
Solvent-free coupling of aryl halides with pyrroles applying visible-light photocatalysis
M.
Obst
,
R. S.
Shaikh
and
B.
König
 *
*
Institut für Organische Chemie, Universität Regensburg, Universitätsstraße 31, 93040 Regensburg, Germany. E-mail: burkhard.koenig@chemie.uni-regensburg.de; Fax: +49 941 943 1717; Tel: +49 941 943 4575
First published on 27th March 2017
Abstract
A novel reactor for solvent-free, visible-light-driven photocatalytic transformations was developed. By rotation of the reaction vessel, the reaction mixture forms a thin film, which allows efficient excitation of the photocatalyst. The reactor was used for the coupling of aryl halides with pyrrole derivatives and phosphites, applying rhodamine 6G as the photocatalyst and DIPEA as the sacrificial electron donor. The necessary amounts of photocatalyst, trapping reagent, and sacrificial electron donor were reduced significantly compared to those for literature known reactions in solution, while isolating the products in moderate to good yields. In general terms, this solvent-free methodology is an interesting alternative to solution photocatalysis due to the presence of high mole fractions of trapping reagent, the exclusion of by-products formed with the solvent, and the reduction of toxic solvent waste.
Introduction
Visible-light photocatalysis applying an organic photoredox catalyst is a valuable approach for mild and efficient transformations.1,2 Harsh reaction conditions and expensive reagents are avoided and the use of light energy often enables reactions, which are kinetically hindered or endothermic. The conversion of light energy into chemical redox power is achieved by the excitation of a visible-light-absorbing photocatalyst and subsequent electron transfer, leading to an organic transformation.2,3 Typically, photocatalytic reactions are performed in solution. However, in many cases, it is helpful to perform reactions under solvent-free conditions as undesired side reactions with the solvent, such as hydrogen atom transfer, can be avoided and the formation of by-products is minimized. Furthermore, the absence of a solvent leads to high reactant and catalyst concentrations, which can be advantageous as the trapping of a reactive intermediate should be favored by the presence of high amounts of trapping reagent. In the case of quantitative product formation, work-up is significantly facilitated.4 Reports on solvent-free visible-light photocatalysis are quite limited, including the oxidation of benzyl alcohol with an organic photocatalyst,5 the oxidation of benzenes to phenols,6 the use of fullerenes linked to silica gel for the oxidation of various organic substrates,7 and the application of semiconducting photocatalysts, such as titanium dioxide and other metal oxides.8–11 Recently, we reported the solvent-free oxidation of benzylic alcohols applying riboflavin tetraacetate as a photocatalyst under blue light irradiation. The reactions were run in a novel milling apparatus, suitable for the conversion of solid substrates.12Herein, we developed a methodology for solvent-free photocatalytic conversions applying an organic photocatalyst in order to combine the advantages of photocatalysis and solvent-free operation. For this purpose, we constructed a novel reactor, which is suitable for efficient photocatalytic transformations of viscous reaction mixtures, realized by the formation of thin films. When searching for an adequate model reaction to establish our methodology, our attention was drawn to C–C coupling reactions. Palladium-catalyzed cross-coupling reactions are widely applied in organic synthesis, including important and valuable standard methods such as the Heck, Suzuki, Stille, Negishi, and Sonogashira coupling reaction.13–16 Due to the sensitivity of palladium-based catalysts to oxygen and water, dry solvents must be used and the reaction must be performed in an inert atmosphere. Therefore, many attempts have been made to perform cross-coupling reactions under solvent-free conditions.17 In this context, mechanochemistry proved to be very useful: Heck, Suzuki, and Sonogashira coupling reactions were performed in a ball mill in an aerobic environment.18–20 Recently, C–C coupling reactions were realized by applying visible-light photocatalysis: aryl radicals (generated from the corresponding halides) were coupled with pyrroles, using rhodamine 6G as the photocatalyst under blue light irradiation and diisopropylethylamine (DIPEA) as the sacrificial electron donor. This system was established by our group and is a very promising approach due to its mild reaction conditions and high functional group tolerance.21 However, high amounts of trapping reagent, DIPEA, and rhodamine 6G are necessary. Thus, through its transfer to solvent-free conditions, we aspired to both the improvement of the catalytic system from an atom-economic point of view and the development of a new photocatalytic methodology.
Results and discussion
Our target reaction was the coupling of aryl halides and pyrroles with rhodamine 6G as the photocatalyst. Aryl halides possessing an electron-withdrawing moiety, such as nitrile or ester, can be applied as substrates to generate an aryl radical. Upon blue light absorption, rhodamine 6G is excited and reduced by a sacrificial electron donor yielding the rhodamine 6G radical anion. This can be excited again by blue light (450 nm = 2.76 eV) transferring an electron to the substrate and regenerating rhodamine 6G in the ground state, the reduction being the rate-determining step as the rate depends on the substituents present in the aryl halide. The aryl halide radical anion cleaves into a halogen anion and an aryl radical, which attacks the pyrrole. The resulting radical is oxidized and deprotonated restoring the heteroaromatic π-system to give the sp2–sp2 coupling product. In general, the crucial point in the catalytic mechanism is the fast trapping of the reactive aryl radical by the pyrrole derivative in order to prevent side reactions such as the formation of the reduced by-product by hydrogen atom abstraction from the solvent. Recently, this was realized by applying high amounts of the trapping reagent, typically 13 to 26 equivalents relative to the substrate.21 However, for effective trapping, not only the amount but also the mole fraction of the trapping reagent, χ(tr), must be considered. It is defined as the molar amount of the trapping reagent, n(tr), divided by the total amount of all substances present in the reaction mixture (sub = substrate; solv = solvent; Rh = rhodamine 6G): | (1) |
Keeping the amount of substrate, rhodamine 6G, and DIPEA constant, a high χ(tr) is equal to a high trapping reagent/substrate ratio, which favors the trapping, which is why we envisaged to maximize the χ(tr) value. In this context, the amounts of both the trapping reagent and the solvent can be varied. The coupling of ortho-bromobenzonitrile to N-methylpyrrole was recently performed applying 10% rhodamine 6G and 1.4 equivalents of DIPEA in DMSO.21 For this system, Fig. 1 shows the mole fraction of the trapping reagent and its dependence on the amounts of the trapping reagent and solvent, with fixed amounts of substrate, rhodamine 6G, and DIPEA. In the literature,21 the reaction of 0.1 mmol of substrate in 1.5 mL (21.1 mmol) of DMSO is reported.
