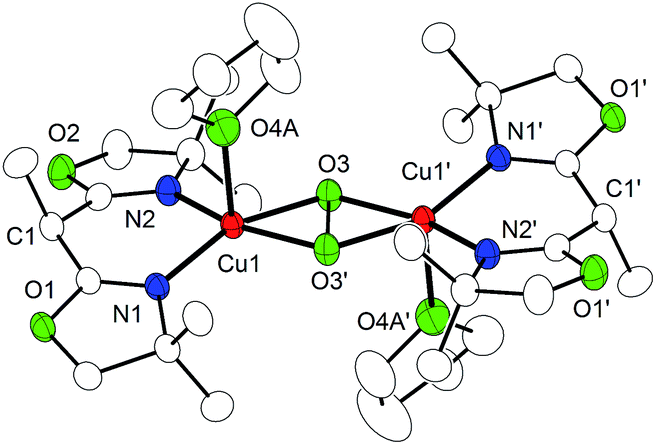 | ||
| Fig. 2 Molecular structure of the cationic part of P5; 30% probability ellipsoids. Hydrogen atoms have been omitted for clarity. | ||
Resonance Raman (rR) spectroscopy (λex = 633 nm) of a THF solution of P4 at 193 K (Fig. 3) and frozen THF solutions of P5 and P6 at 77 K showed an oxygen isotope sensitive feature around 740 cm−1 which shifts to around 700 cm−1 when using 18O2 (P4: 744 cm−1, Δ[18O2] = −40 cm−1; P5: 735 cm−1, Δ[18O2] = −39 cm−1; P6:29 742 cm−1, Δ[18O2] = −39 cm−1). These values fall in the typical range reported for O–O stretching vibrations of P cores (730−760 cm−1 with Δ[18O2] of ca. −40 cm−1,1,39 including oxy-hemocyanin40 and oxy-tyrosinase41). Obviously, the exceptionally long O–O bonds found in P4 and P5 do not translate into unusual rR signatures.42,43
 | ||
| Fig. 3 Resonance Raman spectrum of P4 (left) in THF and O7 (right) in THF/pentane (1:1) at 193 K. The 16O2 spectra are in black and the 18O2 spectra are in red. Residual solvent signals are marked with an asterisk (*). | ||
Experimental data for the magnetic coupling in Cu2/O2 systems determined by SQUID magnetometry are still scarce, mostly because of the thermal lability of such intermediates. Magnetic susceptibility measurements for crystalline material of P4 confirmed the common S = 0 ground state resulting from very strong antiferromagnetic coupling between the cupric ions.44,45 Simulations showed that the lower limit of the exchange coupling is very high, −J ≥ 800 cm−1, based on H = −2JS1S2 (Fig. S3†).46 This is similar to the singlet–triplet splitting observed for oxy-hemocyanin (−J ≥ 300 cm−1)47 and some Cu(μ-η2:η2-O2)Cu model systems.48,49 In summary, the cationic cores of P4–P6 represent genuine Cu(μ-η2:η2-O2)Cu species with all their characteristic structural, spectroscopic and magnetic signatures, though with unusually long O–O bonds. Optical features of these complexes are essentially identical when recorded in THF or in a 1:1 THF:acetone solution, confirming the integrity of the P cores.
Titration of the purple coloured solutions of P5 or P6 in THF with the base diazabicycloundecane (DBU) at low temperatures (193 K) caused a distinct colour change to dark green. Spectral changes were monitored by in situ UV-vis spectroscopy and showed the gradual fading of the bands typical for the P core with the concomitant rise of new bands at 297 nm and 395 nm (Fig. 1a). Full conversion was reached after the addition of about 2 equivalents of DBU per dinuclear copper species, and the resulting products O7 and O8 seemed reasonably stable in the presence of excess DBU. Interestingly, O7 could also be prepared directly from the deprotonated ligand [MeBOX]−, which was obtained as an air sensitive white powder by treating Me,HBOX with one equivalent of nBuLi (see ESI† for details). Reaction of [MeBOX]− with [Cu(MeCN)4]ClO4 in THF gave an air sensitive yellow colored solution of the complex [(MeBOX)CuI] (characterized by NMR spectroscopy and ESI-MS; see ESI†). Subsequent oxygenation in THF at 193 K yielded a dark green coloured solution with optical features identical to those resulting from the titration experiment starting from P5. This corroborates that DBU serves as a base to abstract the backbone protons of the Me,HBOX capping ligands in P5 (Scheme 2).
