
Microfluidic synthesis of α-ketoesters via oxidative coupling of acetophenones with alcohols under metal-free conditions†
Shiyu
Guo
a,
Zhongxue
Dai
a,
Jiawei
Hua
a,
Zhao
Yang
b,
Zheng
Fang
*a and
Kai
Guo
 *ac
*ac
aCollege of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, 30 Puzhu Rd S, Nanjing, 211816, China. E-mail: fzcpu@163.com; guok@njtech.edu.cn; Fax: +862558139935; Tel: +862558139926
bCollege of Engineering, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing, 210003, China
cState Key Laboratory of Materials-Oriented Chemical Engineering, 30 Puzhu Rd S., Nanjing, 211816, China
First published on 10th August 2017
Abstract
An efficient and novel method for the synthesis of α-ketoesters has been developed via oxidative coupling of acetophenones with alcohols under TBHP/I2/DBU conditions in a microfluidic chip reactor, which has a wide substrate scope, uses a lower dosage of iodine and affords higher product yields in only a few seconds. Moreover, a scale-up continuous flow system is also feasible and the plausible reaction mechanism has been proposed based on a series of control experiments.
Introduction
Microfluidics, the technology that processes or manipulates small (10−9 to 10−8 litres) amounts of fluids using channels with dimensions of tens to hundreds of micrometres, has become increasingly popular.1 Microfluidic reactors have a number of advantages over conventional chemical laboratory batch synthesis, such as a shorter molecular diffusion distance, a larger specific surface area, and higher mass and heat transfer capacity, which will be expected to promote highly effective chemical reactions.2 In addition, microfluidic reactors could also provide precise control over different reaction parameters and allow for simple screening and optimization of reaction conditions. The development of microfluidic reactors is not only of great significance for optimizing synthetic procedures, but also helpful for improving related chemical industry processes.3α-Ketoesters as double functional compounds exhibit a broad range of biological and pharmaceutical activities.4 Furthermore, α-ketoesters have been widely applied as useful precursors for functional group transformations in organic synthesis and drug development, such as the synthesis of various enzyme inhibitors and the cholesterol-lowering drug Cystatin C.5 Consequently, research on novel and efficient methods for the synthesis of α-ketoesters has become a hot topic in organic synthesis. Traditionally, the methods for the synthesis of α-ketoesters used to be the esterification of α-ketoacyl halides and α-ketoacids,6 oxidation of α-hydroxy esters7 and double carbopalladative esterification.8 In recent years, Cu-catalyzed aerobic oxidative esterification of alcohols with 1,3-diones,9 α-carbonyl aldehydes10 or methyl ketones11 to obtain α-ketoesters has been developed successfully. Besides, some other methods under metal-free conditions have also been reported.12 Non-metal catalytic oxidation systems have received more and more attention in organic synthesis because they can overcome the drawbacks of the expensive, poisonous and air-sensitive properties of metals or organometallics. For example, Wu and co-workers developed various I2-mediated methods that functionalized the sp3 C–H bond in aryl methyl ketones.13 Jiang and co-workers reported a TBHP (tert-butylhydroperoxide)/I2-mediated domino oxidative cyclization for the one-pot synthesis of polysubstituted oxazoles in 2010.14 Wang and co-workers successfully developed a TBHP/I2-promoted oxidative coupling of acetophenones with amines for the synthesis of α-ketoamides.15
In 2013, Yoshida and co-workers developed reactions of organolithiums with dialkyl oxalates for the synthesis of functionalized α-ketoesters using a two-step flow microreactor.16 As part of our ongoing research on the preparation of α-ketoesters and microfluidic technology, herein, we develop a TBHP/I2-promoted oxidative coupling of acetophenones with alcohols leading to α-ketoesters in a microfluidic chip reactor, which is efficient, safe and of great practical worth (Scheme 1). This methodology which utilizes a microfluidic reactor has many prominent advantages: (1) employs cheap and readily available starting materials with good atom economy; (2) greatly improves the reaction efficiency of iodine and product yields; (3) shortens the reaction time from hours to several seconds with a broad substrate scope; and (4) the system is efficient, tunable, safe and easy to scale up under metal-free conditions.
