
Development of semi-continuous chemo-enzymatic terpene epoxidation: combination of anthraquinone autooxidation and the lipase-mediated epoxidation process†
Sumanth
Ranganathan
 a and
Volker
Sieber
*abc
a and
Volker
Sieber
*abc
aTechnical University of Munich, Chair of Chemistry of Biogenic Resources, Schulgasse 16, Straubing 94315, Germany. E-mail: sieber@tum.de
bTechnical University of Munich, Catalysis Research Center (CRC), Ernst-Otto-Fischer Straße 1, Garching 85748, Germany
cFraunhofer Institute of Interfacial Engineering and Biotechnology (IGB), Bio-, Electro and Chemo Catalysis BioCat Branch Straubing, Schulgasse 11a, Straubing 94315, Germany
First published on 3rd October 2017
Abstract
Lipase has been used for epoxidizing olefins such as monoterpenes for more than two decades. This epoxidation is accomplished by adding hydrogen peroxide (H2O2) to a carboxylic acid in the presence of a lipase such as Candida antartica lipase B (CALB) to produce percarboxylic acid, which then epoxidizes monoterpenes according to the Prilezhaev mechanism. One drawback of this process is the need for continuous addition of hydrogen peroxide to maintain maximum productivity. To overcome this hurdle, the industrial anthraquinone autooxidation process for hydrogen peroxide production was scaled down and coupled with lipase-mediated epoxidation in a semi-continuous manner. Palladium on alumina pellets (5% loading) was used as the catalyst for obtaining high yields of high-concentration hydrogen peroxide (50% weight by volume), followed by epoxidation of 3-carene, (+) limonene, and α-pinene. A total reaction time of 5 h was used for hydrogen peroxide production and 2–3 h for the epoxidation reactions. Pure 3-carene epoxide and α-pinene epoxide were obtained in isolated yields of 88.8 ± 2.8% and 83.8 ± 2.6%, respectively. Limonene epoxide was obtained as a mixture of mono- and di-epoxides in a ratio of 70% and 30%, respectively, with an isolated yield of 71.5 ± 3.1%.
1 Introduction
Epoxides, also known as oxiranes, are cyclic ethers that are industrially significant owing to their high reactivity to form intermediates, which in turn form products of high value. The process is straightforward and involves the addition of a free or substituted oxygen atom to an olefin.1 Epoxides can be produced using pure oxygen or air,2–4 hydroperoxides,5,6 hydrogen peroxide,7,8 and peroxy compounds.9 Industrially, oxygen- or ozone-based epoxidation is practiced as a gas-phase reaction in the presence of a metal catalyst for ethene,10 propene,11 and butene;12,13 however, other olefins in the liquid state are seldom epoxidized in this manner. An alternative to this method is the Prilezhaev reaction, which uses peroxycarboxylic acid in stoichiometric amounts to perform epoxidations. Note that meta-chloroperbenzoic acid (m-CPBA) is the most commonly used peroxycarboxylic acid in these syntheses.14 Generally, handling and cleaning issues coupled with the possibility of an explosion hazard make this process dangerous at industrial levels of production; therefore, in situ generation of peroxycarboxylic acid or slow addition of the compound is recommended. However, slow addition of peroxycarboxylic acid produces huge amounts of waste (equimolar to the amount of product); hence, in situ generation is preferred.15Peroxycarboxylic acid can be generated in situ either chemically or enzymatically. Harsh reaction conditions, such as a strong mineral acid catalyst with carboxylic acid and hydrogen peroxide, are required to produce peroxycarboxylic acid. This leads to waste neutralization issues and unwanted side reactions that contribute to polluting processes, e.g., formation of performic acid. Therefore, the enzymatic method reported by Björkling et al. is preferred.16 The reaction schemes for both chemical and enzymatic means of epoxidation are depicted in Scheme 1. Ever since this report was published in 1992, Prilezhaev-based epoxidation that uses lipases has been the most preferred route for epoxidations17–26 owing to its safer and simpler synthetic conditions.