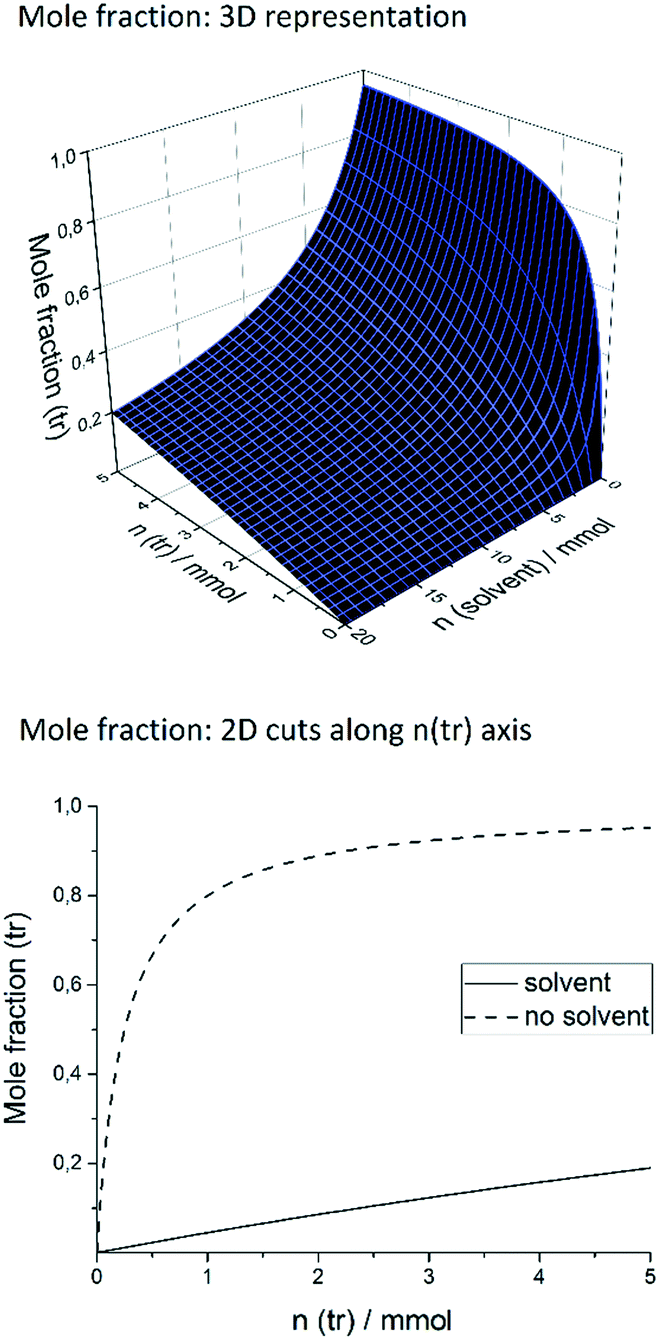 | ||
| Fig. 1 Top: Dependence of the mole fraction of the trapping reagent (N-methylpyrrole) on the amount of the trapping reagent and solvent. Bottom: Dependence on the amount of the trapping reagent with (21.12 mmol, 1.5 mL) and without the solvent. | ||
As illustrated in Fig. 1, an increase of n(tr) at that amount of solvent raises the χ(tr) value only slightly, and high n(tr) values are necessary. A more elegant and simple way to increase χ(tr) is to decrease the amount of solvent. By doing so, χ(tr) reaches its maximum when no solvent is used at all, given a certain n(tr) value. In the solvent-free system, a slight increase of n(tr) causes a strong increase of χ(tr) (dashed graph), whereas it is practically impossible to reach a comparable χ(tr) value when 21.1 mmol of solvent is present. In other words, solvent-free operation achieves much higher χ(tr) values at lower amounts of trapping reagent, compared to reactions in solution. While 1.8 mmol (18 eq.) of trapping reagent corresponds to a χ(tr) of ca. 0.08 in solution, a more than tenfold value (0.88) is achieved in the solvent-free system. Consequently, by transferring the catalytic system to solvent-free conditions, it should be possible to significantly reduce the necessary amount of trapping reagent and, by that, to improve the atom economy. Another issue is the oxidation of the sacrificial electron donor (DIPEA) by the excited rhodamine 6G. In solvent-free operation, the resulting high concentration of DIPEA should favor the effective reduction of rhodamine 6G and hinder the undesired deactivation by fluorescence.
We began the development of the solvent-free photocatalytic methodology by designing a suitable reactor. Solvent-free photocatalysis bears several challenges, excluding the application of conventional laboratory reaction setups. First, the penetration depth of light in heterogeneous systems is low, because of scattering by solid particles and absorption of light by catalyst molecules in the outer part of the reaction mixture, which leads to shielding of the catalyst molecules and impedes their excitation. The latter can be critical even in homogeneous solution, especially at high catalyst concentrations.22 Considering the much higher catalyst concentrations in solvent-free systems, it becomes obvious that this is the main issue, which must be addressed when constructing a reactor. Another problem is the limited diffusion in solvent-free systems: depending on the reactants, more or less viscous pastes are formed and the realization of catalytic cycles is difficult. It is often crucial that charge and energy transfer can occur quickly, which requires mobility of both catalyst and reactant molecules within the reaction mixture. Furthermore, homogenization is a demanding task due to the high viscosity, excluding the use of a magnetic stirrer.
From all these considerations, we draw the conclusion that it is necessary to realize thin films of the reaction mixture in order to achieve a high relative surface area, which is crucial for efficient excitation of the photocatalyst. Based on this, we recently constructed a rod mill apparatus, which is suitable for the conversion of solid substrates. Therein, a film of the reaction mixture is realized, which enables efficient excitation of the photocatalyst. Using the reactor, we could oxidize several benzylic alcohols to their corresponding carbonyl compounds with riboflavin tetraacetate under blue light irradiation with oxygen as the terminal oxidant.12 The novel “rotating film reactor”, suitable for the conversion of paste-like mixtures, is shown in Fig. 2. It consists of a crimp glass vial similar to the ones used for homogeneous photocatalysis. It is fixed to a glass stick, which is linked to a KPG stirrer, and can be closed with a septum to achieve inert conditions, or left open, which is necessary for reactions with oxygen as the terminal oxidant. The glass vial is surrounded by four LEDs installed on an aluminum frame. A reaction is performed by filling the reaction mixture into the vial, followed by transfer to inert conditions, if necessary, and sonication in an ultrasonic bath. The vial is then fixed on the KPG stirrer and the rotation is switched on (1200 rpm).