 | ||
| Scheme 1 The oxidative coupling of acetophenones with alcohols leading to α-ketoesters in a microfluidic chip reactor. | ||
Results and discussion
On the basis of previous works on oxidative coupling of acetophenones with amines to produce α-ketoamides, we initially focused on the study of TBHP/I2/base promoted oxidative esterification of acetophenone 1a with 2-phenylethanol 2a to obtain α-ketoesters. Obviously, the nucleophilicity of alcohols was worse than that of amines. Thus, the base plays an important role in this transformation. Preliminary optimization of the reaction conditions for the synthesis of α-ketoesters was carried out in batch to screen a variety of iodine source promoters, oxidants, bases and some other parameters (the experimental details are listed in the ESI†). Although the I2/TBHP/DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) system was determined to be the best reaction conditions, all of the attempts produced less than 60% yield of the target product 3aa in batch. Besides, the dosage of iodine was very large probably because of sublimation or evaporation of iodine when the reaction was carried out in a high temperature batch reactor. On the other hand, DBU may interfere with the oxidation cycle of I2/TBHP in this reaction, especially in the one-pot batch reactor where all reactants always keep reacting in the sealed tube. The reaction in the presence of DBU and TBHP is strongly exothermic, causing more side reactions which restrict its scale-up in batch.In order to overcome the problems above and improve the yields of α-ketoester products, we continued optimizing the TBHP/I2/DBU reaction conditions using a microfluidic chip reactor. As shown in Scheme 2, the microfluidic chip reactor is composed of syringe pumps (A and B), a controller (C) and a start unit (D). The reaction volume of the chip is only 10 μL, which leads to a high surface-to-volume ratio, as well as efficient mass transfer and heat transfer. The reaction time is modulated by changing the flow rate of the syringes. And the temperature of the chip fixed on the start unit (D) was controlled by the controller (C). Particularly, in this reaction, the reagents were divided into two parts of the syringes: (a) 2.0 mmol acetophenone 1a, 8.0 mmol 2-phenylethanol 2a, iodine and 3.0 mmol DBU in DMF; and (b) 8.0 mmol TBHP in DMF. The results are summarized in Scheme 2. First of all, the dosage of iodine was examined (Scheme 2, entries 1–6). To our delight, even if the dosage of iodine was reduced to 0.4 equiv., 64% yield of product 3aa could be obtained in 30 s at 90 °C (Scheme 2, entry 3). And the results clearly demonstrate that iodine in the chip has a higher reaction efficiency than that in batch as shown in Fig. 1. On this basis, the residence time was adjusted from 10 s to 60 s (Scheme 2, entries 7–11), and the corresponding flow rates were from 30 μL min−1 to 6.0 μL min−1 (flow ratea = flow rateb). A better yield of 71% 3aa was obtained at 40 s (Scheme 2, entry 9). However, further extending the reaction time reduced the product yield, which is a reasonable phenomenon in a continuous-flow system, since a longer residence time results in a smaller average velocity in a fixed reactor and leads to weaker mass transfer. For the purpose of further improving the product yield, the reaction temperature of the chip was also investigated (Scheme 2, entries 12–16). To our delight, the chip at 110 °C afforded the highest yield of 86% 3aa, which led to the optimized reaction conditions of the microfluidic synthesis method (Scheme 2, entry 15).
 | ||
| Scheme 2 Optimization of reaction conditions to α-ketoesters in the microfluidic chip reactor.a a Reaction conditions: solution A: 2.0 mmol 1a, 8.0 mmol 2a, iodine and 3.0 mmol DBU in 3 mL DMF; solution B: 8 mmol TBHP in 3 mL DMF; b volume of chip reactor = 10 μL, flow ratea = flow rateb (μL min−1); c isolated yield. | ||
 | ||
| Fig. 1 Effect of dosage of iodine in different reactors on the yields of product 3aa. The reactions were carried out under the corresponding optimized reaction conditions of batch and the microfluidic chip, respectively. | ||
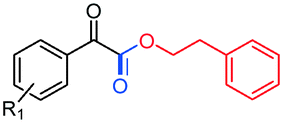
With the optimized reaction conditions in hand, a library of α-ketoesters 3 was synthesized from acetophenones 1 with alcohols 2 using the microfluidic chip reactor. The results are summarized in Table 1. Generally, acetophenones bearing electron-withdrawing or electro-donating groups on different positions of the benzene ring reacted smoothly with 2-phenylethanol 2a, and the corresponding α-ketoester products were obtained in moderate to good yields (Table 1, entries 3aa–3ga). What's more, 2-acetylnaphthalene 3h and 2-acetylthiophene 3i were also tolerated and afforded products 3ha and 3ia in 84% and 73% yields, respectively. Aliphatic methyl ketones such as cyclohexanone, acetone and 2-butanone were also examined, however, they failed to transform to the corresponding target products. Next, a variety of alcohols were also studied under the optimized reaction conditions in the microfluidic chip reactor. The results showed that aromatic and aliphatic, primary and secondary, saturated and unsaturated alcohols were all applicable substrates in this microfluidic synthesis method to generate the corresponding products in good yields (Table 1, entries 3ab–3am). Notably, cholesterol that exists extensively in animal bodies was also subjected to this reaction with acetophenone 1a. And it was found that cholesterol could also undergo this oxidative coupling reaction smoothly to afford the corresponding α-ketoester 3ao in 76% yield, which may serve as an important and practical method for the late-stage modification of bio-active molecules. In addition, some of the substrates above were also examined in batch (3aa, 3ah and 3al), although the corresponding yields were much lower than those in the microfluidic chip reactor system. The reaction time of the batch experiment was 12 h, which was much longer than 40 s in the chip.