 | ||
| Scheme 2 Acid/base mediated interconversion between the P core of P5 (R = Me) and P6 (R = H) and the O core of O7 (R = Me) and O8 (R = H). | ||
In THF solution, O7 and O8 both feature three dominant absorption bands with λmax/nm (ε/M−1 cm−1) around 300 (∼2.6 × 104), 335 (∼7–8 × 103) and 400 (∼10–11 × 103) typical for bis(μ-oxido) dicopper(III) species.50,51 This suggests that peripheral deprotonation of the terminal Me,HBOX ligand in P5 and H,HBOX ligand in P6 triggers conversion of the R,HP to the RO core (Scheme 2). The simulated spectrum of O7 obtained from TD-DFT calculations shows four absorption bands at 310, 340, 400 and 690 nm, which is in pleasant agreement with the experimental spectrum (for a simplified assignment of the two dominant transitions see Fig. 1c, for details the ESI†). Similar optical features have been observed by Herres-Pawlis et al. for a guanidine supported bis(μ-oxido) complex, and these authors arrived at an identical assignment of the electronic transitions underlying the two dominant absorptions.52 The band at 690 nm originates from a ligand-to-metal charge transfer with BOX-centered donor MO (cf. the ESI†).
rR spectroscopy of a solution of O7 at 193 K in a 1:1 mixture of THF and pentane revealed a single oxygen isotope sensitive feature at 598 cm−1 (Δ[18O] = −26 cm−1) typical for the breathing mode of the O core (Fig. 3, right). Final proof came from X-ray diffraction analysis of the single crystals of O7, grown by slow diffusion of Et2O into a 1:1 MeTHF:pentane solution at 193 K. O7 has a molecular structure with crystallographically imposed C2 symmetry, each copper ion being ligated by one bidentate [MeBOX]− ligand and the two bridging oxo atoms. The copper coordination geometry is significantly distorted from square planar (angle between the OCuO and NCuN planes: 24.5°) because the terminal [MeBOX]− ligands are severely twisted with respect to each other (Fig. 4). In line with expectation, the Cu–N and Cu–O bonds (1.91 Å and 1.82 Å, respectively) are significantly shorter than in P4 and P5. The Cu⋯Cu distance contracts to 2.87 Å, and the O⋯O separation of 2.24 Å evidences cleavage of the O–O bond. The combined data clearly indicate that O7 is indeed [(MeBOXCu)2(μ-O)2], an unusual example of a neutral bis(μ-oxido) dicopper(III) complex.53,54
 | ||
| Fig. 4 Molecular structure of O7 (top view and side view) with 30% probability ellipsoids. Hydrogen atoms and solvent molecules have been omitted for clarity. | ||
Deprotonation and transformation of the P into the O core also leads to some notable changes in the ligand backbone. Angles around the bridging carbon atoms (C1) change from 109.2(4)°–114.9(4)° in P5 (in one of the two crystallographically independent cations, the second one is disordered) to 118.6(4)°–120.8(4)° in O7. The sp2 character of this carbon atom is also reflected in the bond lengths (excluding C–CH3) which are approximately 0.1 Å shorter in O7. Furthermore C1 is now located within the plane of its three surrounding carbon atoms (0.02 Å deviation vs. 0.45 Å in the case of P5).
A DFT assessment of the P/O core isomerization for P5 and O7 nicely corroborates the experimental observations (Scheme 3): for P5, the peroxido isomer is found to be more stable than the bis(μ-oxido) isomer O5 by ΔG193 = 4 kcal mol−1, and is separated from the latter by a barrier of Δ‡G193 = 10 kcal mol−1. For the neutral complex O7, a reverse stability order results with O7 being favored by 7 kcal mol−1 over P7, and an interconversion barrier of 11 kcal mol−1 was computed. Moreover, the computational results indicate that a putative singly (de)protonated species (for which we find the bis(μ-oxido) isomer to be more stable, ESI†) should not be present in solution as it is thermodynamically disfavored against decomposition into P5 and O7.