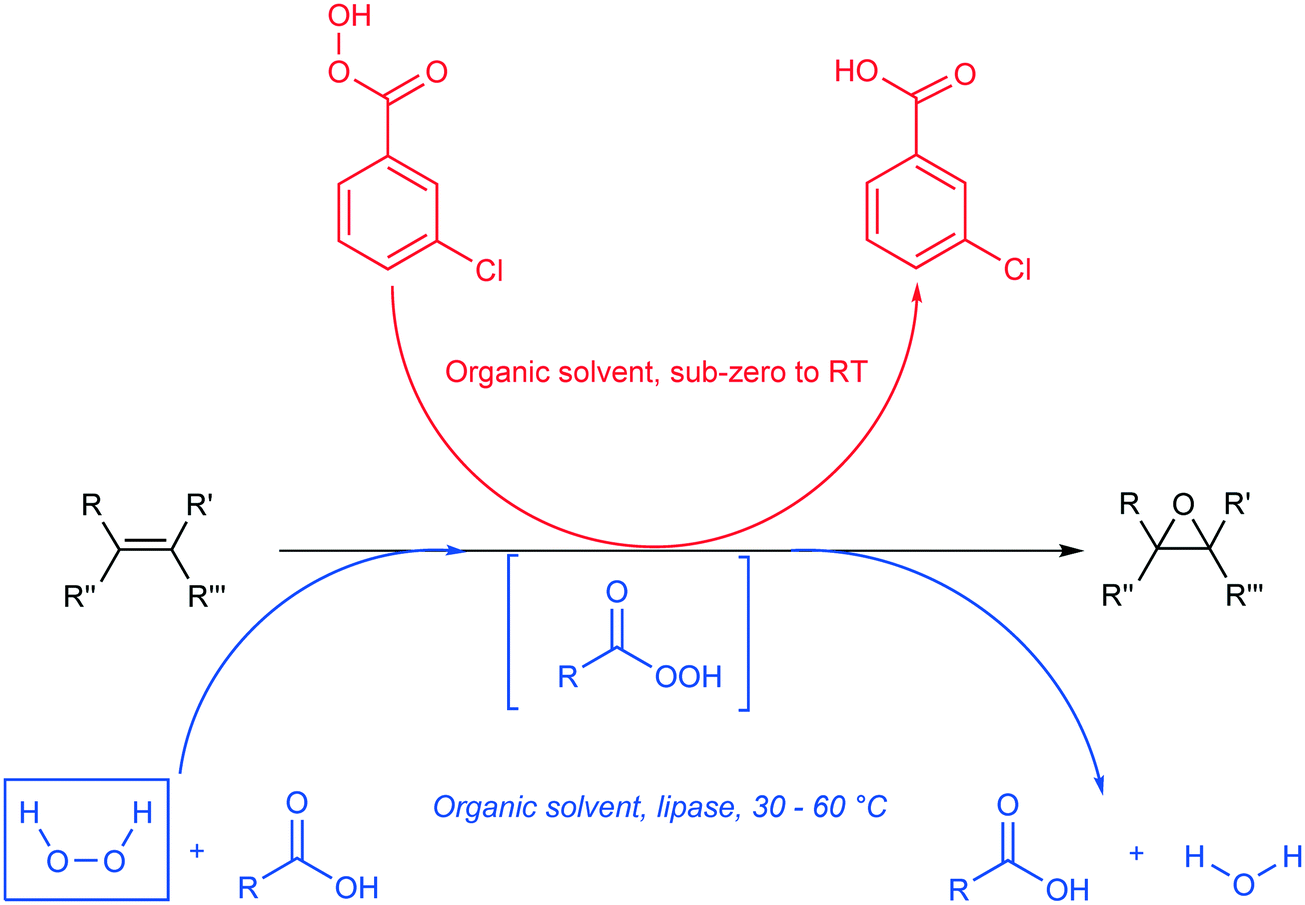 | ||
| Scheme 1 Epoxidation of alkene (black) using the procedure of Prilezhaev (red) and Björkling et al. (blue).16 The rectangular, dashed box implies that the compound was generated in situ. (R, R′, R′′, and R′′′ are functional groups present on the alkene). | ||
However, as evident from the scheme above, lipase-mediated epoxidation has one major drawback, i.e., the exhaustion of hydrogen peroxide, which limits the synthesis to a batch process. This implies the need to add fresh hydrogen peroxide continuously or in situ generation using enzymatic,27–30 electrochemical,31–33 photocatalytic,34–36 or chemical means (Schemes 3 and 4).37–39 Ni et al. reported the use of an enzymatic cascade for producing H2O2 and CO2 from methanol in combination with a peroxidase enzyme for the production of halogenated thymols.40 Holtmann et al. used an electrochemical approach to generate H2O2 from the same reaction.41 Churakova et al. used EDTA in the presence of light to efficiently generate moderate amounts of H2O2 from an aromatic peroxygenase in order to obtain aromatic phenols from their corresponding precursors.42 In addition to these specific examples, several other works have used the combination of in situ H2O2 generation methods with reactions requiring H2O2.43,44 One common trend in these processes is that they are excellent innovations for lab-scale applications when only low concentrations of hydrogen peroxide are required. The feasibility of the industrial use of these methods has not been reported so far or is pending investigation. In industrial applications, hydrogen peroxide is produced chemically using autooxidation45 or direct H2/O2 (ref. 46) processes as well as using the 2-propanol oxidation process for a brief period.47
The focus of this work is the functionalization of renewable resources for the production of value-added fine chemicals. Terpenes are one such resource as they are naturally occurring hydrocarbons predominantly obtained from plants as ethereal oils. Terpenes comprise repeating isoprene units, which are susceptible to biological degradation and can be obtained as waste products from the paper and pulp industry.48–50 Currently, these hydrocarbon reserves are combusted for energy production or used in paints and varnishes; however, functionalizing these terpenes would be beneficial for the fragrance, flavor, fine chemical, and of late, the polymer industry.51–54 Recently, our research group reported a combination of the chemical anthraquinone process and lipase-mediated epoxidation,55 the very first report on using such a combination for producing epoxides. Although innovative, the process could only be used for a single run, i.e., a batch reaction. In this study, terpenes were chosen as the olefins to be epoxidized. This study also focuses on designing a semi-continuous epoxidation method for terpenes and developing a prototype process for the industry. We combined the processes of H2O2 production and epoxidation, which provided us better control on production as well as room for other reactions that require H2O2. H2O2 production was increased by optimizing the catalyst loading. The transfer of gaseous hydrogen to dissolved hydrogen in the working solution was enhanced using high mixing rates. Moreover, a stainless steel mesh container for the palladium catalyst was used to protect the palladium catalysts from shear forces associated with the high mixing rates. The combined effect of both should yield higher H2O2 yields. For epoxidation, ethyl acetate was used as the reaction solvent owing to its greenness and capability as a peroxy acid generator (Scheme 2).