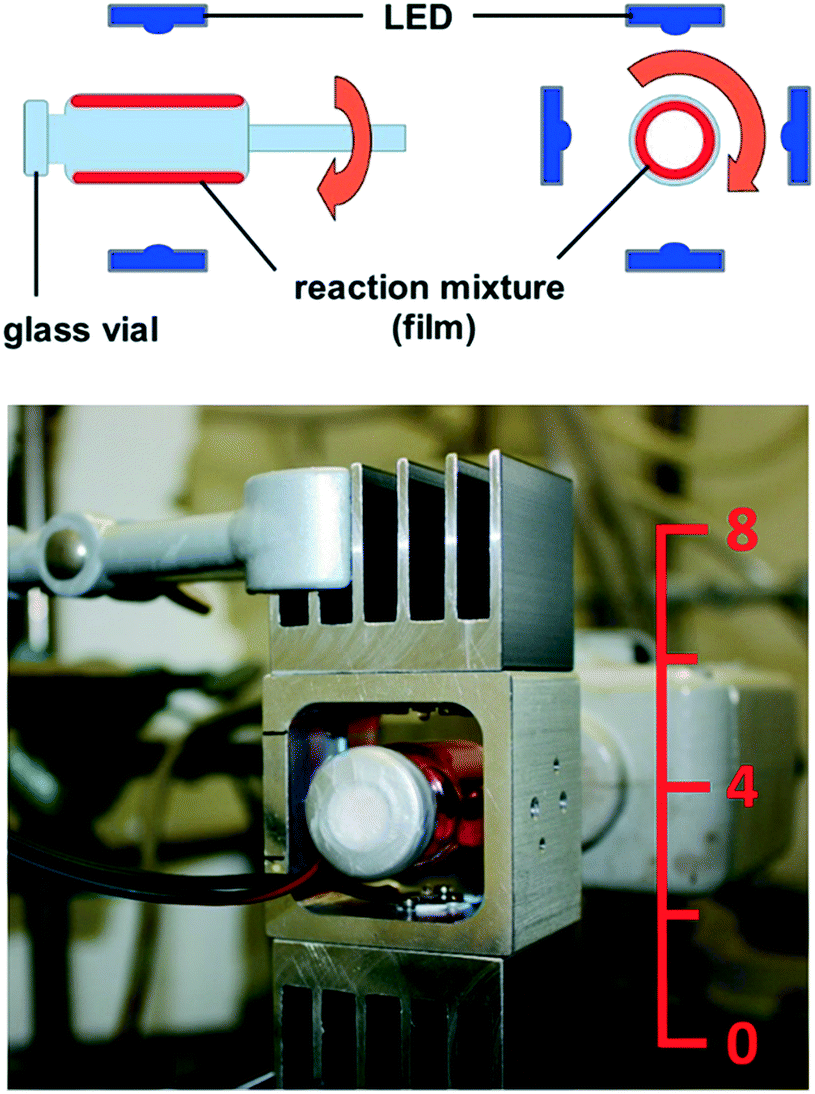 | ||
| Fig. 2 Rotating film reactor, schematic (side and front view) and photograph (numbers on the ruler indicate cm). | ||
The resulting centrifugal force presses the reaction mixture against the inner wall of the vial, forming a film. Eventually, the LEDs are switched on and the reaction can proceed.
In order to evaluate the rotating film reactor and to optimize the reaction conditions, we applied para-bromobenzonitrile as the substrate and N-methylpyrrole as the trapping reagent, leading to the C–C coupling product 1 (Table 1). In an initial trial applying 10% rhodamine 6G, 3 equivalents of N-methylpyrrole, and 1 equivalent of DIPEA, we could isolate the product in 38% yield, showing that the reaction proceeds under solvent-free conditions in the rotating film reactor (Table 1, entry 1). As mentioned above, high catalyst concentrations impede the efficient excitation of the catalyst molecules within the reaction mixture, especially in heterogeneous systems. Therefore, we decreased the catalyst loading to 5%, which increased the yield to 50% (entry 2). However, in both reactions, solid particles and partial phase separation occurred. To overcome this problem, we performed the reaction with 5 equivalents of N-methylpyrrole. Being the only liquid component, N-methylpyrrole acts as an excipient, enabling the formation of a film and the mobility of the catalyst and substrate molecules.
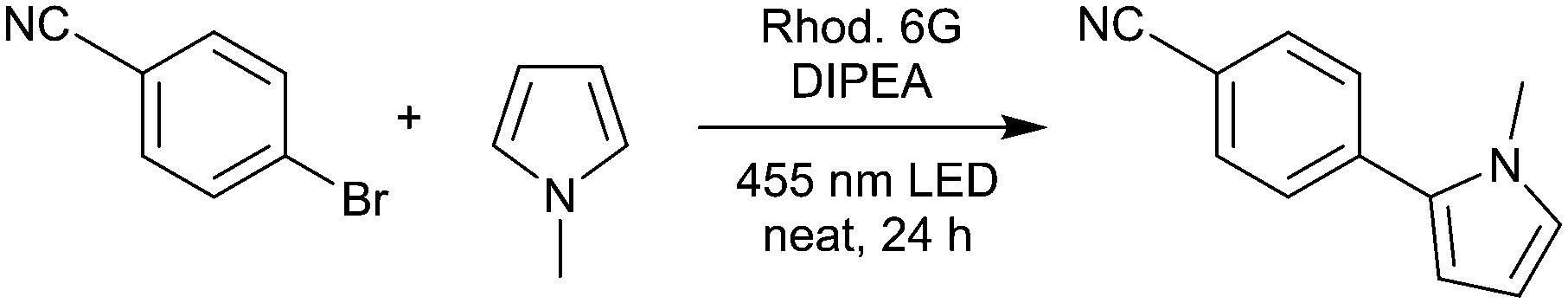
Moreover, we decreased the catalyst loading to 2% to further enhance the excitation efficiency (entry 3). To our delight, this increased the yield to 60%. We then varied the amount of the sacrificial electron donor, DIPEA. Raising the DIPEA amount to 1.5 equivalents decreased the yield to 49% (entry 4). In this reaction, bleaching of rhodamine 6G was observed. Thus, we applied 10% rhodamine 6G and 1.2 as well as 1.4 equivalents of DIPEA (entries 5 and 6). By doing so, we obtained the products in 60% and 58% yield, which is similar to entry 3. Thus, more than one equivalent of DIPEA seems to bleach parts of the catalyst, which is why more rhodamine 6G is necessary to compensate for this effect. To summarize, we obtained the optimized reaction conditions as 2% rhodamine 6G, 5 equivalents of N-methylpyrrole, and 1 equivalent of DIPEA. For entry 3, the thickness of the film formed from the reaction mixture is 0.26 mm (calculated using the volume of the reaction mixture and the reactor geometry). The control reactions showed that only traces of the product were formed without rhodamine 6G (entry 7), whereas 21% conversion was observed in the absence of DIPEA (entry 8). In the dark, no product was formed (entry 9). For the optimized conditions (entry 3), the mole fraction, χ(tr), of N-methylpyrrole is 0.71, applying 5 equivalents. In the literature,21 by applying 18 equivalents of N-methylpyrrole, χ(tr) was only 0.08 for the same reaction. Thus, by solvent-free operation, we achieved a ninefold χ(tr) value with less than one third of the equivalents of the trapping reagent compared to the reaction in solution.