 | ||
| Scheme 3 Isomerization reactions for P5BS2 (a) and P7 (b) computed at the BLYP-D3/def2TZVP(SDD)/COSMO(THF)//PBE-D3/def2TZVP(SDD)/SMD(THF) level of DFT. | ||
Addition of common Brönstedt acids ([LuH]OTf, [LuH]BF4 and HBF4·Et2O where LuH = lutidinium) to solutions of O7 at 193 K did not trigger back-isomerization to the μ-η2:η2-peroxido species P5, but led to decomposition instead. However, the reversibility of the equilibrium shown in Scheme 2 was demonstrated by the following experiment: reaction of two equivalents of H,HBOX with one equivalent of [Cu(MeCN)4]ClO4 in THF in the presence of O2 directly led to the formation of the bis(μ-oxido) complex O8. In this case, deprotonation is accomplished by excess H,HBOX acting as a Brönstedt base.55 Similarly, in situ UV-vis monitoring of H,HBOX titration into a solution of P6 (P core) indicated the formation of O8 (O core) (Fig. 5 and S7†). Subsequent titration of [Cu(MeCN)4]ClO4 into the reaction mixture in the presence of excess O2, in turn, resulted in the conversion back to P6 (Fig. 5 and S8†). Thus, re-protonation of the ligand backbone in O8 is accomplished by [HH,HBOX]+. This procedure enables complete control of the P/O interconversion without the need of adding exogenous acids. UV-vis monitoring of the reaction sequence (Fig. 5) confirmed that close to 1 equivalent of O8 is formed during the first step, and a further 0.25 equivalents of P6 result after addition of the second equivalent of [Cu(MeCN)4]ClO4 (Fig. 5).56
 | ||
| Fig. 5 UV-vis monitoring of the reaction sequence demonstrating the interconversion of P6 to O8 using H,HBOX as a base and subsequent conversion of O8 back to P6 upon addition of [Cu(MeCN)4]ClO4 in the presence of O2 (baseline correction and dilution factor applied). | ||
On warming from 193 K to room temperature, the intensely colored solutions of P4, P5, and O7 gradually changed to light blue, indicating decay of the dicopper/dioxygen complexes. IR spectra of the light blue solid material isolated thereafter showed a characteristic OH stretching vibration around 3480 cm−1 (Fig. S4†). Single crystals of the material were obtained employing Cu(I) triflate as the metal source, and X-ray crystallography revealed the formation of the dihydroxido complex [(Me,MeBOXCu)2(μ-OH)2](CF3SO3)2 (9, Cu⋯Cu separation: 3.00 Å) (Fig. 6). No degradation or oxygenation of the BOX ligands was observed.
 | ||
| Fig. 6 Molecular structure of 9 set at 30% probability. Most hydrogen atoms have been omitted for clarity. | ||
Conclusions
In conclusion, we have shown that the peroxido/bis(μ-oxido) interconversion in a [Cu2O2] complex supported by proton-responsive BOX ligands can be controlled by peripheral (de)protonation events on the ligand backbone. We suggest this system as a bioinorganic mimic for type III dicopper proteins (or the dicopper active site of pMMO) in which the Cu ions are supported by histidine imidazoles, which offer a backside N atom amenable to potential (de)protonation equilibria in response to changes in local pH. In fact, (de)protonation of histidine imidazole ligands in metalloproteins is widely used for tuning redox potentials and electronic structures of the metallocofactors,57–60 and it is an integral part of biologically important proton coupled electron transfer (PCET) reactivity (such as in the Rieske proteins).61 It is an interesting perspective to introduce, via proton-responsive ligands, PCET reactivity to Cux/O2 intermediates. We are currently pursuing further studies in this direction.Acknowledgements
Financial support by the DAAD (PhD fellowship for V. E. G.) and the DFG (IRTG 1422 Metals Sites in Biomolecules: Structures, Regulation and Mechanisms) is gratefully acknowledged. Quantum chemical calculations have been performed at the Center of Scientific Computing (CSC) Frankfurt on the FUCHS and LOEWE-CSC high-performance compute clusters.Notes and references
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. F. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659 CrossRef CAS PubMed.
- J. Y. Lee and K. D. Karlin, Curr. Opin. Chem. Biol., 2015, 25, 184 CrossRef CAS PubMed.
- E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047 CrossRef CAS PubMed.