 | ||
| Scheme 2 Lipase-mediated (CALB) epoxidation of 3-carene, limonene and α-pinene in ethyl acetate. | ||
 | ||
| Scheme 3 Reduction of 2-ethyl anthraquinone (2-EAQ) in the presence of 2.5 mol% palladium at 60 °C to produce 2-ethyl anthraqhydroquinone (2-EAH2Q). | ||
 | ||
| Scheme 4 Auto oxidation of 2-EAH2Q to 2-EAQ and hydrogen peroxide (H2O2) production at 22 °C in the presence of air. | ||
2 Results & discussion
First, the anthraquinone-based autooxidation process was optimized to obtain maximum H2O2 production. Lipase-mediated epoxidation has already been optimized in our previous studies24,25 and will be used herein with a single change in the reaction medium. Ethyl acetate was used in this work as opposed to toluene and deep eutectic solvents, which were used previously.2.1 Optimization of palladium loading
The first part of the developmental stage of the H2O2 production process was determination of the amount of palladium required for the hydrogenation reaction in the anthraquinone process. Previously, we had determined that 10 mol% was necessary to successfully convert 2-ethyl anthraquinone (2-EAQ) to 2-ethyl anthrahydroquinone (2-EAH2Q). The catalyst used previously was a fine commercial powder of Pd/C as opposed to Pd/Al2O3, which was used in this study.55 Hence, optimization was performed using the one variable at a time (OVAT) strategy, wherein one of the parameters is varied whilst keeping the others constant. Here, the parameter of interest was the percentage of palladium used with respect to the amount of 2-EAQ used. Tests were performed by mixing 2 × 10−2 mol 2-EAQ with a 100 cm3 working solution (60% mesitlene and 40% tributyl phosphate) at 250 rpm, as explained in section 4.2.1. The H2O2 concentration obtained at the end of the reaction was calculated using the ABTS assay, which has been described in section 4.3.1. The isolated yields obtained at the end of the run for various palladium-to-2-EAQ ratios are given in Table 1, which shows that 2.5 mol% yielded the best results for hydrogen peroxide production.| S. no. | % Pd/Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2-EAQ (mol:mol) :2-EAQ (mol:mol) |
Isolated yield (%) |
|---|---|---|
| 1 | 0.5 | — |
| 2 | 1.25 | 94.3 |
| 3 | 2.5 | 97.2 |
| 4 | 5 | 93.8 |
| 5 | 10 | 90 |
According to Table 1, a loading of 0.5 mol% did not produce any 2-EAH2Q; as a result, no H2O2 was produced. Using 1.25 and 2.5 mol% resulted in yields of 94.3% and 97.2%, respectively; however, when using 5 and 10 mol%, the yields were less than 97%. This behavior could be the result of nonspecific hydrogenation of the aromatic rings of the 2-EAQ molecule, reduction of a single keto function of 2-EAQ, or dimerization of nonspecific products.38
Interestingly, when using scaled-up conditions, i.e., 1.25 mol% Pd/Al2O3, 6.5 × 10−2 mol 2-EAQ, 60% mesitylene, 40% tributyl phosphate (working-solution volume of 150 cm3), 60 °C temperature, and mixing at 250 rpm, there was a substantial reduction in the isolated yield of H2O2 (∼75%). The reason for the decrease in the H2O2 yield was poisoning of the catalyst, as confirmed when the catalysts were transferred from the working solution to a glass beaker and left overnight in the fume hood to dry after washing with 2-propanol. The following day, yellowish crystals were visible on both the beaker and the catalyst, which we hypothesize was 2-EAQ. Increasing the washing steps with 2-propanol did not help with the removal of this impurity; however, such a behavior was not observed when using 2.5 mol% Pd/Al2O3 under scaled-up conditions. We presume that this is because of an appropriate ratio of Pd-to-2-EAQ was being used for the hydrogenation reaction. At the end of the reaction, a similar H2O2 yield was obtained. Hence, in the interest of scaling up, the decision to use 2.5 mol% of Pd/Al2O3 with respect to 2-EAQ was made.
2.2 Reusability tests for palladium
Once the palladium loading for H2O2 production was finalized, the next step was to check for the number of cycles for which the catalyst could be used. All tests were performed in triplicate. The first tests were performed without washing between runs. The H2O2 concentration was tested using the ABTS assay (section 4.3.1), and the results are given in Table 2. The reaction conditions used in this test are given in section 4.2.1.| % Isolated H2O2 yield | ||
|---|---|---|
| Run 1 | Run 2 | Run 3 |
| 87.5 ± 2.2 | 58.7 ± 4.3 | 18.5 ± 3.8 |
According to Table 2, the amount of H2O2 decreased drastically over three cycles possibly because of catalyst inactivation, leaching of the palladium on account of the high shear and grinding forces associated with mixing, or residual reactants sticking to the catalyst. First, the catalyst was washed in an attempt to increase reusability. For this purpose, several solvents were screened, such as acetone, acetone:water (1:1 v/v), water, 2-propanol, and n-hexane:acetone (1:1 v/v). None of the solvents improved the H2O2 yield and are therefore not discussed here. A literature survey was conducted, and, finally, the washing protocol reported by Wang et al. in 2004 (ref. 56) was selected. The palladium on alumina catalyst was first washed with 15 cm3 of ethanol and then with 15 cm3 water; this process was repeated two times. A detailed account of the procedure can be found in section 4.2.2.