Having optimized the reaction conditions using N-methylpyrrole, we applied other radical trapping reagents, including pyrrole, 2,4-dimethylpyrrole, and N-benzylpyrrole (Scheme 1). With para-bromobenzonitrile as the substrate, we obtained the products 2, 3, and 4 in moderate yields. The application of ortho-bromobenzonitrile with N-methylpyrrole as the trapping reagent led to a significantly higher yield (77%, product 5b), which is possibly due to the better stabilization of the radical anion when the nitrile group is located in the ortho-position. This reaction was recently performed in solution, resulting in 78% yield.21 Using ortho-iodobenzonitrile as the substrate resulted in a good yield (product 5c). However, for ortho-chlorobenzonitrile, bleaching of the photocatalyst and little conversion of the substrate in GC occurred. Therefore, we increased the catalyst loading to 10% and the reaction time to 48 h and isolated the product 5a in good yield. The best-performing substrate ortho-bromobenzonitrile was then coupled with pyrrole, 2,4-dimethylpyrrole, and N-benzylpyrrole. The products 6, 7, and 8 were isolated in moderate to good yields (60%, 55%, and 62%); in solution, product 6 had been isolated in 64% yield.21
 | ||
| Scheme 1 Coupling of aryl halides with pyrrole derivatives. | ||
Hence, for the products 5b and 6, we were able to achieve similar yields to those in solution, but with lower amounts of the trapping reagent, photocatalyst, and DIPEA. Similar to para-bromobenzonitrile, N-methylpyrrole as the trapping reagent resulted in the highest yields when comparing all pyrrole derivatives used. The reaction of methyl 4-bromobenzoate with N-methylpyrrole led to the formation of product 9 in moderate yield.
We then moved to nitrogen heteroaryl bromides as substrates; in the literature, their coupling with pyrrole derivatives using the rhodamine 6G system has already been reported.21 First, we applied 3-bromopyridine as the substrate (Scheme 2). However, with 2% rhodamine 6G, we observed bleaching and low conversion, so we increased the catalyst loading to 10%, which resulted in moderate yield (product 10, 49%); in the literature, the reported yield for this product is 59%.21 The same conditions were then used for the