|
|
||
|---|---|---|
| a Reaction conditions: solution A: 2.0 mmol acetophenones 1, 8.0 mmol alcohols 2, 0.8 mmol iodine, and 3.0 mmol DBU in 3 mL DMF; solution B: 8 mmol TBHP in 3 mL DMF; flow ratea = flow rateb= 7.5 μL min−1, residence time = 40 s, T = 110 °C. Yields given for isolated products after chromatography. b Reaction conditions: 0.5 mmol 1, 2.0 mmol 2, 0.75 mmol iodine, 0.75 mmol DBU and 2 mmol TBHP in 3 mL DMF were heated for 10 h at 110 °C in sealed batch mode. | ||
|
R1 = H, 3aa, 86%, (59%)b |
 3ha, 84%
3ha, 84% |
| R1 = p-NO2, 3ba, 90% | ||
| R1 = p-F, 3ca, 89% | ||
| R1 = p-Cl, 3da, 87% | ||
| R1 = p-OMe, 3ea, 82% | ||
| R1 = p-Me, 3fa, 80% | ||
| R1 = m-Me, 3ga, 79% | ||
 3ia, 73%
3ia, 73% |

|
R2 = p-Cl, 3aa, 76% |
| R2 = o-Cl, 3ab, 78% | ||
| R2 = p-Br, 3ad, 80% | ||
| R2 = o-Me, 3ae, 88% | ||
| R2 = p-OMe, 3af, 89% | ||
 3ag, 65%
3ag, 65% |

|
R3 = H, 3ah, 84%, (50%)b |
| R3 = p-Cl, 3aa, 76% | ||
| R3 = p-Me, 3aa, 76% | ||
 3ak, 80%
3ak, 80% |
 3al, 73%, (36%)b
3al, 73%, (36%)b |
 3am, 76%
3am, 76% |
 3an, 70%
3an, 70% |
 3an, 70%
3an, 70% |
|

In order to verify the practical value of this method, we next designed a scale-up assembled continuous flow system, which has greater flux, with 1.0 mm inner diameter (ID) and a larger reaction volume, with a 20 mL coiled tube reactor. To our delight, the reaction between acetophenone 1a and 2-phenylethanol 2a proceeded effectively under I2/TBHP/DBU conditions in this assembled continuous flow reactor system, affording a good yield of product 3aa, which implies its good practical applied foreground (Scheme 3).
 | ||
| Scheme 3 Scale-up assembled continuous flow reactor system.a a Reaction conditions: solution A: 10.0 mmol 1a, 40.0 mmol 2a, 4.0 mmol I2 and 15.0 mmol DBU in 25 mL DMF, flow rate = 1.0 mL min−1; solution B: 40 mmol TBHP in 25 mL DMF, flow rate = 1.0 mL min−1; T = 110 °C, reaction time = 10 min. | ||
For the purpose of probing the mechanism of this reaction, a series of control experiments were carried out as shown in Scheme 4. First, almost no product 3aa was produced when I2, DBU or TBHP was removed from the optimized conditions (Scheme 4, eqn (1)). This result indicated that these reactants play an essential role in this reaction. Furthermore, it also turned out that when 1.5 equiv. of the radical inhibitor (TEMPO or BHT) was added into the reaction system, the generation of product 3aa was hindered badly, which suggested that a possible radical pathway was involved in the mechanism (Scheme 4, eqn (2)). Finally, when α-iodo acetophenone B or 2-phenethoxy-1-phenylethan-1-one C was subjected to the optimized conditions with 2-phenylethanol 2a, excellent yields of the desired product 3aa could be obtained respectively, which implies the intermediacy of compounds B and C in the oxidative coupling transformations (Scheme 4, eqn (3) and (4)).