- S. Itoh and S. Fukuzumi, Acc. Chem. Res., 2007, 40, 592 CrossRef CAS PubMed.
- L. Q. Hatcher and K. D. Karlin, J. Biol. Inorg Chem., 2004, 9, 669 CrossRef CAS PubMed.
- C. Citek, S. Herres-Pawlis and T. D. P. Stack, Acc. Chem. Res., 2015, 48, 2424 CrossRef CAS PubMed.
- L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013 CrossRef CAS PubMed.
- S. Schindler, Eur. J. Inorg. Chem., 2000, 2311 CrossRef CAS.
- M. Rolff, J. Schottenheim and F. Tuczek, Chem. Soc. Rev., 2011, 40, 4077 RSC.
- E. I. Solomon, Inorg. Chem., 2016, 55, 6364 CrossRef CAS PubMed.
- A. Hoffmann, C. Citek, S. Binder, A. Goos, M. Rübhausen, E. C. Wasinger, T. D. P. Stack, O. Troeppner, I. Ivanovic and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2013, 52, 5398 CrossRef CAS PubMed.
- J. Schottenheim, C. Gernert, B. Herzigkeit, J. Krahmer and F. Tuczek, Eur. J. Inorg. Chem., 2015, 3501 CrossRef CAS.
- J. N. Hamann and F. Tuczek, Chem. Commun., 2014, 50, 2298 RSC.
- J. N. Hamann, B. Herzigkeit, R. Jurgeleit and F. Tuczek, Coord. Chem. Rev., 2017, 334, 54 CrossRef CAS.
- J. A. Halfen, S. Mahapatra, E. C. Wilkinson, S. Kaderli, V. G. Young, L. Que, A. D. Zuberbühler and W. B. Tolman, Science, 1996, 271, 1397 CAS.
- S. Itoh, M. Taki, H. Nakao, P. L. Holland, W. B. Tolman, L. Que Jr and S. Fukuzumi, Angew. Chem., Int. Ed., 2000, 39, 398 CrossRef CAS PubMed.
- J. B. Gary, C. Citek, T. A. Brown, R. N. Zare, E. C. Wasinger and T. D. P. Stack, J. Am. Chem. Soc., 2016, 138, 9986 CrossRef CAS PubMed.
- J. Becker, P. Gupta, F. Angersbach, F. Tuczek, C. Naether, M. C. Holthausen and S. Schindler, Chem. – Eur. J., 2015, 21, 11735 CrossRef CAS PubMed.
- H.-C. Liang, M. J. Henson, L. Q. Hatcher, M. A. Vance, C. X. Zhang, D. Lahti, S. Kaderli, R. D. Sommer, A. L. Rheingold, A. D. Zuberbühler, E. I. Solomon and K. D. Karlin, Inorg. Chem., 2004, 43, 4115 CrossRef CAS PubMed.
- X. Ottenwaelder, D. J. Rudd, M. C. Corbett, K. O. Hodgson, B. Hedman and T. D. P. Stack, J. Am. Chem. Soc., 2006, 128, 9268 CrossRef CAS PubMed.
- J. Cahoy, P. L. Holland and W. B. Tolman, Inorg. Chem., 1999, 38, 2161 CrossRef CAS PubMed.
- L. Q. Hatcher, M. A. Vance, A. A. Narducci Sarjeant, E. I. Solomon and K. D. Karlin, Inorg. Chem., 2006, 45, 3004 CrossRef CAS PubMed.
- S. Itoyama, K. Doitomi, T. Kamachi, Y. Shiota and K. Yoshizawa, Inorg. Chem., 2016, 55, 2771 CrossRef CAS PubMed.
- M. A. Culpepper, G. E. Cutsail, B. M. Hoffman and A. C. Rosenzweig, J. Am. Chem. Soc., 2012, 134, 7640 CrossRef CAS PubMed.
- C. Citek, J. B. Gary, E. C. Wasinger and T. D. P. Stack, J. Am. Chem. Soc., 2015, 137, 6991 CrossRef CAS PubMed.
- C. Wilfer, P. Liebhäuser, A. Hoffmann, H. Erdmann, O. Grossmann, L. Runtsch, E. Paffenholz, R. Schepper, R. Dick and S. Herres-Pawlis, Chem. – Eur. J., 2015, 21, 17639 CrossRef CAS PubMed.