coupling of 5-bromopyrimidine with N-methylpyrrole and pyrrole, resulting in good and moderate yields (products 11 and 12, 67% and 52%). Recently, the rhodamine 6G system was also applied in solution (DMSO) for the coupling of aryl bromides with phosphites, yielding aryl phosphonates.23 We performed the solvent-free coupling of para-bromobenzonitrile with diethylphosphite and diisopropylphosphite, applying 10% rhodamine 6G, resulting in the products 13 and 14 in good yields (Scheme 3); the yields reported in the literature23 are 76% for product 13 and 80% for product 14.
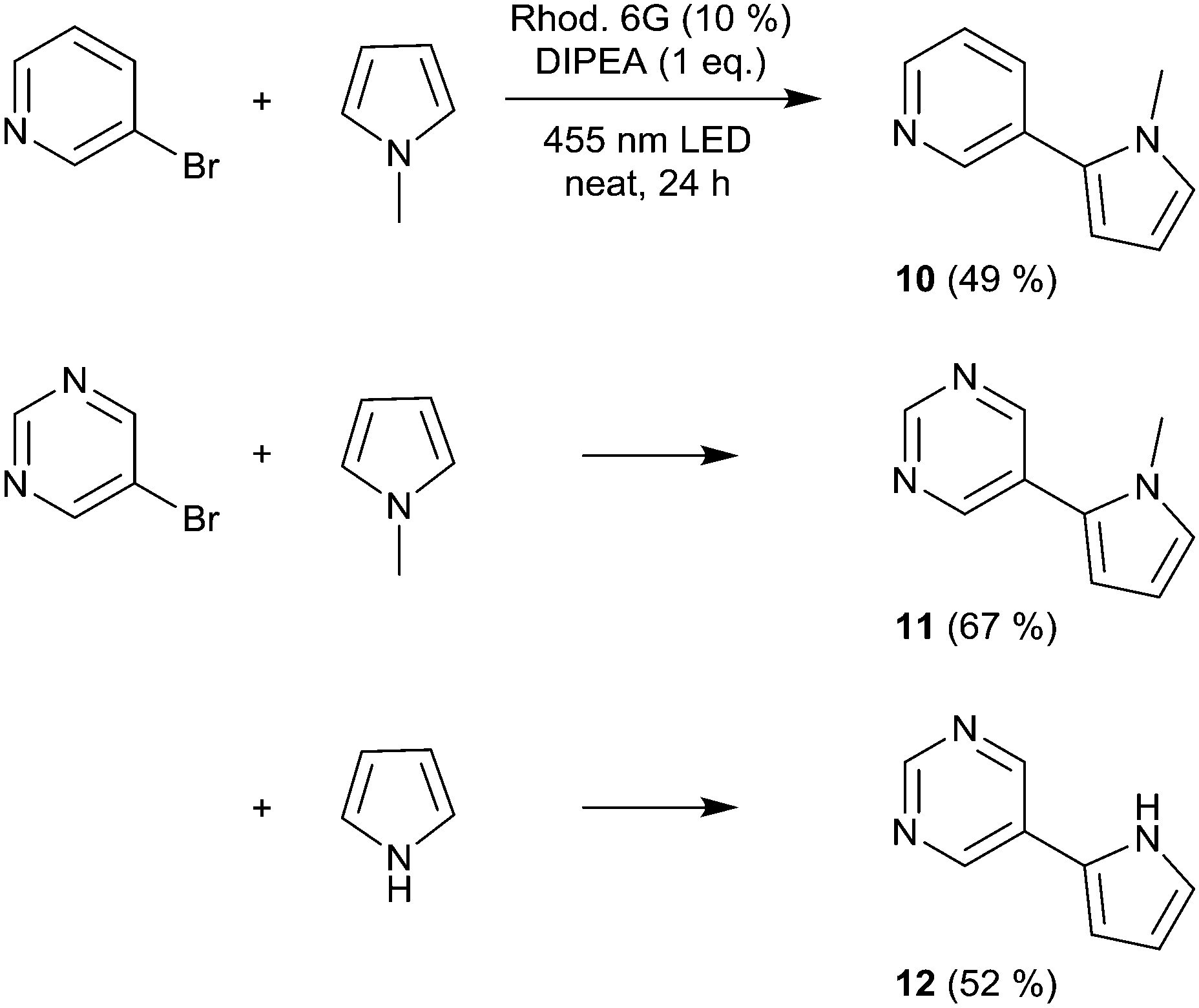 | ||
| Scheme 2 Coupling of nitrogen heteroaryl bromides with pyrroles. | ||
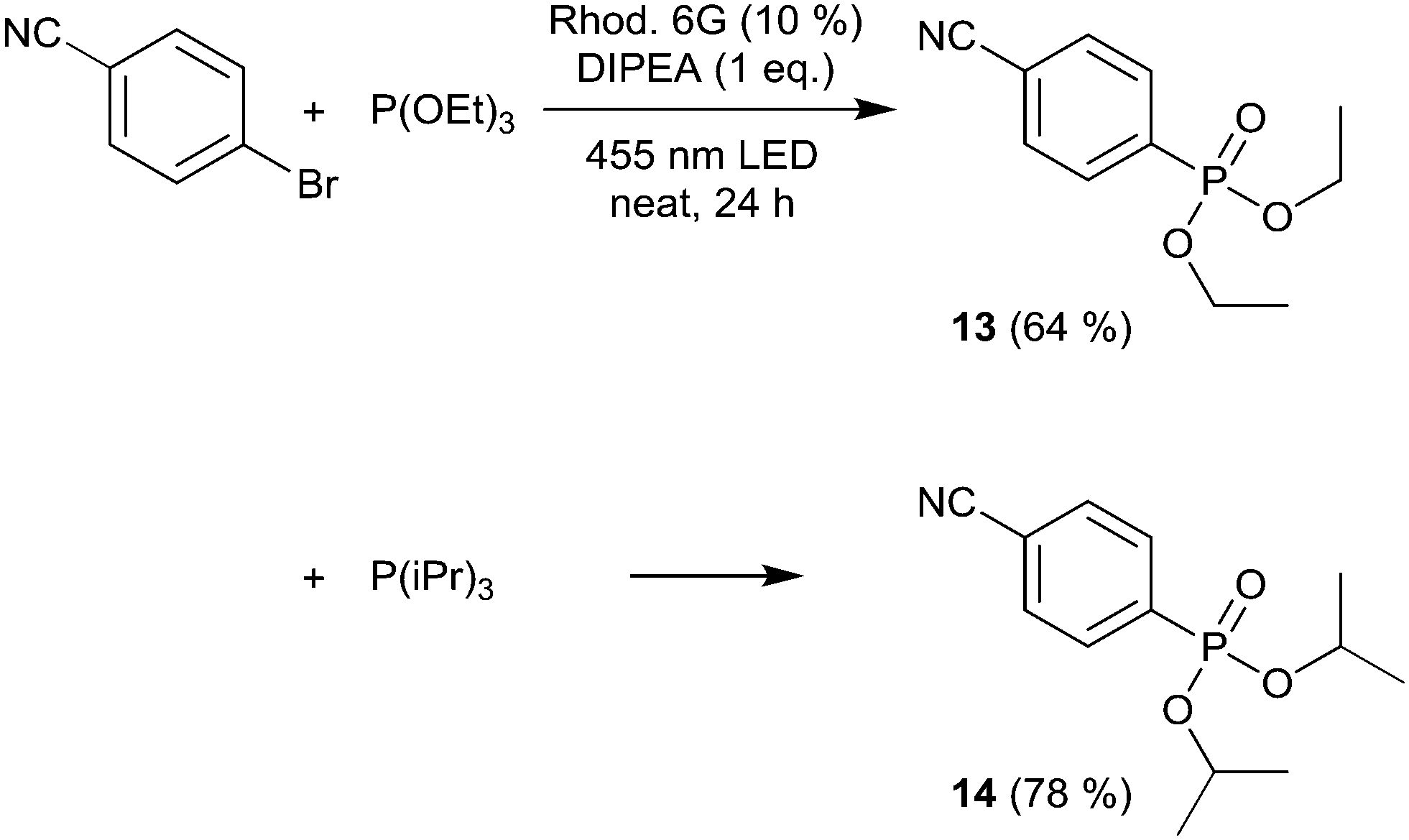 | ||
| Scheme 3 Coupling of para-bromobenzonitrile with diethylphosphite and diisopropylphosphite. | ||
Conclusions
We have developed a novel rotating film reactor for solvent-free, visible-light-driven photocatalytic transformations. In this reactor, rotation of the reaction vial generates a thin film, which enables efficient excitation of the photocatalyst due to the high relative surface area. The applicability of the reactor was shown for the solvent-free coupling of aryl halides with pyrroles and phosphites, applying rhodamine 6G as the photocatalyst and DIPEA as the sacrificial electron donor; the products were isolated in moderate to good yields. Compared to reactions recently performed in solution, we could significantly decrease the amount of trapping reagent, rhodamine 6G, and DIPEA. The solvent-free operating conditions lead to much higher mole fraction values of the trapping reagent than those in solution, allowing lower amounts of the trapping reagent to be used and thus improving the overall atom economy.To summarize, this solvent-free methodology is a promising alternative to conventional photocatalysis performed in solution. It may be beneficial for reactions where undesired side reactions with the solvent lead to by-product formation. Moreover, reactions may profit from high substrate and catalyst concentrations, facilitating the trapping of radical intermediates. Furthermore, solubility problems, which are caused by the different polarity of reactants and catalyst, are overcome.