 | ||
| Scheme 4 Control experiments. | ||
On the basis of the control experiments above and previous reports,17 a plausible mechanism of this microfluidic synthesis of α-ketoesters from acetophenones and alcohols was proposed as shown in Scheme 5. Initially, α-iodo acetophenone B was generated from iodination of acetophenone 1. Subsequently, the reaction due to nucleophilic attack of alcohol on the carbonyl α-position was promoted by DBU with the release of HI, forming the key intermediate C. Then, the methylene of intermediate C underwent free radical substitution with the tert-butylperoxy free radical generated from the reaction of tert-butylperoxy, OH− and I2via homolytic cleavage of tert-butyl hydroperoxide to generate intermediate D,18 which underwent further oxidation by TBHP to produce the target α-ketoester products 3.
 | ||
| Scheme 5 Plausible reaction mechanism. | ||
Conclusions
In conclusion, we have developed a highly efficient and practical microfluidic synthetic method to produce α-ketoesters using a microfluidic chip reactor. With this methodology, a wide variety of α-ketoesters have been obtained in moderate to good yields from acetophenones and alcohols under I2/TBHP/DBU conditions. Moreover, higher yields and shorter reaction times are achieved by means of microfluidic technology, which is more convenient, moderate and safe. It also clearly implies the major advantages of continuous systems which allow chemical or reaction parameters to be independently adjusted, hence providing a larger design space for developing chemical reactions or systems. Further studies on the scale-up experiment and application of this strategy are ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been supported by the National Natural Science Foundation of China (Grant No. 21522604, U1463201 and 21402240); the Jiangsu Province Natural Science Fund (Grant No. BK20150031); a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD); and the National Key Research and Development Program of China (2016YFB0301500).Notes and references
- (a) K. S. Elvira, X. C. i. Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed; (b) N.-N. Deng, M. Yelleswarapu, L. Zheng and W. T. S. Huck, J. Am Chem. Soc., 2017, 139, 587–590 CrossRef CAS PubMed; (c) R. E. Fernandez, A. Rohani, V. Farmehini and N. S. Swami, Anal. Chim. Acta, 2017, 966, 11–33 CrossRef CAS PubMed; (d) S. Gholizadeh, M. S. Draz, M. Zarghooni, A. Sanati-Nezhad, S. Ghavami, H. Shafiee and M. Akbari, Biosens. Bioelectron., 2017, 91, 588–605 CrossRef CAS PubMed; (e) W. Lan, S. Jing, X. Guo and S. Li, Chem. Eng. Sci., 2017, 158, 58–63 CrossRef CAS; (f) F. M. Moeller, F. Kriegel, M. Kiss, V. Sojo and D. Braun, Angew. Chem., Int. Ed., 2017, 56, 2340–2344 CrossRef CAS PubMed; (g) R. Ortiz, J. L. Chen, D. C. Stuckey and T. W. J. Steele, ACS Appl. Mater. Interfaces, 2017, 9, 13801–13811 CrossRef CAS PubMed; (h) M.-H. Park, E. Reategui, W. Li, S. N. Tessier, K. H. K. Wong, A. E. Jensen, V. Thapar, D. Ting, M. Toner, S. L. Stott and P. T. Hammond, J. Am. Chem. Soc., 2017, 139, 2741–2749 CrossRef CAS PubMed; (i) I. Pereiro, S. Tabnaoui, M. Fermigier, O. du Roure, S. Descroix, J.-L. Viovy and L. Malaquin, Lab Chip, 2017, 17, 1603–1615 RSC; (j) X. Xie, W. Zhang, A. Abbaspourrad, J. Ahn, A. Bader, S. Bose, A. Vegas, J. Lin, J. Tao, T. Hang, H. Lee, N. Iverson, G. Bisker, L. Li, M. S. Strano, D. A. Weitz and D. G. Anderson, Nano Lett., 2017, 17, 2015–2020 CrossRef CAS PubMed.
- (a) H. Hisamoto, T. Saito, M. Tokeshi, A. Hibara and T. Kitamori, Chem. Commun., 2001, 2662–2663 RSC; (b) G. Veser, Chem. Eng. Sci., 2001, 56, 1265–1273 CrossRef CAS; (c) K. Jahnisch, V. Hessel, H. Lowe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406–446 CrossRef PubMed; (d) B. P. Mason, K. E. Price, J. L. Steinbacher, A. R. Bogdan and D. T. McQuade, Chem. Rev., 2007, 107, 2300–2318 CrossRef CAS PubMed; (e) Q. Feng, J. Liu, X. Li, Q. Chen, J. Sun, X. Shi, B. Ding, H. Yu, Y. Li and X. Jiang, Small, 2017, 13, 1603109 CrossRef PubMed; (f) P. Kluson, P. Stavarek, V. Penkavova, H. Vychodilova, S. Hejda and M. Bendova, Chem. Eng. Process., 2017, 115, 39–45 CrossRef CAS; (g) K. Meller, M. Szumski and B. Buszewski, Sens. Actuators, B, 2017, 244, 84–106 CrossRef CAS; (h) B. Swain, M. H. Hong, L. Kang, B. S. Kim, N.-H. Kim and C. G. Lee, Chem. Eng. J., 2017, 308, 311–321 CrossRef CAS.
- (a) B. Ahmed-Omer, J. C. Brandt and T. Wirth, Org. Biomol. Chem., 2007, 5, 733–740 RSC; (b) E. R. Murphy, J. R. Martinelli, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 1734–1737 CrossRef CAS PubMed; (c) D. M. Roberge, B. Zimmermann, F. Rainone, M. Gottsponer, M. Eyholzer and N. Kockmann, Org. Process Res. Dev., 2008, 12, 905–910 CrossRef CAS; (d) J. B. Wacker, I. Lignos, V. K. Parashar and M. A. M. Gijs, Lab Chip, 2012, 12, 3111–3116 RSC; (e) J.-C. Galas, A.-M. Haghiri-Gosnet and A. Estevez-Torres, Lab Chip, 2013, 13, 415–423 RSC; (f) J. Wang, S.-S. Gu, H.-S. Cui, X.-Y. Wu and F.-A. Wu, Bioresour. Technol., 2014, 158, 39–47 CrossRef CAS PubMed; (g) E. Baeten, B. Verbraeken, R. Hoogenboom and T. Junkers, Chem. Commun., 2015, 51, 11701–11704 RSC; (h) G. Vile, N. Almora-Barrios, N. López and J. Peŕez-Ramírez, ACS Catal., 2015, 5, 3767–3778 CrossRef CAS; (i) G. Vilé, P. Dähler, J. Vecchietti, M. Baltanás, S. Collins, M. Calatayud, A. Bonivardi and J. Pérez-Ramírez, J. Catal., 2015, 324, 69–78 CrossRef.
- (a) M. Wessels, G. M. Konig and A. D. Wright, J. Nat. Prod., 2001, 64, 1556–1558 CrossRef CAS PubMed; (b) Y. Nie, R. Xiao, Y. Xu and G. T. Montelione, Org. Biomol. Chem., 2011, 9, 4070–4078 RSC; (c) B. A. Boughton, L. Hor, J. A. Gerrard and C. A. Hutton, Bioorg. Med. Chem., 2012, 20, 2419–2426 CrossRef CAS PubMed; (d) Y. Aoki, S. Tanimoto, D. Takahashi and K. Toshima, Chem. Commun., 2013, 49, 1169–1171 RSC.
- (a) A. J. Cooper, J. Z. Ginos and A. Meister, Chem. Rev., 1983, 83, 321–358 CrossRef CAS; (b) M. Hayashi and S. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 2249–2252 CrossRef CAS PubMed; (c) B. Eftekhari-Sis and M. Zirak, Chem. Rev., 2015, 115, 151–264 CrossRef CAS PubMed; (d) W. Zhao, P. K. Yan and A. T. Radosevich, J. Am. Chem. Soc., 2015, 137, 616–619 CrossRef CAS PubMed.
- (a) M. V. Dealmeida, D. H. R. Barton, I. Bytheway, J. A. Ferreira, M. B. Hall, W. S. Liu, D. K. Taylor and L. Thomson, J. Am. Chem. Soc., 1995, 117, 4870–4874 CrossRef CAS; (b) P. Wipf and C. R. J. Stephenson, Org. Lett., 2003, 5, 2449–2452 CrossRef CAS PubMed.