- L. Chiang, W. Keown, C. Citek, E. C. Wasinger and T. D. P. Stack, Angew. Chem., Int. Ed., 2016, 55, 10453 CrossRef CAS PubMed.
- M. Rolff, J. Schottenheim and J. Tuczek, J. Coord. Chem., 2010, 63, 2383 CrossRef.
- A. Walli, S. Dechert, M. Bauer, S. Demeshko and F. Meyer, Eur. J. Inorg. Chem., 2014, 4660 CrossRef CAS.
- A. Walli, S. Dechert and F. Meyer, Eur. J. Org. Chem., 2013, 7044 CrossRef CAS.
- Density functional calculations were performed at the RI-BLYP-D3/def2-TZVP(SDD)/COSMO(THF)//RI-PBE-D3/def2-TZVP(SDD)/SMD(THF) level. The accuracy of this approach has been established by comparison to explicitly correlated CCSD(T)-F12b/CBS(T/Q) benchmark results (see the ESI† for further details). A similarly favorable performance of the BLYP functional for related chemistry has been documented recently: P. Gupta, M. Diefenbach, M. C. Holthausen and M. Förster, Chem. – Eur. J., 2017, 23, 1427 CrossRef CAS PubMed.
- N. C. Eickman, R. S. Himmelwright and E. I. Solomont, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 2094 CrossRef CAS.
- G. Y. Park, M. F. Qayyum, J. Woertink, K. O. Hodgson, B. Hedman, A. Sarjeant, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2012, 2, 8513 CrossRef PubMed.
- B. M. Lam, J. A. Halfen, V. G. Young, J. R. Hagadorn, P. L. Holland, A. Lledós, L. Cucurull-Sánchez, J. J. Novoa, S. Alvarez and W. B. Tolman, Inorg. Chem., 2000, 39, 4059 CrossRef CAS PubMed.
- M. Kodera, K. Katayama, Y. Tachi, K. Kano, S. Hirota and S. Fujinami, J. Am. Chem. Soc., 1999, 121, 11006 CrossRef CAS.
- N. Kitajima, K. Fujisawa, Y. Moro-oka and K. Toriumi, J. Am. Chem. Soc., 1989, 111, 8975 CrossRef CAS.
- N. Kitajima, K. Fujisawa, C. Fujimoto, Y. Moro-oka, S. Hashimoto, T. Kitagawa, K. Toriumi, K. Tatsumi and A. Nakamura, J. Am. Chem. Soc., 1992, 114, 1277 CrossRef CAS.
- Y. Funahashi, T. Nishikawa, Y. Wasada-Tsutsui, Y. Kajita, S. Yamaguchi, H. Arii, T. Ozawa, K. Jitsukawa, T. Tosha, S. Hirota, T. Kitagawa and H. Masuda, J. Am. Chem. Soc., 2008, 130, 16444 CrossRef CAS PubMed.
- M. J. Henson, P. Mukherjee, D. E. Root, T. D. P. Stack and E. I. Solomon, J. Am. Chem. Soc., 1999, 121, 10332 CrossRef CAS.
- C. R. Andrew, H. Yeom, S. Valentine, B. G. Karlsson, N. Bonander, G. Van Pouderoyen, G. W. Canters, T. M. Loehr and J. Sanders-Loehr, J. Am. Chem. Soc., 1994, 116, 11489 CrossRef CAS.
- N. C. Eickman, E. I. Solomon, J. A. Larrabee, T. G. Spiro and K. Lerch, J. Am. Chem. Soc., 1978, 100, 6529 CrossRef CAS.
- Dependent on the particular choice of functional, basis set and relativistic approach, DFT optimized structures for the peroxido isomer show significant variations in the O–O bond length, overall underestimating the experimentally determined value by −0.13 to −0.24 Å. Consistently, the O–O stretching vibration is substantially overestimated by 80–355 cm−1 compared to experiment, again depending on the DFT approach chosen. As detailed in the ESI,† the strong method dependence goes back to a very shallow stretching potential along the O–O bond: for example, a relaxed scan of the O–O bond length in P5 performed at the BP86/SVP level of DFT from RO–O = 1.46 Å (optimized minimum structure) to 1.58 Å (experimental bond length) results in an energy increase of merely 2.2 kcal mol−1. Harmonic O–O stretching frequencies, however, computed for these two points vary by an enormous 260 cm−1. These findings, together with the fact that the neglect of crystal packing effects and vibrational anharmonicities further complicate comparison with experiment, forced us to conclude that a quantitative modeling of structural and vibrational characteristics of the peroxido species studied here is outside the scope of presently available density functional methods.
- While having a small amount of the O isomer in the crystals (see E. Pidcock, S. DeBeer, H. V. Obias, B. Hedman, K. O. Hodgson, K. D. Karlin and E. I. Solomon, J. Am. Chem. Soc., 1999, 121, 1870 CrossRef CAS ) cannot be completely ruled out, this seems unlikely because (i) free refinement turned out to be unstable when trying to model an O–O disorder in the crystallographic structures; (ii) similar long O–O bonds are observed for both complexes, P4 and P5, the latter lacking the ligand backbone proton required for deprotonation-induced transformation to the corresponding bis(μ-oxido) species; and (iii) the rR spectra of both solid materials do not show any isotope sensitive bands at ∼600 cm−1 that could be assigned to the O isomer.
- K. E. Dalle, T. Gruene, S. Dechert, S. Demeshko and F. Meyer, J. Am. Chem. Soc., 2014, 136, 7428 CrossRef CAS PubMed.
- N. Kindermann, E. Bill, S. Dechert, S. Demeshko, E. J. Reijerse and F. Meyer, Angew. Chem., Int. Ed., 2015, 54, 1738 CrossRef CAS PubMed.
- In good agreement with this experimentally determined lower limit, a broken-symmetry DFT assessment of P5 resulted in −J = 1080 cm−1 (see Scheme 3 and the ESI†).
- E. I. Solomon, Chem. Rev., 1992, 92, 521 CrossRef CAS.
- K. D. Karlin, Z. Tyeklár, A. Farooq, R. R. Jacobson, E. Sinn, D. W. Lee, J. E. Bradshaw and L. J. Wilson, Inorg. Chim. Acta, 1991, 182, 1 CrossRef CAS.
- M. J. Baldwin, D. E. Root, J. E. Pate, K. Fujisawa, N. Kitajima and E. I. Solomon, J. Am. Chem. Soc., 1992, 114, 10421 CrossRef CAS.
- P. L. Holland, C. J. Cramer, E. C. Wilkinson, S. Mahapatra, K. R. Rodgers, S. Itoh, M. Taki, S. Fukuzumi, L. Que Jr and W. B. Tolman, J. Am. Chem. Soc., 2000, 122, 792 CrossRef CAS.
- For O8 the relative intensity of the bands around 335 and 400 nm is inverted.
- S. Herres-Pawlis, P. Verma, R. Haase, P. Kang, C. T. Lyons, E. C. Wasinger, U. Floerke, G. Henkel and T. D. P. Stack, J. Am. Chem. Soc., 2009, 131, 1154 CrossRef CAS PubMed.
- D. J. E. Spencer, N. W. Aboelella, A. M. Reynolds, P. L. Holland and W. B. Tolman, J. Am. Chem. Soc., 2002, 124, 2108 CrossRef CAS PubMed.
- B. F. Straub, F. Rominger and P. Hofmann, Chem. Commun., 2000, 3, 1611 RSC.
- S. Milione and V. Bertolasi, Tetrahedron Lett., 2011, 52, 3570–3574 CrossRef CAS.
- Some decomposition occurs, and hence the re-formation of P6 is not quantitative.
- N. Nakanishi, F. Takeuchi and M. Tsubaki, J. Biochem., 2007, 142, 553 CrossRef CAS PubMed.
- E. Fadda, N. Chakrabarti and R. Pomes, J. Phys. Chem. B, 2005, 109, 22629 CrossRef CAS PubMed.
- Y. Lin and C. Lim, J. Am. Chem. Soc., 2004, 126, 2602 CrossRef CAS PubMed.
- J. J. Warren and J. M. Mayer, Biochemistry, 2015, 54, 1863 CrossRef CAS PubMed.
- A. Albers, S. Demeshko, S. Dechert, C. T. Souma, J. M. Mayer and F. Meyer, J. Am. Chem. Soc., 2014, 136, 3946 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1511427–1511431. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04820j |
| This journal is © The Royal Society of Chemistry 2017 |