Experimental
Materials and methods
The solid starting materials (except rhodamine 6G) were ground in a pestle and mortar before use. NMR spectra were recorded on a Bruker Avance 300.Technical data of the rotating film reactor
Four LEDs (455 nm, 700 mA) on a square aluminum frame (inner diameter = 35 mm) were used. The outer diameter of the glass vial was 22 mm, the inner diameter was 19 mm and the LED–vial distance was 3 mm. The inner height of the vial was 28 mm. The power of LED irradiation was 28.1 mW cm−2.Synthesis
Compounds 1, 2, 5, 6, 10, 13, and 14 are reported in the literature. For NMR data of compounds 1, 5, 6, and 10, see the literature.21 For compound 2, see the literature.24 For compounds 13 and 14, see the literature.23
4-(1-Methyl-1H-pyrrol-2-yl)benzonitrile (1). Procedure for entry 3 (Table 1): 4-bromobenzonitrile (127.4 mg, 0.7 mmol) was reacted with 1-methylpyrrole (311 μL, 3.5 mmol, 5 eq.) applying rhodamine 6G (6.7 mg, 0.014 mmol, 2%) and DIPEA (122 μL, 0.7 mmol, 1 eq.). Yield: 76.9 mg (60%). 1H NMR (300 MHz, CDCl3): 7.72–7.63 (m, 2H), 7.56–7.47 (m, 2H), 6.84–6.77 (m, 1H), 6.37 (dd, J = 3.7, 1.8 Hz, 1H), 6.25 (dd, J = 3.7, 2.7 Hz, 1H), 3.73 (s, 3H). MS: 182.0838 [M]+.
4-(1H-Pyrrol-2-yl)benzonitrile (2). 4-Bromobenzonitrile (127.4 mg, 0.7 mmol) was reacted with pyrrole (245 μL, 3.5 mmol, 5 eq.) applying rhodamine 6G (6.7 mg, 0.014 mmol, 2%) and DIPEA (122 μL, 0.7 mmol, 1 eq.). Yield: 60.9 mg (52%). 1H NMR (300 MHz, CDCl3): 8.91 (bs, 1H), 7.66–7.50 (m, 4H), 7.00–6.92 (m, 1H), 6.72–6.65 (m, 1H), 6.39–6.32 (m, 1H). MS: 169.0772 [MH]+.
4-(3,5-Dimethyl-1H-pyrrol-2-yl)benzonitrile (3). 4-Bromobenzonitrile (109.2 mg, 0.6 mmol) was reacted with 2,4-dimethylpyrrole (309 μL, 3.0 mmol, 5 eq.) applying rhodamine 6G (5.7 mg, 0.012 mmol, 2%) and DIPEA (105 μL, 0.6 mmol, 1 eq.). Yield: 67.5 mg (57%). 1H NMR (300 MHz, CDCl3): 8.26 (bs, 1H), 7.64–7.57 (m, 2H), 7.51–7.43 (m, 2H), 5.92–5.86 (m, 1H), 2.32 (s, 3H), 2.29 (s, 3H). 13C NMR (300 MHz, CDCl3): 138.0, 132.5, 130.0, 125.1, 124.8, 120.0, 119.5, 111.6, 107.5, 13.2, 13.1. MS: 197.1077 [MH]+.
4-(1-Benzyl-1H-pyrrol-2-yl)benzonitrile (4). 4-Bromobenzonitrile (127.4 mg, 0.7 mmol) was reacted with 1-benzylpyrrole (540 μL, 3.5 mmol, 5 eq.) applying rhodamine 6G (6.7 mg, 0.014 mmol, 2%) and DIPEA (122 μL, 0.7 mmol, 1 eq.). Yield: 68.2 mg (37%). 1H NMR (300 MHz, CDCl3): 7.63–7.55 (m, 2H), 7.46–7.38 (m, 2H), 7.37–7.25 (m, 3H), 7.06–6.98 (m, 2H), 6.90–6.83 (m, 1H), 6.43 (dd, J = 3.6, 1.7 Hz, 1H), 6.37–6.31 (m, 1H), 5.21 (s, 2H). 13C NMR (300 MHz, CDCl3): 138.1, 137.7, 132.9, 132.3, 128.9, 128.5, 127.7, 126.2, 125.3, 119.0, 111.0, 110.0, 109.3, 51.0. MS: 259.1228 [MH]+.
2-(1-Methyl-1H-pyrrol-2-yl)benzonitrile (5). Characterization: 1H NMR (300 MHz, CDCl3): 7.78–7.71 (m, 1H), 7.66–7.58 (m, 1H), 7.48–7.36 (m, 2H), 6.81 (dd, J = 2.6, 1.8 Hz, 1H), 6.43 (dd, J = 3.7, 1.8 Hz, 1H), 6.26 (dd, J = 3.7, 2.7 Hz, 1H), 3.62 (s, 3H). 13C NMR (300 MHz, CDCl3): 136.9, 133.6, 132.5, 130.9, 129.9, 127.5, 124.9, 118.7, 112.7, 111.5, 108.3, 34.9. MS: 182.1188 [M]+.
5a . 2-Chlorobenzonitrile (96.3 mg, 0.7 mmol) was reacted with 1-methylpyrrole (311 μL, 3.5 mmol, 5 eq.) applying rhodamine 6G (33.5 mg, 0.07 mmol, 10%) and DIPEA (122 μL, 0.7 mmol, 1 eq.). The reaction time was 48 h. Yield: 83.2 mg (64%).