- (a) M. Tanaka, T. Kobayashi and T. Sakakura, Angew. Chem., Int. Ed. Engl., 1984, 23, 518 CrossRef; (b) T. Sakakura, H. Yamashita, T. Kobayashi, T. Hayashi and M. Tanaka, J. Org. Chem., 1987, 52, 5733–5740 CrossRef CAS; (c) E. V. Johnston, E. A. Karlsson, L.-H. Tran, B. Akermark and J.-E. Backvall, Eur. J. Org. Chem., 2010, 1971–1976 CrossRef CAS.
- (a) R. Hosseinzadeh, M. Mohadjerani, M. Tajbakhsh and M. Nouzarian, Synth. Commun., 2011, 41, 1725–1732 CrossRef CAS; (b) S. R. Reddy, S. Stella and A. Chadha, Synth. Commun., 2012, 42, 3493–3503 CrossRef CAS.
- C. Zhang, P. Feng and N. Jiao, J. Am. Chem. Soc., 2013, 135, 15257–15262 CrossRef CAS PubMed.
- C. Zhang and N. Jiao, Org. Chem. Front., 2014, 1, 109–112 RSC.
- (a) X. Huang, X. Li, M. Zou, J. Pan and N. Jiao, Org. Chem. Front., 2015, 2, 354–359 RSC; (b) X. Xu, W. Ding, Y. Lin and Q. Song, Org. Lett., 2015, 17, 516–519 CrossRef CAS PubMed.
- (a) A. Sagar, S. Vidyacharan and D. S. Sharada, RSC Adv., 2014, 4, 37047–37050 RSC; (b) S. Battula, N. Battini, D. Singh and Q. N. Ahmed, Org. Biomol. Chem., 2015, 13, 8637–8641 RSC; (c) Y. Xie, J. Liu, Y. Huang and L. Yao, Tetrahedron Lett., 2015, 56, 3793–3795 CrossRef CAS; (d) S. Guo, Z. Dai, Z. Yang, N. Zhu, L. Wan, X. Li, C. Liu, Z. Fang and K. Guo, RSC Adv., 2016, 6, 98422–98426 RSC; (e) K. S. Vadagaonkar, H. P. Kalmode, S. L. Shinde and A. C. Chaskar, ChemistrySelect, 2017, 2, 900–903 CrossRef CAS.
- (a) Q. Gao, X. Wu, S. Liu and A. Wu, Org. Lett., 2014, 16, 1732–1735 CrossRef CAS PubMed; (b) Q. Gao, S. Liu, X. Wu, J. Zhang and A. Wu, J. Org. Chem., 2015, 80, 5984–5991 CrossRef CAS PubMed; (c) S. Liu, H. Xi, J. Zhang, X. Wu, Q. Gao and A. Wu, Org. Biomol. Chem., 2015, 13, 8807–8811 RSC; (d) J. Zhang, X. Wu, Q. Gao, X. Geng, P. Zhao, Y.-D. Wu and A. Wu, Org. Lett., 2017, 19, 408–411 CrossRef CAS PubMed.
- H. Jiang, H. Huang, H. Cao and C. Qi, Org. Lett., 2010, 12, 5561–5564 CrossRef CAS PubMed.
- X. Zhang and L. Wang, Green Chem., 2012, 14, 2141–2145 RSC.
- A. Nagaki, D. Ichinari and J.-I. Yoshida, Chem. Commun., 2013, 49, 3242–3244 RSC.
- (a) M. Lamani and K. R. Prabhu, Chem. – Eur. J., 2012, 18, 14638–14642 CrossRef CAS PubMed; (b) W. Wei, Y. Shao, H. Hu, F. Zhang, C. Zhang, Y. Xu and X. Wan, J. Org. Chem., 2012, 77, 7157–7165 CrossRef CAS PubMed; (c) G. Majji, S. Guin, S. K. Rout, A. Behera and B. K. Patel, Chem. Commun., 2014, 50, 12193–12196 RSC.
- J. Zhang, Z. Wang, Y. Wang, C. Wan, X. Zheng and Z. Wang, Green Chem., 2009, 11, 1973–1978 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, LC-ESI-MS/MS analysis, analytical data and NMR spectra of products. See DOI: 10.1039/c7re00107j |
| This journal is © The Royal Society of Chemistry 2017 |