2.3 Use of a stainless steel mesh container to enhance Pd/Al2O3 reusability
Once the washing protocol was finalized, the washing process was performed again; consequently, the reusability of the catalyst was slightly higher (Fig. 2) than the previous tests performed (Table 2) without washing. However, the loss of activity over four cycles was still high. Catalyst pellets were recovered via filtration. During this step, the filter paper showed black spots of palladium particles, possibly resulting from leaching of the catalyst caused by shear forces or grinding operation of the magnetic stirrer.To overcome this drawback, the hydrogenation step was repeated using a stainless-steel mesh container for Pd (Fig. 1). In this step, the catalyst was loaded into a stainless steel mesh designed in the form of a pouch. Upon performing hydrogenation using this setup, the loss of activity was minimal (Fig. 2). As shown in the figure, the experiment with the container that houses the catalyst yielded better results than the reaction without it. Unfortunately, there was a considerable loss in the activity of the palladium catalyst despite the washing and protection from shear and grinding forces; the reason for this could be due to the leaching of the catalyst from the support or the formation of intermediates during hydrogenations that could not produce H2O2. The exact reason needs to be investigated further.
 | ||
| Fig. 1 Use of a stainless-steel container to reduce catalyst leaching and to increase the miscibility of hydrogen with 2-EAQ. a – 2-EAQ in the working solution before hydrogenation. b – 2-EAH2Q, the product of hydrogenation of 2-EAQ. | ||
 | ||
| Fig. 2 Isolated H2O2 yields obtained using a stainless-steel container (squares) and that obtained without it (diamonds). Reaction conditions: 0.02 mol 2-EAQ, 65 cm3 mesitylene, 35 cm3 tributyl phosphate, and 2.5 mol% Pd/Al2O3 (5% loading). | ||
2.4 Comparison of H2O2 production in the industry with that achieved this work
H2O2 is produced worldwide on an industrial scale exclusively via the anthraquinone autooxidation process.57 One cycle of the optimized anthraquinone process comprises hydrogenation, catalyst separation, oxidation, and extraction/concentration steps.37–39 Traditionally, a slurry-type reactor is used for the hydrogenation step,57 as in the case of our previous study.55 This leads to leaching of the palladium catalyst into the working solution, thereby catalyzing the decomposition of H2O2. As a result, fixed-bed reactors are now used in the industry to avoid the tedious unit operation of filtration.58 This study combines the advantages of both reactor types and presents an initial case of a “hybrid reactor” for this purpose. Pd/Al2O3 pellets are placed in a self-assembled stainless-steel container with a mesh size of 0.2 mm, which is advantageous for preventing any shear loss due to mixing whilst ensuring proper diffusion of reactants to the catalytic surface. The mixing of reactants in this work was performed at 1000 rpm, which ensures maximum mass transfer.2.5 Development of the combined semi-continuous approach for the epoxidation of terpenes
A semi-continuous approach based on the combined anthraquinone process and a lipase process was used to epoxidize monoterpenes. The scheme of this setup is shown in Fig. 3. | ||
| Fig. 3 Semi-continuous approach for combining the industrial anthraquinone process (blue-box contents) for hydrogen peroxide production and lipase-mediated epoxidation (red-box contents) of monoterpenes. | ||
First, the anthraquinone process was performed for hydrogen peroxide formation. It was followed by lipase-mediated epoxidation of monoterpenes. After 5 h, a membrane pump was used to transfer the working solution (dark red to brown) from the hydrogenation chamber to a sparging chamber. A flow rate of 100 cm3 min−1 (maximum capacity of the pump) was used throughout this process for transferring liquids. The reduced working solution was then transferred from the sparging chamber to the oxygenation chamber, and, after oxidation for 15–30 min, the working solution (oxidized, yellow) was transferred to a separating funnel to which water was added for extraction. The working solution was then transferred to the hydrogenation chamber and the reaction was repeated.
The hydrogen peroxide solution produced using the anthraquinone autooxidation process (50% w/v) was collected in a reservoir until further use (Fig. 3). The epoxidation chamber comprised two parts: a reaction chamber and a purification chamber. The reaction chamber contained ethyl acetate, terpene, and CALB. To this mixture, an appropriate amount (with respect to terpene) of H2O2 was added to start the reaction, which was monitored by sampling at regular intervals and subjecting the samples to chromatography-mass spectrometry (GC-MS) analysis. After confirming 100% conversion of the starting material, the reaction components were transferred to a purification chamber and pure epoxide was obtained according to a protocol published previously.24
2.6 Epoxidation results
Lipase-mediated epoxidation of monoterpenes, namely 3-carene, limonene, and α-pinene, was performed according to the procedure mentioned in section 4.2.4. Samples were withdrawn at regular intervals, and the conversion profile was followed using gas GC-MS, as reported previously.55 The kinetics of the epoxidation of the three reactants is shown in the Fig. 4. | ||
| Fig. 4 Conversion profile of 5 × 10−3 mol reactant (3-carene – diamond; limonene – square; α-pinene – triangle), 7.5 × 10−3 mol H2O2 (50% w/v), and 0.1 g CALB in 25 × 10−3 L ethyl acetate at 45 °C. | ||
From the figure, it can be inferred that limonene (square) converted to its corresponding mono- and di-epoxide within 2 h and that 3-carene (diamond) and α-pinene (triangle) underwent full conversion within 3 h. It is to be noted that the limonene reaction was ca. 67% selective for the mono epoxide and 30% selective for the di-epoxide. Additionally, all three reactions yielded diols (≤3%) because of epoxide ring opening (ESI;† GC chromatograms and mass spectra). Comparing this result with that obtained using our previous system with toluene,24 the reaction time reduced by a factor of 4 for limonene and 3.33 for 3-carene and α-pinene. The GC chromatograms and mass spectra are attached in the supplementary information. Comparing the results of the epoxidation in this study with the choline chloride:urea·H2O2 DES system25 demonstrates that the reaction shows similar results of total turnover of reactants within 3 h.25 The epoxides produced were then purified using a procedure developed previously.24 Isolated yields of 88.8 ± 2.8, 71.5 ± 3.1, and 83.8 ± 2.6% were obtained for 3-carene, limonene, and α-pinene, respectively.
A prototype of this process was recently published by our research group.55 Previously, a working solution of toluene and ethyl acetate (3:2 v/v) was used with a catalyst loading of 10 mol%. Additionally, a fine powder of commercially available palladium on carbon (Pd/C) was used as the catalyst. The use of Pd/C was problematic during the filtration step as fine particles passed through the filter, leading to multiple filtration steps. The oxidation step was coupled with epoxidation using CALB as a catalyst. Lipase converted ethyl acetate into ethanol and acetic acid, thereby restricting the process to be operated strictly in the batch mode. The process used in the present study overcomes all the challenges discussed above and is capable of being operated in a semi-continuous mode. The working solution was replaced by mesitylene:tributyl in a ratio of 1:1 (v/v). Although not essential, this was done to facilitate better solubility of 2-EAQ, 2-EAH2Q, and hydrogen gas. Moreover, this liquid combination is one of the preferred solvents in the industry.39 To overcome the issues related with filtration, palladium on alumina pellets was used so that the filtration step could be avoided; however, there were issues regarding catalyst leaching that need to be solved. To this end, a stainless-steel mesh container designed in-house was used. A minor change was made in the epoxidation part of the process by using an increased amount of terpene compared with the previous work.55
3 Conclusion
This study describes the coupling of chemical and enzymatic processes, i.e., the anthraquinone autooxidation synthesis of hydrogen peroxide and lipase-mediated epoxidation. To the best of our knowledge, such a design is the first of its kind. We used a stainless-steel mesh container to prevent the shear and mechanical grinding forces, thereby enhancing hydrogenation reactions by employing high mixing rates. In other words, this setup is a combination of the continuous stirred tank reactor (CSTR) and a fixed-bed reactor, making it a hybrid reactor that incorporates the advantages of both. High mixing rates ensure maximum mass transfer, catalyst reuse of up to five cycles with minimal loss of activity, and low-temperature operation, which are innovations in hydrogen peroxide production. Owing to the combination of the two processes, there is the opportunity to use the hydrogen peroxide reservoir as feed for other reactions that require H2O2. Additionally, this combination gives the option of diluting H2O2 according to demand. Since lipase-mediated epoxidation has been studied exclusively for a variety of reactants, the range of this combined process is broad. Moreover, the conversion profiles of the three tested compounds suggest that compared with toluene, the time taken to achieve complete conversion in ethyl acetate was 4 times lower for limonene and 3.33 times lower for 3-carene and α-pinene. To summarize, we believe that this new semi-continuous approach can be scaled up to industrial standards with relative ease and extended to other olefins as well.4 Materials and methods
4.1 Materials
2-Ethyl anthraquinone (2-EAQ) (Lot#: E12206-100G, 97% purity), mesitylene (Lot#: M7200-500ML, 98% purity), tributyl phosphate (Lot#: 158615-1L, 97% purity), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonsäure diammonium-salz (ABTS) (Lot#: A1888-2G, 98% purity), peroxidase from horseradish (HRP) (Lot#: P6782-50MG), 950–2000 units per mg by ABTS assay), α-pinene (Lot#: 147524-250ML, 98% purity), and (+) 3-carene (Lot#: 115576-1L, 90% purity) were purchased from Sigma-Aldrich. Palladium on alumina (Pd/Al2O3) pellets were obtained from VWR Chemicals (Lot#: 41825.06, 5% loading). (+) Limonene (Lot#: 179395000, 96% purity) was purchased from Fischer Scientific. Potassium phosphate buffer (KPi) (1 × 10−4 mol L−1, pH 5.0) was prepared in the laboratory according to standard buffering procedure and used as such. Ethanol (Lot#: T171.2-25L, 96% purity) was purchased from Carl Roth. Candida antartica lipase B (CALB) was obtained from c-LEcta (Lot#: 20606-4, 17000 PLU g−1). LC-MS grade ethyl acetate (EtoAc) was obtained from Th. Geyer (Lot: 2278-1L, 99.95% purity). Disposable cuvettes (Ref. #: 67.742, polystyrene material, 10 × 4 × 45 mm3) were bought from Sarstedt. Membrane pumps (KNF SIMDOS®10) fitted with a PTFE membrane to specifically handle organic solvents were used. Tygon® F-4040-A tubing (Lot#: 224-0525, inner diameter 3.2 × 10−3 m, outer diameter 6.4 × 10−3 m, thickness 1.6 × 10−3 m) was purchased from VWR Chemicals and used for liquid transfer. A UV-Vis spectrophotometer from Shimadzu (UV-1800) was used to measure the absorbance during the ABTS assay. A stainless-steel mesh (wire diameter 0.12 mm; mesh size 0.20 mm) was purchased from Metallwaren-Riffert, Austria.
4.2 Synthetic methods
4.3 Analytical methods
• GC: QP 2010, Shimadzu.
• Autoinjector: AOC-5000 by Jain, Compipal.
• MS: GC-MS QP2010 Plus, Shimadzu).
A 30 m long BPX5 with dimensions of 0.25 mm diameter and 0.25 μm thickness was used as the GC column. Helium was used as the carrier gas at a flowrate of 13.2 ml min−1. Temperature profile used for GC and MS for optimal separation of compounds was:
i. ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif)
![[s with combining low line]](https://www.rsc.org/images/entities/char_0073_0332.gif) c
c![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif)
![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif)
![[o with combining low line]](https://www.rsc.org/images/entities/char_006f_0332.gif)
![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif)
![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif)
![[g with combining low line]](https://www.rsc.org/images/entities/char_0067_0332.gif)
![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) : start at 60 °C and hold the temperature for 1 minute. Increase the temperature at the rate of 10 °C min−1 until 170 °C, after which the temperature was further increased to 270 °C at the rate of 70 °C min−1. This temperature was then held for 3 minutes.
: start at 60 °C and hold the temperature for 1 minute. Increase the temperature at the rate of 10 °C min−1 until 170 °C, after which the temperature was further increased to 270 °C at the rate of 70 °C min−1. This temperature was then held for 3 minutes.
ii. ![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif) s
s![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif)
![[c with combining low line]](https://www.rsc.org/images/entities/char_0063_0332.gif) : the ion source temperature was 200 °C and the interface temperature was maintained at 250 °C.
: the ion source temperature was 200 °C and the interface temperature was maintained at 250 °C.
The software program “GC-MS Postrun Analysis” from Shimadzu was used to analyze the reaction components and the mass to charge ratio (m/Q) ratio were compared to the database of National Institute of Standard and Technology (NIST) library; version 14. Ethyl acetate was used as the solvent and to avoid huge signals form this compound, a solvent cut was introduced at 3.9 min with the help of the software. All GC-MS chromatograms obtained for 3-carene/epoxide, limonene/mono- and di-epoxide, α-pinene/epoxide and octanoic acid are given in the supplemental information.
Conflicts of interest
There are no conflicts to declare.References
- G. Sienel, R. Rieth and K. T. Rowbottom, Epoxides, In: Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, pp. 1–16 Search PubMed.
- J. W. Brown, Q. T. Nguyen, T. Otto, N. N. Jarenwattananon, S. Glöggler and L. S. Bouchard, Epoxidation of alkenes with molecular oxygen catalyzed by a manganese porphyrin-based metal-organic framework, Catal. Commun., 2015, 59, 50–54 CrossRef CAS.
- L. Vanoye, J. Wang, M. Pablos, C. de Bellefon and A. Favre-Réguillon, Epoxidation using molecular oxygen in flow: facts and questions on the mechanism of the Mukaiyama epoxidation, Catal. Sci. Technol., 2016, 6(13), 4724–4732 CAS.
- J. Dou and F. Tao, Selective epoxidation of cyclohexene with molecular oxygen on catalyst of nanoporous Au integrated with MoO3 nanoparticles, Appl Catal A Gen., 2017, 529, 134–142 CrossRef CAS.
- Z. Wang, Sharpless Epoxidation, In: Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., 2010, pp. 2580–2585 Search PubMed.
- S. El-Korso, I. Khaldi, S. Bedrane, A. Choukchou-Braham, F. Thibault-Starzyk and R. Bachir, Liquid phase cyclohexene oxidation over vanadia based catalysts with tert-butyl hydroperoxide: Epoxidation versus allylic oxidation, J. Mol. Catal. A: Chem., 2014, 394, 89–96 CrossRef CAS.
- B. S. Lane and K. Burgess, Metal-catalyzed epoxidations of alkenes with hydrogen peroxide, Chem. Rev., 2003, 103(7), 2457–2473 CrossRef CAS PubMed.
- T. Michel, M. Cokoja, V. Sieber and F. E. Kühn, Selective epoxidation of (+)-limonene employing methyltrioxorhenium as catalyst, J. Mol. Catal. A: Chem., 2012, 358, 159–165 CrossRef CAS.
- H. Klenk, P. H. Götz, R. Siegmeier and W. Mayr, Peroxy Compounds, Organic, In: Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, pp. 325–360 Search PubMed.
- M. L. Bocquet, A. Michaelides, D. Loffreda, P. Sautet, A. Alavi and D. A. King, New insights into ethene epoxidation on two oxidized Ag{111} surfaces, J. Am. Chem. Soc., 2003, 125(19), 5620–5621 CrossRef CAS PubMed.
- D. M. Perez Ferrandez, M. H. J. M. De Croon, J. C. Schouten and T. A. Nijhuis, Gas-phase epoxidation of propene with hydrogen peroxide vapor, Ind. Eng. Chem. Res., 2013, 52(30), 10126–10132 CrossRef CAS.
- R. Atkinson, S. M. Aschmann, J. Arey and E. C. Tuazon, Formation yields of epoxides and O(3P) atoms from the gas-phase reactions of O3 with a series of alkenes, Int. J. Chem. Kinet., 1994, 26(9), 945–950 CrossRef CAS.
- T. Berndt, A. Kreipl and U. Lenski, Epoxidation of 2-Butene from Raffinate 2 Feedstock, Ind. Eng. Chem. Res., 2010, 49(8), 3957–3960 CrossRef CAS.
- Z. Wang, Prilezhaev Reaction, In: Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., 2010, pp. 2270–2274 Search PubMed.
- A. S. Rao, H. R. Mohan and A. Charette, m-Chloroperbenzoic Acid, In: Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2001 Search PubMed.
- F. Björkling, H. Frykman, S. E. Godtfredsen and O. Kirk, Lipase catalyzed synthesis of peroxycarboxylic acids and lipase mediated oxidations, Tetrahedron, 1992, 48(22), 4587–4592 CrossRef.
- F. P. Cuperus, S. T. Bouwer, G. F. H. Kramer and J. T. P. Derksen, Lipases Used for the Production of Peroxycarboxylic Acids, Biocatal. Biotransform., 1994, 9(1–4), 89–96 CrossRef CAS.
- M. Rüsch gen Klaas and S. Warwel, Complete and partial epoxidation of plant oils by lipase-catalyzed perhydrolydsis, Ind. Crops Prod., 1999, 9, 125–132 CrossRef.
- C. Orellana-Coca, J. M. Billakanti, B. Mattiasson and R. Hatti-Kaul, Lipase mediated simultaneous esterification and epoxidation of oleic acid for the production of alkylepoxystearates, J. Mol. Catal. B: Enzym., 2007, 44(3–4), 133–137 CrossRef CAS.
- M. A. Moreira and M. G. Nascimento, Chemo-enzymatic epoxidation of (+)-3-carene, Catal. Commun., 2007, 8(12), 2043–2047 CrossRef CAS.
- Y. Xu, N. R. B. J. Khaw and Z. Li, Efficient epoxidation of alkenes with hydrogen peroxide, lactone, and lipase, Green Chem., 2009, 11(12), 2047–2051 RSC.
- A. A. Tzialla, E. Kalogeris, A. Enotiadis, A. A. Taha, D. Gournis and H. Stamatis, Effective immobilization of Candida antarctica lipase B in organic-modified clays: Application for the epoxidation of terpenes, Mater. Sci. Eng. B Solid State Mater. Adv. Technol., 2009, 165(3), 173–177 CrossRef CAS.
- F. D. A. Corrêa, F. K. Sutili, L. S. M. Miranda, S. G. F. Leite, R. O. M. A. De Souza and I. C. R. Leal, Epoxidation of oleic acid catalyzed by PSCI-Amano lipase optimized by experimental design, J. Mol. Catal. B: Enzym., 2012, 81, 7–11 CrossRef.
- S. Ranganathan, J. Tebbe, L. O. Wiemann and V. Sieber, Optimization of the lipase mediated epoxidation of monoterpenes using the design of experiments — Taguchi method, Process Biochem., 2016, 51(10), 1479–1485 CrossRef CAS.
- S. Ranganathan, S. Zeitlhofer and V. Sieber, Development of a lipase-mediated epoxidation process for monoterpenes in choline chloride-based deep eutectic solvents, Green Chem., 2017, 19(11), 2576–2586 RSC.
- P. Zhou, X. Wang, B. Yang, F. Hollmann and Y. Wang, Chemoenzymatic epoxidation of alkenes with Candida antarctica lipase B and hydrogen peroxide in deep eutectic solvents, RSC Adv., 2017, 7(21), 12518–12523 RSC.
- L. P. Nelles, J. A. Arnold and D. S. Willman, Enzymatic production of hydrogen peroxide and acetaldehyde in a pressure reactor, Biotechnol. Bioeng., 1990, 36(8), 834–838 CrossRef CAS PubMed.
- C. López and A. Cavaco-Paulo, In-situ enzymatic generation of hydrogen peroxide for bleaching purposes, Eng. Life Sci., 2008, 8(3), 315–323 CrossRef.
- T. Tzanov, S. A. Costa, G. M. Gübitz and A. Cavaco-Paulo, Hydrogen peroxide generation with immobilized glucose oxidase for textile bleaching, J. Biotechnol., 2002, 93(1), 87–94 CrossRef CAS PubMed.
- M. Aghbolaghy and A. Karimi, Simulation and optimization of enzymatic hydrogen peroxide production in a continuous stirred tank reactor using CFD-RSM combined method, J. Taiwan Inst. Chem. Eng., 2014, 45(1), 101–107 CrossRef CAS.
- T. Jeric, R. J. M. Bisselink, W. van Tongeren and A. M. Le Marechal, Decolorization and Mineralization of Reactive Dyes, by the H2O2/UV Process With Electrochemically Produced H2O2, Acta Chim. Slov., 2013, 60(3), 666–672 CAS.
- J. M. Peralta-Hernández and L. A. Godínez, Electrochemical Hydrogen Peroxide Production in Acidic Medium Using a Tubular Photo-reactor: Application in Advanced Oxidation Processes, J. Mex. Chem. Soc., 2014, 58(3), 348–355 Search PubMed.
- V. Viswanathan, H. A. Hansen and J. K. Nørskov, Selective Electrochemical Generation of Hydrogen Peroxide from Water Oxidation, J. Phys. Chem. Lett., 2015, 6(21), 4224–4228 CrossRef CAS PubMed.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa and S. Tanaka, et al., Sunlight-driven hydrogen peroxide production from water and molecular oxygen by metal-free photocatalysts, Angew. Chem., Int. Ed., 2014, 53(49), 13454–13459 CrossRef CAS PubMed.
- H. Zhuang, L. Yang, J. Xu, F. Li, Z. Zhang and H. Lin, et al., Robust Photocatalytic H2O2 Production by Octahedral Cd3(C3N3S3)2 Coordination Polymer under Visible Light, Sci. Rep., 2015, 5, 16947 CrossRef CAS PubMed.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Efficient Photocatalytic Production of Hydrogen Peroxide from Water and Dioxygen with Bismuth Vanadate and a Cobalt(II) Chlorin Complex, ACS Energy Lett., 2016, 1(5), 913–919 CrossRef CAS.
- G. Goor, Hydrogen Peroxide - Manufacture and Industrial Use for Production of Organic Chemicals, In: Catalytic Oxidations with Hydrogen Peroxide as Oxidant, 1992, pp. 13–43 Search PubMed.
- G. Goor, J. Glenneberg and S. Jacobi, Hydrogen Peroxide, In: Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process, Angew. Chem., Int. Ed., 2006, 45(42), 6962–6984 CrossRef CAS PubMed.
- Y. Ni, E. Fernández-Fueyo, A. G. Baraibar, R. Ullrich, M. Hofrichter and H. Yanase, et al., Peroxygenase-Catalyzed Oxyfunctionalization Reactions Promoted by the Complete Oxidation of Methanol, Angew. Chem., Int. Ed., 2016, 55(2), 798–801 CrossRef CAS PubMed.
- D. Holtmann, T. Krieg, L. Getrey and J. Schrader, Electroenzymatic process to overcome enzyme instabilities, Catal. Commun., 2014, 51, 82–85 CrossRef CAS.
- E. Churakova, M. Kluge, R. Ullrich, I. Arends, M. Hofrichter and F. Hollmann, Specific photobiocatalytic oxyfunctionalization reactions, Angew. Chem., Int. Ed., 2011, 50(45), 10716–10719 CrossRef CAS PubMed.
- J. Rocha-Martin, S. Velasco-Lozano, J. M. Guisán and F. López-Gallego, Oxidation of phenolic compounds catalyzed by immobilized multi-enzyme systems with integrated hydrogen peroxide production, Green Chem., 2014, 16(1), 303–311 RSC.
- K. Köninger, M. Grote, I. Zachos, F. Hollmann and R. Kourist, Light-driven Enzymatic Decarboxylation, J. Visualized Exp., 2016,(111), 5–9 Search PubMed.
- H.-J. Riedl and G. Pfleiderer, Production of Hydrogen Peroxide, US Pat., 2215883, 1940, p. 2 Search PubMed.
- J. K. Edwards, B. E. Solsona, P. Landon, A. F. Carley, A. Herzing and C. J. Kiely, et al., Direct synthesis of hydrogen peroxide from H2 and O2 using TiO2-supported Au-Pd catalysts, J. Catal., 2005, 236(1), 69–79 CrossRef CAS.
- F. F. Rust, Manufacture of Hydrogen Peroxide, 1959, pp. 1–14 Search PubMed.
- D. S. Seigler, Introduction to Terpenes, In: Plant Secondary Metabolism, 1998, pp. 312–323 Search PubMed.
- J. L. Bicas, A. P. Dionisio and G. M. Pastore, Bio-oxidation of terpenes: An approach for the flavor Industry, Chem. Rev., 2009, 109(9), 4518–4531 CrossRef CAS PubMed.
- R. Mewalal, D. K. Rai, D. Kainer, F. Chen, C. Külheim and G. F. Peter, et al., Plant-Derived Terpenes: A Feedstock for Specialty Biofuels, Trends Biotechnol., 2017, 35(3), 227–240 CrossRef CAS PubMed.
- J. L. F. Monteiro and C. O. Veloso, Catalytic Conversion of Terpenes into Fine Chemicals, Top. Catal., 2004, 27(1–4), 169–180 CrossRef CAS.
- L. Caputi and E. Aprea, Use of Terpenoids as Natural Flavouring Compounds in Food Industry, Recent Pat. Food, Nutr. Agric., 2011, 3(1), 9–16 CrossRef CAS.
- W. Schwab, C. Fuchs and F. C. Huang, Transformation of terpenes into fine chemicals, Eur. J. Lipid Sci. Technol., 2013, 115(1), 3–8 CrossRef CAS.
- S. Kandi, V. Godishala, P. Rao and K. V. Ramana, Biomedical Significance of Terpenes: An Insight, Biomed. Biotechnol., 2015, 3(1), 8–10 CAS.
- S. Ranganathan, T. Gärtner, L. O. Wiemann and V. Sieber, A one pot reaction cascade of in situ hydrogen peroxide production and lipase mediated in situ production of peracids for the epoxidation of monoterpenes, J. Mol. Catal. B: Enzym., 2015, 114, 72–76 CrossRef CAS.
- Q. Wang, L. Wang, Y. Wang, F. He, Z. Li and Z. Mi, Study on deactivation and regeneration of Pd/Al 2 O 3 catalyst in hydrogen peroxide production by the anthraquinone process, React. Kinet. Catal. Lett., 2004, 81(2), 297–304 CrossRef CAS.
- J. H. Teles, I. Hermans, G. Franz and R. A. Sheldon, Oxidation, In: Ullmann's Encyclopedia of Industrial Chemistry, WILEY-VCH Verlag, 2015, pp. 1–103 Search PubMed.
- R. Ciriminna, L. Albanese, F. Meneguzzo and M. Pagliaro, Hydrogen Peroxide: A Key Chemical for Today's Sustainable Development, ChemSusChem, 2016, 9(24), 3374–3381 CrossRef CAS PubMed.
- H. Bisswanger, Enzyme Assays, In: Practical Enzymology, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 93–264 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00112f |
| This journal is © The Royal Society of Chemistry 2017 |