5b . 2-Bromobenzonitrile (127.4 mg, 0.7 mmol) was reacted with 1-methylpyrrole (311 μL, 3.5 mmol, 5 eq.) applying rhodamine 6G (6.7 mg, 0.014 mmol, 2%) and DIPEA (122 μL, 0.7 mmol, 1 eq.). Yield: 98.8 mg (77%).
5c . 2-Iodobenzonitrile (160.3 mg, 0.7 mmol) was reacted with 1-methylpyrrole (311 μL, 3.5 mmol, 5 eq.) applying rhodamine 6G (6.7 mg, 0.014 mmol, 2%) and DIPEA (122 μL, 0.7 mmol, 1 eq.). Yield: 77.9 mg (60%).
2-(1H-Pyrrol-2-yl)benzonitrile (6). 2-Bromobenzonitrile (127.4 mg, 0.7 mmol) was reacted with pyrrole (245 μL, 3.5 mmol, 5 eq.) applying rhodamine 6G (6.7 mg, 0.014 mmol, 2%) and DIPEA (122 μL, 0.7 mmol, 1 eq.). Yield: 71.1 mg (60%). 1H NMR (300 MHz, CDCl3): 9.23 (bs, 1H), 7.68–7.50 (m, 3H), 7.30–7.18 (m, 1H), 7.01–6.92 (m, 1H), 6.88–6.81 (m, 1H), 6.39–6.31 (m, 1H). 13C NMR (300 MHz, CDCl3): 135.7, 134.1, 133.2, 128.2, 126.7, 125.9, 120.9, 120.2, 110.4, 106.0. MS: 168.1010 [M]+.
2-(3,5-Dimethyl-1H-pyrrol-2-yl)benzonitrile (7). 2-Bromobenzonitrile (109.2 mg, 0.6 mmol) was reacted with 2,4-dimethylpyrrole (309 μL, 3.0 mmol, 5 eq.) applying rhodamine 6G (5.7 mg, 0.012 mmol, 2%) and DIPEA (105 μL, 0.6 mmol, 1 eq.). Yield: 65.2 mg (55%). 1H NMR (300 MHz, CDCl3): 8.29 (s, 1H), 7.71–7.63 (m, 1H), 7.61–7.52 (m, 1H), 7.51–7.43 (m, 1H), 7.33–7.23 (m, 1H), 5.92–5.85 (m, 1H), 2.30 (s, 3H), 2.20 (s, 3H). 13C NMR (400 MHz, CDCl3): 137.1, 133.9, 132.6, 129.6, 129.4, 125.9, 123.1, 120.1, 119.6, 110.5, 109.3, 13.1, 12.8. MS: 196.1366 [M]+.
2-(1-Benzyl-1H-pyrrol-2-yl)benzonitrile (8). 2-Bromobenzonitrile (127.4 mg, 0.7 mmol) was reacted with 1-benzylpyrrole (539 μL, 3.5 mmol, 5 eq.) applying rhodamine 6G (6.7 mg, 0.014 mmol, 2%) and DIPEA (122 μL, 0.7 mmol, 1 eq.). Yield: 114.6 mg (62%). 1H NMR (300 MHz, CDCl3): 7.73–7.66 (m, 1H), 7.58–7.48 (m, 1H), 7.43–7.33 (m, 2H), 7.32–7.19 (m, 3H), 6.99–6.90 (m, 2H), 6.98–6.83 (m, 1H), 6.50 (dd, J = 3.6, 1.7 Hz, 1H), 6.39–6.31 (m, 1H), 5.12 (s, 2H). 13C NMR (300 MHz, CDCl3): 138.2, 136.9, 133.5, 132.4, 130.9, 130.0, 128.7, 127.7, 127.6, 126.5, 124.4, 118.5, 113.2, 112.0, 109.0, 51.12. MS: 258.1617 [M]+.
Methyl 4-(1-methyl-1H-pyrrol-2-yl)benzoate (9). Methyl 4-bromobenzoate (150.5 mg, 0.7 mmol) was reacted with 1-methylpyrrole (311 μL, 3.5 mmol, 5 eq.) applying rhodamine 6G (6.7 mg, 0.014 mmol, 2%) and DIPEA (122 μL, 0.7 mmol, 1 eq.). Yield: 69.3 mg (46%). 1H NMR (300 MHz, CDCl3): 8.11–8.03 (m, 2H), 7.53–7.45 (m, 2H), 6.80–6.74 (m, 1H), 6.36 (dd, J = 3.7, 1.8 Hz, 1H), 6.24 (dd, J = 3.6, 2.7 Hz, 1H), 3.94 (s, 3H), 3.72 (s, 3H). 13C NMR (300 MHz, CDCl3): 167.0, 137.8, 133.5, 129.8, 127.9, 125.2, 110.1, 108.3, 52.1, 35.4. MS: 215.1332 [M]+.
3-(1-Methyl-1H-pyrrol-2-yl)pyridine (10). Rhodamine 6G (33.5 mg, 0.07 mmol, 10%) was weighed into the reaction vial, which was transferred to inert conditions, followed by the addition of 3-bromopyridine (67 μL, 0.7 mmol), DIPEA (122 μL, 0.7 mmol, 1 eq.), and 1-methylpyrrole (311 μL, 3.5 mmol, 5 eq.). Yield: 54.7 mg (49%). 1H NMR (300 MHz, CDCl3): 8.74–8.63 (m, 1H), 8.58–8.46 (m, 1H), 7.73–7.65 (m, 1H), 7.32 (dd, J = 7.8, 4.8 Hz, 1H), 6.80–6.72 (m, 1H), 6.30 (dd, J = 3.6, 1.8 Hz, 1H), 6.23 (dd, J = 3.6, 2.7 Hz, 1H), 3.67 (s, 3H). MS: 158.0842 [M]+.
5-(1-Methyl-1H-pyrrol-2-yl)pyrimidine (11). 5-Bromopyrimidine (111.3 mg, 0.7 mmol) was reacted with 1-methylpyrrole (311 μL, 3.5 mmol, 5 eq.) applying rhodamine 6G (33.5 mg, 0.07 mmol, 10%) and DIPEA (122 μL, 0.7 mmol, 1 eq.). Yield: 75.0 mg (67%). 1H NMR (300 MHz, CDCl3): 9.12 (s, 1H), 8.77 (s, 2H), 6.83–6.75 (m, 1H), 6.35 (dd, J = 3.7, 1.8 Hz, 1H), 6.26–6.19 (m, 1H), 3.69 (s, 3H). 13C NMR (300 MHz, CDCl3): 156.5, 155.4, 127.5, 127.0, 126.0, 110.9, 108.8, 35.3. MS: 159.1125 [M]+.
5-(1H-Pyrrol-2-yl)pyrimidine (12). 5-Bromopyrimidine (111.3 mg, 0.7 mmol) was reacted with pyrrole (245 μL, 3.5 mmol, 5 eq.) applying rhodamine 6G (33.5 mg, 0.07 mmol, 10%) and DIPEA (122 μL, 0.7 mmol, 1 eq.). Yield: 52.7 mg (52%). 1H NMR (300 MHz, DMSO-d6): 11.63 (s, 1H), 9.05 (s, 2H), 8.93 (s, 1H), 7.05–6.97 (m, 1H), 6.82–6.73 (m, 1H), 6.24–6.15 (m, 1H). 13C NMR (300 MHz, CDCl3): 154.9, 151.0, 126.8, 124.4, 121.3, 109.7, 107.8. MS: 146.0718 [MH]+.
Diethyl (4-cyanophenyl)phosphonate (13). 4-Bromobenzonitrile (72.8 mg, 0.4 mmol) was reacted with triethylphosphite (346 μL, 2.0 mmol, 5 eq.) applying rhodamine 6G (19.2 mg, 0.04 mmol, 10%) and DIPEA (68 μL, 0.4 mmol, 1 eq.). Flash column chromatography was performed using a gradient of ethyl acetate in dichloromethane. Yield: 60.9 mg (64%). 1H NMR (300 MHz, CDCl3): 7.92–7.80 (m, 2H), 7.75–7.65 (m, 2H), 4.20–3.95 (m, 4H), 1.27 (t, J = 7.1 Hz, 6H). 13C NMR (300 MHz, CDCl3): 133.9 (d, J = 187.7 Hz), 132.2 (d, J = 9.9 Hz), 132.0 (d, 14.9 Hz), 117.8, 115.9 (d, J = 3.6 Hz), 62.7 (d, J = 6.7 Hz), 16.3 (d, J = 6.3 Hz). MS: 239.0696 [M]+.
Diisopropyl (4-cyanophenyl)phosphonate (14). 4-Bromobenzonitrile (54.6 mg, 0.3 mmol) was reacted with triisopropylphosphite (370 μL, 1.5 mmol, 5 eq.) applying rhodamine 6G (14.4 mg, 0.03 mmol, 10%) and DIPEA (51 μL, 0.3 mmol, 1 eq.). Yield: 62.5 mg (78%). 1H NMR (300 MHz, CDCl3): 7.92–7.79 (m, 2H), 7.72–7.63 (m, 2H), 4.75–4.58 (m, 4H), 1.31 (d, J = 6.2 Hz, 3H), 1.17 (d, J = 6.2 Hz, 3H). 13C NMR (300 MHz, CDCl3): 135.4 (d, J = 188.3 Hz), 132.2 (d, J = 9.8 Hz), 131.9 (d, J = 14.9 Hz), 117.9, 115.6 (d, J = 3.6 Hz), 71.6 (d, J = 5.8 Hz), 24.0 (d, J = 4.1 Hz), 23.8 (d, J = 4.8 Hz). MS: 252.0780 [M]+–CH3.
References
- J. J. Douglas, M. J. Sevrin and C. R. J. Stephenson, Org. Process Res. Dev., 2016, 20, 1134 CrossRef CAS.
- M.-Y. Cao, X. Ren and Z. Lu, Tetrahedron Lett., 2015, 56, 3732 CrossRef CAS.
- M. Reckenthaeler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727 CrossRef CAS.
- R. Trotzki, M. M. Hoffmann and B. Ondruschka, Green Chem., 2008, 10, 767 RSC.
- K. Ohkubo, K. Suga and S. Fukuzumi, Chem. Commun., 2006, 2018 RSC.
- K. Ohkubo, K. Hirose and S. Fukuzumi, Chem. – Eur. J., 2015, 21, 2855 CrossRef CAS PubMed.
- T. Hino, T. Anzai and N. Kuramoto, Tetrahedron Lett., 2006, 47(9), 1429 CrossRef CAS.
- W. Feng, G. Wu, L. Li and N. Guan, Green Chem., 2011, 13, 3265 RSC.
- R. Yuan, S. Fan, H. Zhou, Z. Ding, S. Lin, Z. Li, Z. Zhang, C. Xu, L. Wu, X. Wang and X. Fu, Angew. Chem., Int. Ed., 2013, 52, 1035 CrossRef CAS PubMed.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473 RSC.
- R. Yuan, S. Fan, H. Zhou, Z. Ding, S. Lin, Z. Li, Z. Zhang, C. Xu, L. Wu, X. Wang and X. Fu, Angew. Chem., Int. Ed., 2013, 52, 1035 CrossRef CAS PubMed.
- M. Obst and B. König, Beilstein J. Org. Chem., 2016, 12, 2358 CrossRef CAS PubMed.
- A. Molnar, Chem. Rev., 2011, 111, 2251 CrossRef CAS PubMed.
- C. Barnard, Platinum Met. Rev., 2008, 52(1), 38 CrossRef CAS.
- A. Biffis, M. Zecca and M. Basato, J. Mol. Catal. A: Chem., 2001, 249 CrossRef CAS.
- A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413 RSC.
- R. Thorwirth, A. Stolle and B. Ondruschka, Green Chem., 2010, 12, 985 RSC.
- E. Tullberg, D. Peters and T. Frejd, J. Organomet. Chem., 2004, 689, 3778 CrossRef CAS.
- S. F. Nielsen, D. Peters and O. Axelsson, Synth. Commun., 2000, 30, 3501 CrossRef CAS.
- I. Ghosh and B. König, Angew. Chem., Int. Ed., 2016, 55(27), 7676 CrossRef CAS PubMed.
- D. Cambie, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noel, Chem. Rev., 2016, 116(17), 10276 CrossRef CAS PubMed.
- R. S. Shaikh, S. J. S. Düsel and B. König, ACS Catal., 2016, 6, 8410 CrossRef CAS.
- F. Jafarpour, S. Rahiminejadan and H. Hazrati, J. Org. Chem., 2010, 75, 3109 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |