
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral and non-conjugated fluorescent salen ligands: AIE, anion probes, chiral recognition of unprotected amino acids, and cell imaging applications†
Guangyu
Shen
,
Fei
Gou
,
Jinghui
Cheng
,
Xiaohong
Zhang
,
Xiangge
Zhou
 and
Haifeng
Xiang
*
and
Haifeng
Xiang
*
College of Chemistry, Sichuan University, Chengdu, 610041, China. E-mail: xianghaifeng@scu.edu.cn; Fax: +86-28-8541-2291
First published on 21st August 2017
Abstract
Natural products are usually non-conjugated and chiral, but organic luminescent materials are commonly polycyclic aromatic molecules with extended π-conjugation. In the present work, we combine with the advantages of non-conjugation and chirality to prepare a series of novel and simple salen ligands (41 samples), which have a non-conjugated and chiral (S,S) and (R,R) cyclohexane or 1,2-diphenylethane bridge but display strong blue, green, and red aggregation-induced emission (AIE) with large Stokes shifts (up to 186 nm) and high fluorescence quantum yields (up to 0.35). Through hydrogen and halogen bonds, these flexible salen ligands can be used as universal anion probes and chiral receptors of unprotected amino acids (enantiomeric selectivity up to 0.11) with fluorescence quantum yields up to 0.29 and 0.27, respectively. Moreover, the effects of different chiral bridges on the molecule arrangement, AIE, and anion and chiral recognition properties are also explored, which provide unequivocal insights for the design of non-conjugated chiral and soft fluorescent materials.
Introduction
Chirality is a universal phenomenon throughout nature.1 Not only many biologically active molecules, such as the naturally occurring amino acids and sugars, are chiral, but also many key chemical and biological processes are profoundly influenced by chiral structures. Therefore, chiral recognition2 through fluorescence method have attracted increasing attention due to its unique advantages, including high sensitivity, good selectivity, short response time, real-time and parallel monitoring, cell imaging, and in situ and non-invasive measurement. Up to date, a lot of different fluorescent chiral receptors have been designed and prepared for enantioselective recognition of chiral amines, acids, or modified amino acids. In the literature, however, there are only a few 1,1′-bi-2-naphthol3 (BINOL)- and tetraphenylethylene4 (TPE)-based fluorescent receptors for chiral recognition of unprotected amino acids, because zwitterionic amino acids have not only a poor solubility in organic solvents but also little interaction with organic receptors in water.5Organic luminescent (fluorescent or phosphorescent) materials6 are commonly polycyclic aromatic molecules with extended π-electron conjugation, but these rigid materials might suffer some disadvantages, such as the synthesis difficulty, poor solubility, and fluorescence aggregation-caused quenching7 (ACQ). On the other hand, non-conjugated soft materials that might have a better solubility, lower cost, higher flexibility, lower cytotoxicity, better biocompatibility, and naturally occurring,8 are usually taken for granted as non-emissive materials. Until recently, some non-conjugated materials, such as 1,1,2,2-tetraphenylethane,9 poly[(maleic anhydride)-alt-(vinyl acetate)],10 polyisobutene succinic anhydrides and imides,11 and steroidal saponin digitonin,12 were reported to show amazing ultraviolet (UV) or blue aggregation-induced emission (AIE).9,13 Our research work continuously focus on the synthesis, optical properties, and sensing applications of N,N′-bis(salicylidene)ethylenediamine (salen), a particular class of salicylaldehyde-based bis-Schiff base (Fig. 1), owing to its facile preparation, good stability, and rich optical properties.7b,14 Our recently work demonstrated that step-like salen ligands14g (R-Cn) and tripod-like tri-Schiff bases15 (TSBs, Fig. 1a) linking with non-conjugated long alkyl bridges display strong red-green-blue (RGB) AIE. If chiral (S,S) and (R,R) cyclohexane or 1,2-diphenylethane bridges are employed, it is easy to prepare chiral salen ligands (Fig. 1b). These non-conjugated chiral ligands are widely used as asymmetric catalysts,16 and they are deemed to be non-emissive and used as turn-on fluorescence probes for detecting metal ions.17 Moreover, non-conjugated soft materials might be used as anion probes, because they are flexible and might have multiple and strong intermolecular interactions with anions through hydrogen bonds.15 Particularly, the effects of different chiral bridges on the molecule arrangement, AIE, and chiral recognition properties are still unexplored. Herein, we demonstrate a novel and simple class of colorful chiral salen ligands (41 samples, Fig. 1b) that are linked by a non-conjugated cyclohexane (Cy) or 1,2-diphenylethane (diPh) bridge but have strong AIE and potential applications in cell imaging and recognition of anion and chiral unprotected amino acids.
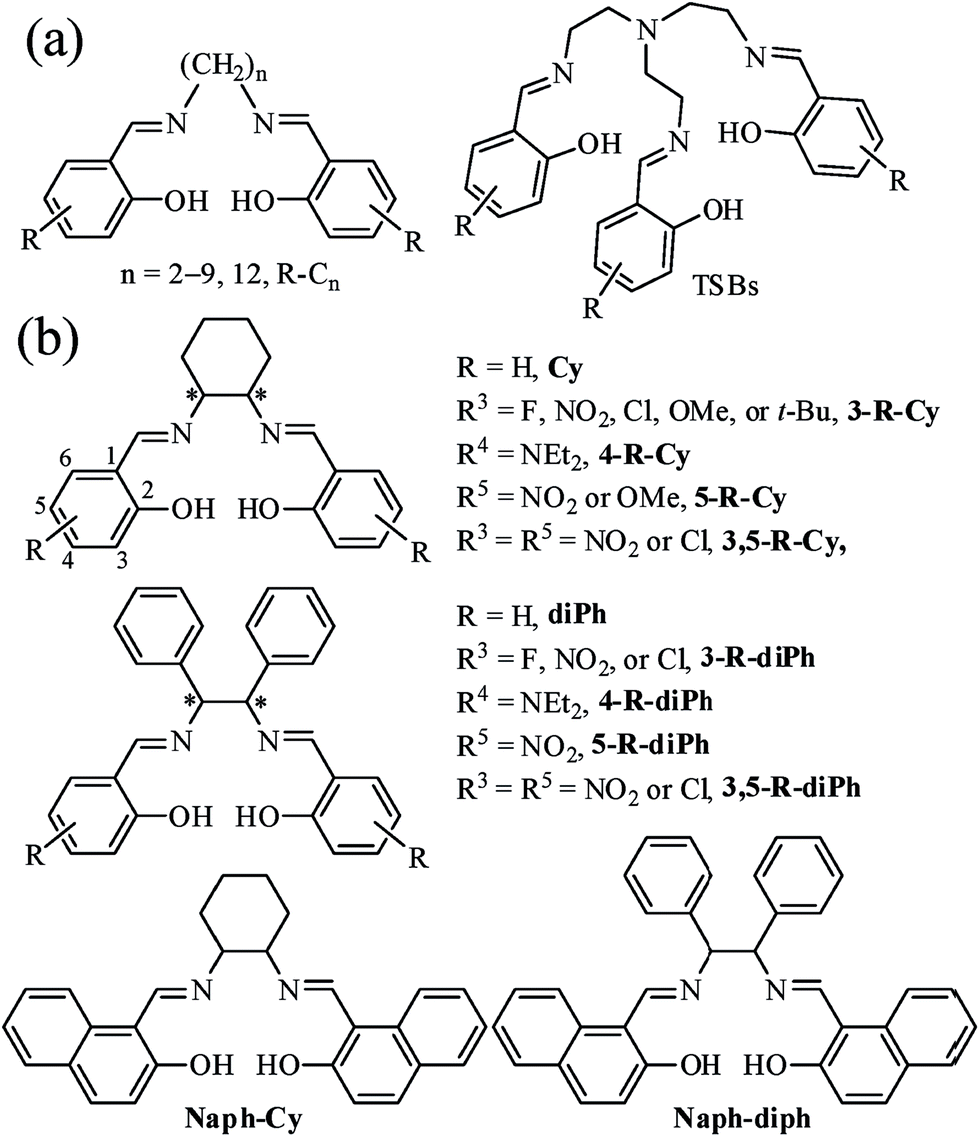 | ||
| Fig. 1 Non-conjugated fluorescent salicylaldehyde-based Schiff bases: (a) step-like R-Cn and tripod-like TSBs in our previous works; (b) chiral V-type Cys and propeller-type diPhs in this work. | ||
Results and discussion
Synthesis and characterization
All salen ligands were reasonably easy to synthesize by the condensation of primary diamine with 2 equivalents of salicylaldehyde precursor in ethanol under refluxing condition.14 Most salen ligands have a bad solubility in petroleum ether, hexane, and water but a good solubility in CH3CN, CH2Cl2, DMF, and DMSO. All salen ligands either in solution or in solid state are stable within several months under air. For most salen ligands, good-quality single crystals can be obtained by the method of slow solvent diffusion/evaporation (CH2Cl2/hexane). For the purpose of comparison, the reference substance C2 (ref. 14g) (Fig. 1a) was prepared as well.Photophysical and AIE properties
The UV/visible absorption and fluorescence data of all synthesized Cys and diPhs at room-temperature are listed Tables S1, S2, Fig. S1, and S2 (in ESI†). Since the photophysical and AIE properties of (S,S), (R,R), and racemic salen ligands are similar, only racemic salen ligands are discussed in this section. As we expected, all salen ligands exhibit weak fluorescence in pure organic solvent of MeCN or DMSO (Tables S1–S3†). For example, racemic 3,5-Cl-Cy shows very weak blue fluorescence (Ф = 0.010) in the dilute MeCN, because the dynamic intramolecular rotations (IRs) of central cyclohexane bridge in 3,5-Cl-Cy molecules provide a possible way to non-radiatively annihilate its excited states and result in the absence of fluorescence consequently. However, if water is added into MeCN, the green fluorescence of 3,5-Cl-Cy is strengthened (Fig. 2), because 3,5-Cl-Cy molecules cannot be dissolved in water, which causes 3,5-Cl-Cy molecules to precipitate and aggregate. At the same time, adding water would red shift its absorption spectrum (Fig. S3†), which further confirms this aggregation. Its green fluorescence reaches the maximum (λem = 511 nm, Ф = 0.064) with a large Stokes shift of 117 nm, when the volume fraction (f) of water is increased to 90%. Furthermore, solid 3,5-Cl-Cy displays strong green-yellow fluorescence (λem = 532 nm, Ф = 0.32) under 360 nm UV lamp (Fig. 2 and 3). All these findings reveal the fact of its AIE nature.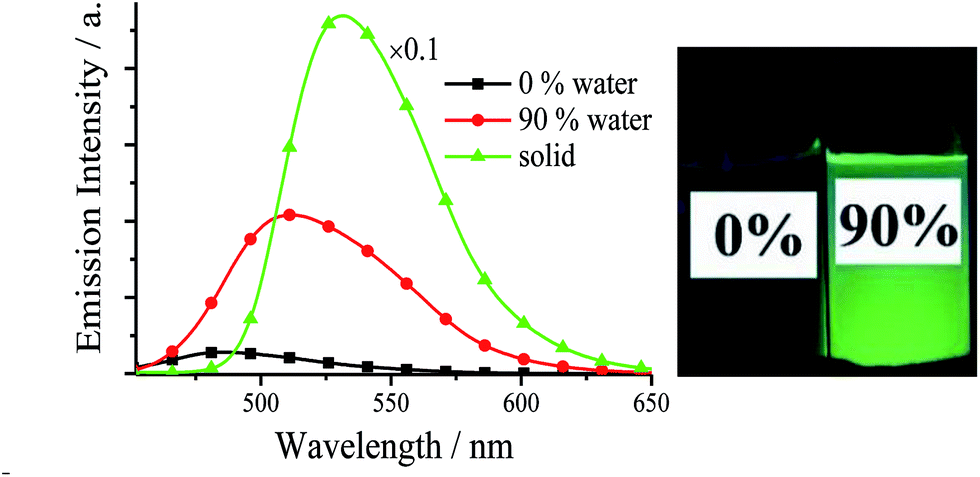 | ||
| Fig. 2 Emission spectra and photographs (under 360 nm UV light) of 3,5-Cl-Cy in MeCN–H2O with different f values (1.0 × 10−5 mol dm−3) and solid. | ||
 | ||
| Fig. 3 Photographs (top: under room light, bottom: under 360 nm UV light) of Cys (a) and diPhs (b) 1 = diPh, 2 = 3-F-(R,R)-diPh, 3 = 3-F-(S,S)-diPh, 4 = 3-Cl-diPh, 5 = 3-Cl-(R,R)-diPh, 6 = 3-Cl-(S,S)-diPh, 7 = 3,5-Cl-diPh, 8 = 3,5-Cl-(R,R)-diPh, 9 = 3,5-Cl-(S,S)-diPh, 10 = 4-NEt2-diPh; 11 = Naph-diPh, 12 = 3-NO2-diPh, 13 = 5-NO2-diPh, 14 = 3,5-NO2-diPh) powders. | ||
All the salen ligands have stronger fluorescence in solid state or water than in organic solvents (Tables S1 and S2†). Most solid samples, such as crystals, powders, and casting films (Fig. S4†), exhibit not only red-shifted but also much stronger fluorescence compared with water samples (Fig. 3, Tables S1, and S2†), and thus we will focus on their solid AIE characteristics. As shown in Fig. 4, Tables S1, and S2,† for solid salen ligands, different substituents have different effects on the emission peaks (λem: 474–565 nm) and colors (blue, green, and red). The presence of a π-extended system (Naph-Cy, λem = 486 nm) to the simplest Cy (λem = 502 nm) leads to a blue shift in fluorescence spectra. On the other hand, electron-donating –OMe (3-OMe-Cy, λem = 523 nm; 5-OMe-Cy, λem = 548 nm) or electron-accepting –F (3-F-Cy, λem = 509), –Cl (3-Cl-Cy, λem = 520 nm; 3,5-Cl-Cy, λem = 532 nm), or –NO2 (3-NO2-Cy, λem = 565 nm; 3,5-NO2, λem = 550 nm) substituents result in red-shifted emission (Fig. 4 and Table S1†). A similar substituent effect was observed for solid diPhs (Fig. 4 and Table S2†), R-Cn,14g and TSBs15 (Fig. 1a) as well, indicating that a substituent has a very obvious effect on λem of these non-conjugated AIE-active materials.
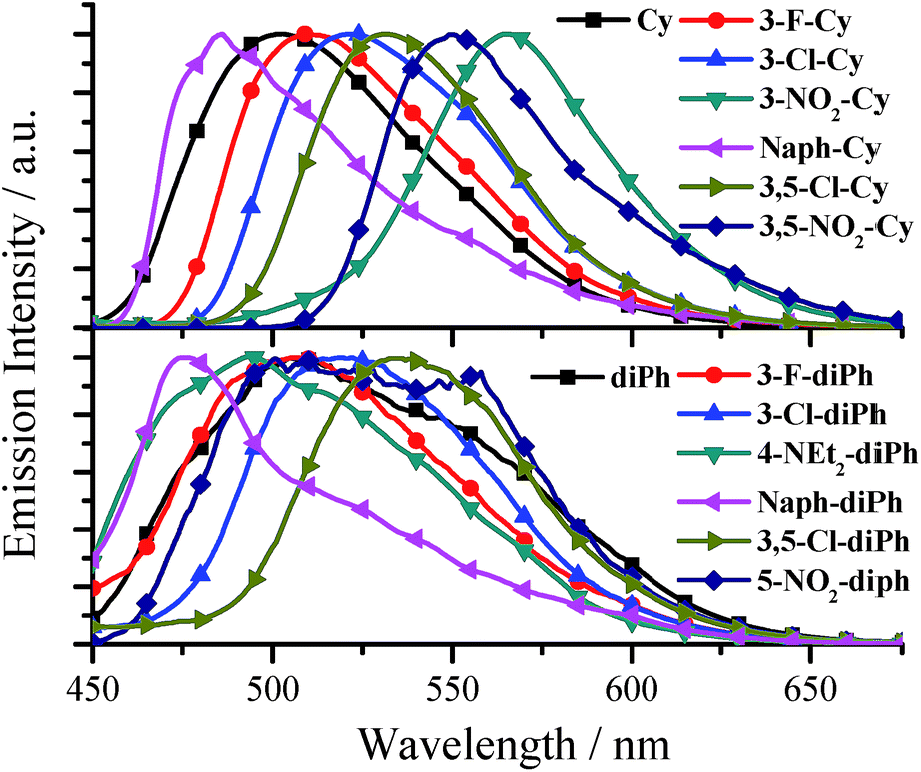 | ||
| Fig. 4 Normalized emission spectra of some Cys and diPhs solids. | ||
All fluorescence quantum yields (Ф) (Tables S1 and S2†) of the aggregated salen ligands in water and solid state were measured by the optical dilute method of Demas and Crosby18 with a standard of quinine sulfate (Фr = 0.55, quinine in 0.05 mol dm−3 sulfuric acid) and an integrating sphere, respectively. Although the salen ligands with a non-conjugated linker have a small π-conjugated system, the fluorescence quantum yields of some salen ligands are unexpected high (up to 0.35 and 0.22 for Cys and diPhs, respectively, Tables S1 and S2†). In general, the introduction of –F (3-F-Cy, Ф = 0.060; 3-F-diPh, Ф = 0.12), –Cl (3-Cl-Cy, Ф = 0.14; 3,5-Cl-Cy, Ф = 0.32; 3-Cl-diPh, Ф = 0.16; 3,5-Cl-diPh, Ф = 0.20) substituents to Cy (Ф = 0.018) or diPh (Ф = 0.10) would improve its fluorescence quantum yield, but the presence of other substituents would bring positive or negative effects on fluorescence quantum yield. Combining with the previous results,14g,15 we can draw a conclusion that chlorination or fluorination is an much more efficient way to improve the fluorescence quantum yields of non-conjugated materials than those of π-conjugated materials.
Mechanism of AIE
The molecular structures and arrangements play a key role in AIE. For most AIE-active materials, they are π-conjugated molecules and stack closely with not only a short interplanar distance (d, for plane molecules14f) or intermolecular aromatic HAr⋯π hydrogen bonds (for non-plane molecules, such as silole19 or TPE20) but also weak intermolecular face-to-face π–π interactions. The former can ensure to eliminate the molecular rotation, the later would prevent the formation of excimer. For non-conjugated AIE-active R-Cn (ref. 14g) and TSBs,15 however, the restriction of C–C single bond rotations in the central alkyl chain bridges through some other non-covalent intermolecular interactions, such as N⋯H, O⋯H, C⋯H, H⋯H, F⋯H, Cl⋯H, and Cl⋯Cl must be considered as well.In order to investigate the effect of molecule arrangements, the X-ray single crystal structures of Cy,213-F-Cy (CCDC 1551151), 3-F-(S,S)Cy (CCDC 1496280), 3-F-(R,R)Cy (CCDC 1496279), 3,5-Cl-Cy,223-NO2-(R,R)Cy,233-F-diPh (CCDC 1551150), and 3-Cl-diPh (CCDC 1551147) are depicted in Fig. 5–7 and S5–S18.† Unlike step-like R-Cn (ref. 14g) and tripod-like TSBs15 molecules, these Cys and diPhs molecules are V- and propeller-type, respectively.
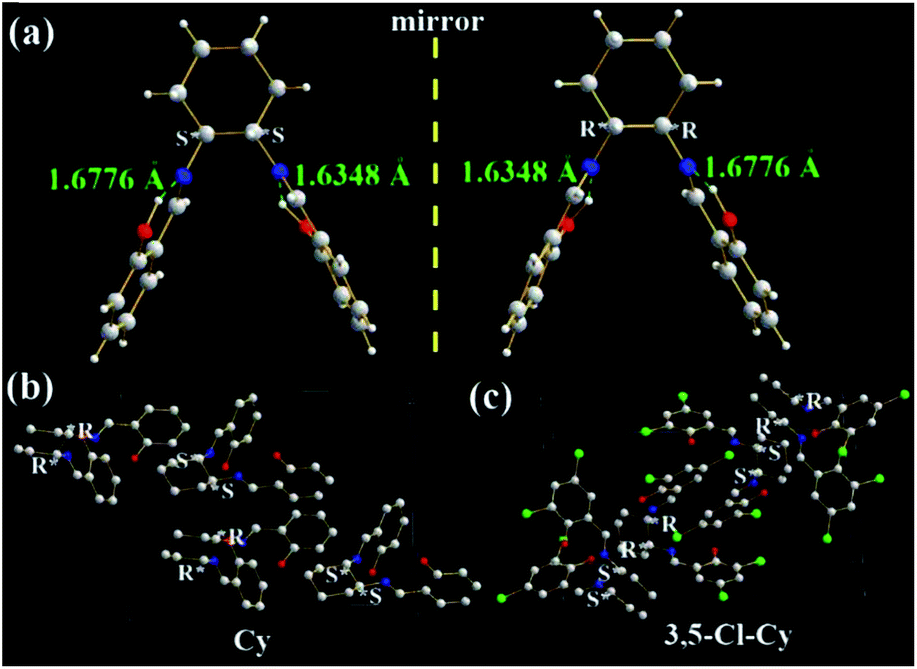 | ||
| Fig. 5 X-ray single crystal structures and packing of Cy and 3,5-Cl-Cy molecules: (a) (S,S) and (R,R) enantiomers of Cy; (b) and (c) packing in a unit cell (H atoms are omitted). | ||
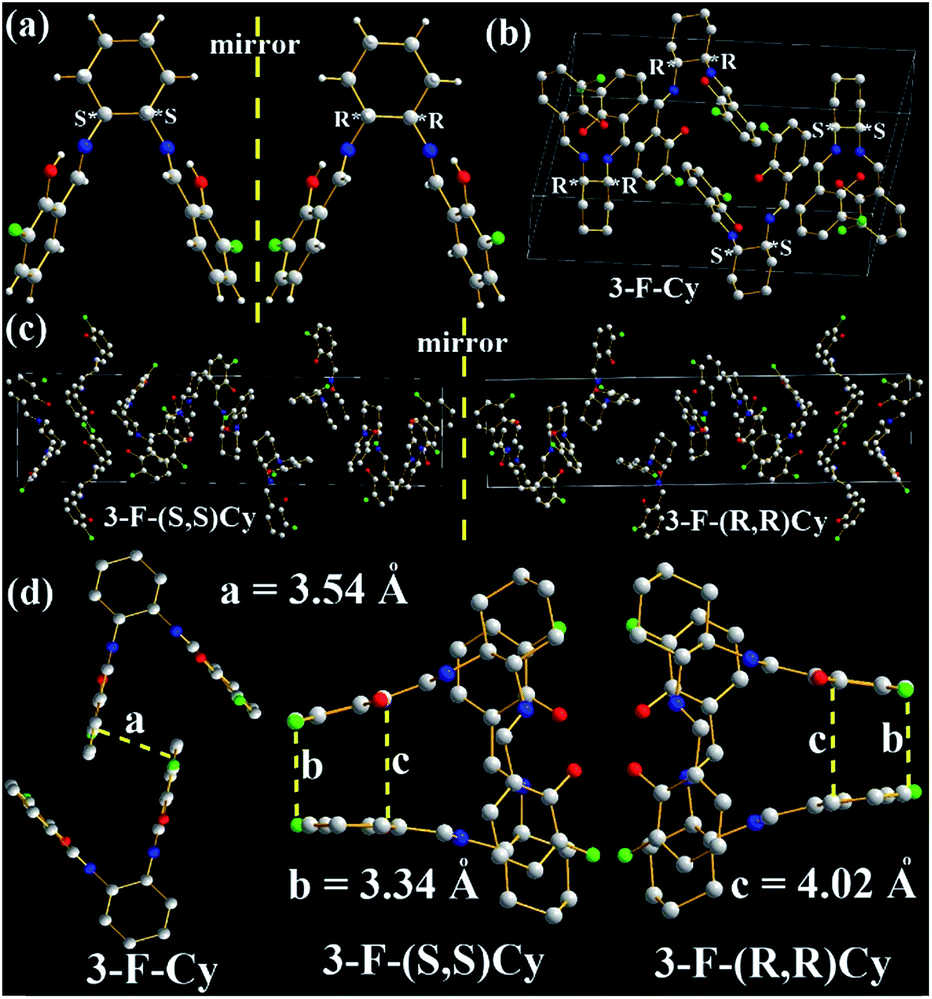 | ||
| Fig. 6 X-ray single crystal structures and packing of 3-F-Cy, 3-F-(S,S)Cy, and 3-F-(R,R)Cy molecules: (a) (S,S) and (R,R) enantiomers; (b) and (c) packing in a unit cell (H atoms are omitted); (d) intermolecular interactions of the two closest molecules (H atoms are omitted). | ||
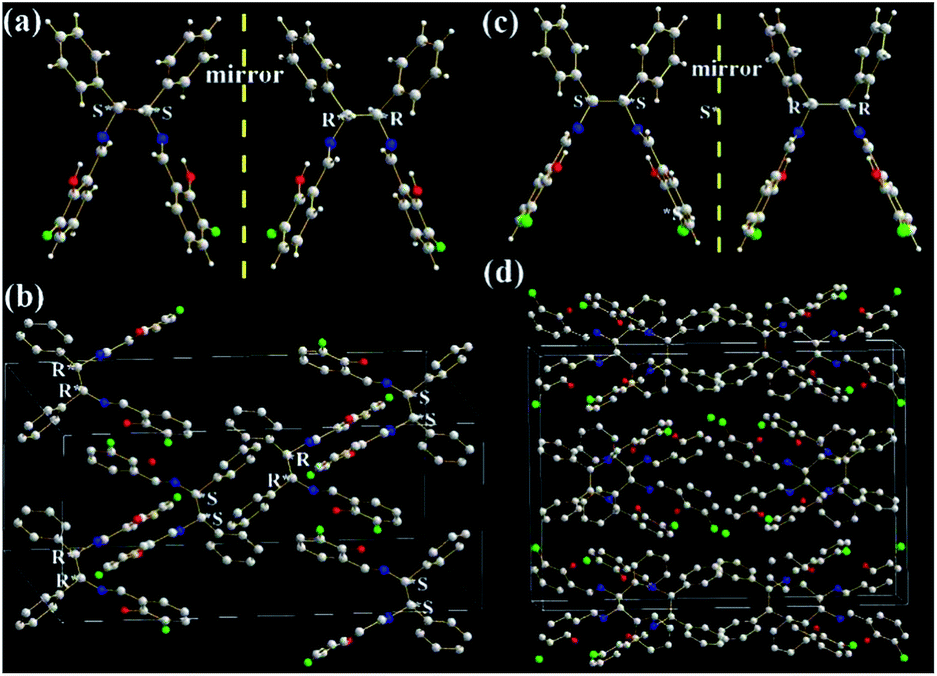 | ||
| Fig. 7 X-ray single crystal structures and packing of 3-F-diPh (a, b) and 3-Cl-diPh (c, d) molecules: (a) and (c) (S,S) and (R,R) enantiomers; (b) and (d) packing in a unit cell (H atoms are omitted). | ||
As expected, two intramolecular N⋯H hydrogen bonds (1.6776 and 1.6348 Å) between H (–OH) and N (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) atoms are found in Cy (Fig. 5a), which would eliminate free rotations of phenol rings. The dihedral angles between two panels of π-conjugated phenol rings are about 56.6°, resulting in its V-type molecular configuration and no intramolecular face-to-face π–π interactions in one Cy molecule. In dilute organic solution, it is obvious that the rotatable C–N and C–C single bonds in the central cyclohexane bridge afford a possible way to non-radiatively annihilate its excited states and quench the fluorescence consequently. In the solid state, however, Cy molecules exhibit a head (cyclohexane)-to-tail (phenol ring) orientation (Fig. 5b). No intermolecular face-to-face π–π interactions (Fig. 5a, b, and S5†) are found between any neighboring Cy molecules. On the contrary, many other strong intermolecular interactions including H⋯H (2.4578, 2.5302, 2.6280, 2.7014, 2.7544, 2.7770, 2.7896, 2.8042, 2.8886, 2.9240, and 2.9792 Å), C⋯H (2.8077 and 2.8296 Å), and O⋯H (2.8111 Å) interactions are found in the two adjacent Cy molecules (Fig. S5†). These above intramolecular and intermolecular interactions would help to restrain the IRs and lead to the presence of AIE consequently. Owing to the existence of chiral carbon atoms, two (S,S) and (R,R) enantiomers (1
N) atoms are found in Cy (Fig. 5a), which would eliminate free rotations of phenol rings. The dihedral angles between two panels of π-conjugated phenol rings are about 56.6°, resulting in its V-type molecular configuration and no intramolecular face-to-face π–π interactions in one Cy molecule. In dilute organic solution, it is obvious that the rotatable C–N and C–C single bonds in the central cyclohexane bridge afford a possible way to non-radiatively annihilate its excited states and quench the fluorescence consequently. In the solid state, however, Cy molecules exhibit a head (cyclohexane)-to-tail (phenol ring) orientation (Fig. 5b). No intermolecular face-to-face π–π interactions (Fig. 5a, b, and S5†) are found between any neighboring Cy molecules. On the contrary, many other strong intermolecular interactions including H⋯H (2.4578, 2.5302, 2.6280, 2.7014, 2.7544, 2.7770, 2.7896, 2.8042, 2.8886, 2.9240, and 2.9792 Å), C⋯H (2.8077 and 2.8296 Å), and O⋯H (2.8111 Å) interactions are found in the two adjacent Cy molecules (Fig. S5†). These above intramolecular and intermolecular interactions would help to restrain the IRs and lead to the presence of AIE consequently. Owing to the existence of chiral carbon atoms, two (S,S) and (R,R) enantiomers (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) are found in the single crystals of racemic Cy (Fig. 5a and b). The R/S-chirality-induced interactions between two enantiomers (Fig. S5†) would help Cy molecules arrange in a tight head-to-tail orientation packing.
:1) are found in the single crystals of racemic Cy (Fig. 5a and b). The R/S-chirality-induced interactions between two enantiomers (Fig. S5†) would help Cy molecules arrange in a tight head-to-tail orientation packing.
Similar two (S,S) and (R,R) enantiomers (1:1) are also found in the single crystals of racemic 3,5-Cl-Cy (Fig. 5c, S6, and S7†), but 3,5-Cl-Cy molecules exhibit a different tail-to-tail orientation from Cy molecules (Fig. 5c). The R/S-chirality of two enantiomers induce their phenol rings to pack together with a d of 3.46 Å (Fig. S6a†) and little intermolecular face-to-face π–π interactions (overlaps in two benzene rings). At the same time, many other strong intermolecular interactions including H⋯H (2.6687, 2.6899, 2.8377, 2.8587, and 2.8978 Å), O⋯H (2.4244, 2.6160, 2.6374, 2.6849 Å), and Cl⋯H (2.8689 and 2.8843 Å) interactions are found in the two adjacent 3,5-Cl-Cy molecules (Fig. S6c, d, and S7†). Moreover, strong Cl⋯Cl interactions (3.2848 Å, Fig. S7c†) are observed, because two benzene rings are packed together. The F and Cl atoms would help to induce intermolecular F⋯H, F⋯F, Cl⋯O, Cl⋯H, or Cl⋯Cl interactions (halogen bond24) that is one possible reason why chlorination or fluorination can improve fluorescence quantum yield (see later discussion). All these data are consistent with the fact that solid 3,5-Cl-Cy and its enantiomers have the highest Ф values (0.32–0.35).
In order to evaluate the effects of different chiral (R,R) and (S,S) bridges on the molecule arrangement, molecular packing of 3-F-Cy, 3-F-(S,S)Cy, and 3-F-(R,R)Cy single crystal structures are shown in Fig. 6 and S8–S15.† Like 3,5-Cl-Cy molecules, racemic 3-F-Cy molecules exhibit a tight R/S-chirality-induced tail-to-tail orientation packing with little intermolecular face-to-face π–π interactions (d = 3.54 Å, Fig. 6a, b, S8, and S9†). Many other strong intermolecular interactions including H⋯H (2.6159, 2.6679, 2.6804, 2.7083, 2.8033, 2.8395, 2.8640, 2.8837, and 2.9128 Å), C⋯H (2.7592, 2.8238, and 2.9546 Å), O⋯H (2.4532, 2.4701, 2.6238, 2.6691, 2.7395, and 2.8609 Å), and F⋯H (2.7008, 2.7167, 2.7771, 2.8422, and 2.8584 Å) interactions are found in the two adjacent 3-F-Cy molecules. The photophysical and AIE properties of 3-F-Cy, 3-F-(S,S)Cy, and 3-F-(R,R)Cy are similar, but 3-F-Cy has a totally different molecule arrangement from 3-F-(S,S)Cy and 3-F-(R,R)Cy (Fig. 6b and c). The molecule arrangements of 3-F-(S,S)Cy and 3-F-(R,R)Cy are similar and mirrored. The two closest molecules in 3-F-(S,S)Cy and 3-F-(R,R)Cy single crystals overlap to form a triplex tail-to-tail, head-to-tail, and head-to-tail arrangement. No intermolecular π–π interactions but many similar strong intermolecular interactions (H⋯H: 2.4496–2.9179 Å, C⋯H: 2.8408–2.9800 Å, O⋯H: 2.3803–2.8672 Å, HAr⋯π: 2.54–2.57 Å, and F⋯H: 2.4464–2.9903 Å) are found in the two adjacent 3-F-(S,S)Cy or 3-F-(R,R)Cy molecules (Fig. S10–S15†). However, there is no intermolecular F⋯F interactions in 3-F-Cy, 3-F-(S,S)Cy, and 3-F-(R,R)Cy, which would be contributed to the fact that their Ф values (0.059–0.062) are lower than that of 3,5-Cl-Cy (0.32).
Our previous work demonstrated that the introduction of strong electron-accepting –NO2 substituents would be an efficient way to red shift fluorescence bands of non-conjugated AIE-active materials.14g,15 In this work, 3-NO2-Cy also shows a red-shifted fluorescence band up to 565 nm (Fig. 4). We failed to grow good-quality single crystals for X-ray analysis in the previous and present work. It is fortunate that the X-ray single crystal structure of 3-NO2-(R,R)Cy is obtained in the literature.23 Unlike the above results, there are strong intermolecular face-to-face π–π interactions in 3-NO2-(R,R)Cy (d = 3.31 Å, Fig. S16†), according with the fact that of 3-NO2-Cy has a red-shifted fluorescence band with a not-so-high Ф value (0.051).
As shown in Fig. 7, two phenol rings and two benzene rings in 3-F-diPh and 3-Cl-diPh molecules are cis form rather than trans form, due to the existence of two chiral (R,R) or (S,S) carbon atoms. Two phenol rings and two benzene rings are separated each other to form their propeller structures, just like another well-known AIE-active material of TPE.9,13,20 Therefore, 3-F-diPh (2.4888–2.8510 Å) and 3-Cl-diPh (2.3507–2.8279 Å) molecules can tightly packed without any face-to-face π–π interactions in their single crystals (Fig. S17 and S18†). It should be noted that strong intermolecular F⋯F (3.2922 Å) and F⋯O (3.1760 Å) interactions are found in 3-F-diPh and 3-Cl-diPh molecules, respectively. All these factors enable solid 3-F-diPh and 3-Cl-diPh have a high Ф value of 0.12 and 0.16, respectively.
Having small π-conjugated systems, these pure organic salen ligands emit strong solid-state emission along with large Stokes shifts (up to 186 nm). This would enable us to doubt that their emission originates from phosphorescence25 rather than fluorescence. Time-resolved emission decay spectra (Fig. S19†) reveal that the emission decay lifetime of 3-F-Cy, 3-F-(S,S)Cy, 3-F-(R,R)Cy, 3-F-diPh, 3-F-(S,S)diPh, and 3-F-(R,R)diPh powders is 0.70, 0.66, 0.66, 1.32, 1.72, and 0.93 ns, respectively. These data are in the range of ns, indicating that their emission is not phosphorescence but fluorescence. The large Stokes shifts might be contributed to the intramolecular hydrogen bonds which would lead to keto and enol tautomers through an ultrafast excited state intramolecular-proton transfer (ESIPT) process.26 With the same R substituent, R-Cy and R-diPh have the similar fluorescence properties, because the low-energy transition is mainly be assigned to the π → π* transition involving molecular orbitals essentially localized on the iminomethylphenol units with little contribution from the non-conjugated cyclohexane and 1,2-diphenylethane bridges (see the later discussion). Moreover, our previous work revealed that both the multi-Schiff base structure and –OH groups are of the greatest importance for AIE.14g
Anion probe properties
As shown in our previous work,15 TSBs (Fig. 1a) are tripod-like side-single-opening cages with three flexible arms of phenol ring and can be used as anion hosts through hydrogen bond. In this work, the two phenol rings of these salen ligands are cis form with a small dihedral angles, and thus they acting as clamps might react with anion through hydrogen bonds and detect anion consequently.In general, (S,S), (R,R), and racemic salen ligands have similar anion responses, and thus racemic salen ligands are discussed in this section. The anion probe properties of Cy, 3-F-Cy, 3-Cl-Cy, 3,5-Cl-Cy, diPh, 3-F-diPh, 3-Cl-diPh, and 3,5-Cl-diPh are demonstrated in Table S3, Fig. 8, and S20–S28.† As example, upon adding many anions, including OH−, F−, Cl−, Br−, I−, NO3−, CO32−, HCO3−, SO42−, S2−, SO32−, P2O74−, PO43−, and HPO42−, to dilute 3,5-Cl-diPh DMSO solution would turn on the blue fluorescence (up to 41.1-fold enhancement) of 3,5-Cl-diPh (Fig. 8a). Alkaline OH− (Fig. S20 and S21†) and neutral F− (Fig. 8) were used to investigate the concentration effect. The fluorescence of 3,5-Cl-diPh exhibits a linear enhancement (correlation coefficient R2 = 0.970, n = 15) upon the addition of 0–40 equivalent of F− and then reaches maximum upon the addition of 100 equivalent of F− (Ф = 0.24). If adding F− further, the fluorescence would reduce and finally reach saturation (Ф = 0.21). A similar fluorescence enhancement is observed upon adding OH− to dilute 3,5-Cl-diPh solution (Ф up to 0.29). The probe performance would be much worse if other solvents, such as MeCN, EtOH, and MeOH, are used.15
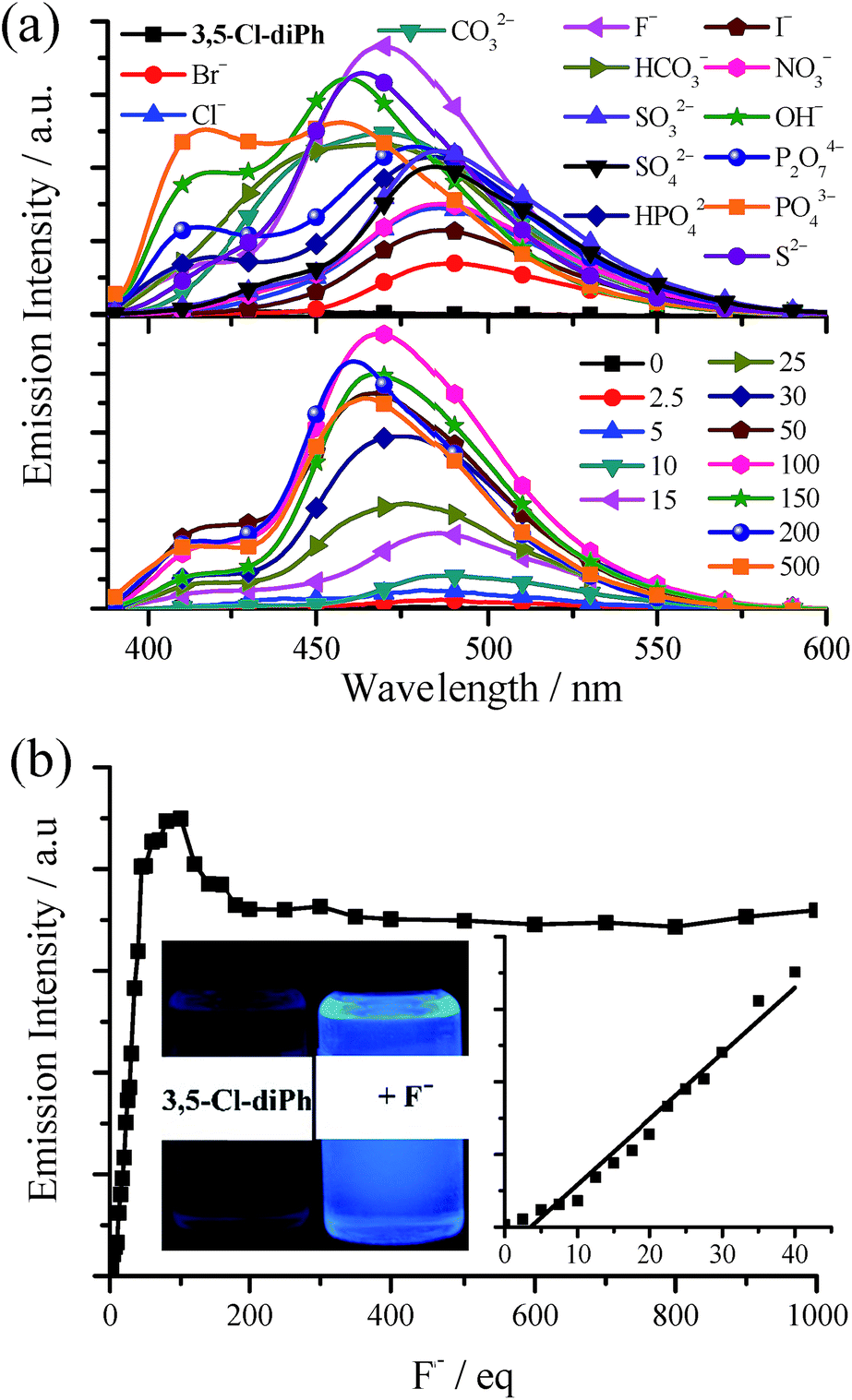 | ||
| Fig. 8 (a) Top: emission spectra of 3,5-Cl-diPh upon the addition of 100 equivalent of different anions. Bottom: emission spectra of 3,5-Cl-diPh upon the addition of different equivalent of F−. (b) Plot of emission intensity of 3,5-Cl-diPh at 468 nm) as a function of F− concentration. Insert: photographs of 3,5-Cl-diPh, 3,5-Cl-diPh + 100 equivalent of F− under 360 nm UV light). In all cases, the probe is 1.0 × 10−5 mol dm−3 in DMSO and is excited at 380 nm. | ||
Cy, 3-F-Cy, 3-Cl-Cy, 3,5-Cl-Cy, diPh, 3-F-diPh, and 3-Cl-diPh have similar anion effects with 3,5-Cl-diPh that they are very sensitive to F−, OH−, S2− and PO43− (Table S3†). On the contrary, step-like C4 (ref. 15) and C2 have much less anion effect on fluorescence (Fig. S29†). Our previous work15 revealed that the reaction between TSBs and X− is not a normal chemical reaction but intermolecular hydrogen bonding (–O–H⋯X−). The two phenol rings of Cy, 3-F-Cy, 3,5-Cl-Cy, 3-F-diPh, and 3-Cl-diPh are cis and diagonal form with a small dihedral angle of 56.6, 51.5, 53.8, 54.6, and 47.5°, respectively. The intramolecular distance O⋯O of Cy, 3-F-Cy, 3,5-Cl-Cy, 3-F-diPh, and 3-Cl-diPh is 6.081, 6.494, 6.352, 5.342, and 5.602 Å, respectively. However, C4 and C2 are step-like and their two phenol rings are parallelly separated with much longer intramolecular distance O⋯O of 8.314 and 10.52 Å, respectively.14g It means that, like TSBs,15Cy, 3-F-Cy, 3,5-Cl-Cy, 3-F-diPh, and 3-Cl-diPh are easier to act as tetradentate chelating agents (see the later discussion) and consequently react with an anion more tightly than C4 and C2 (Fig. S30†).
Chirality and chiral recognition
The chirality properties of 3-F-Cy, 3-F-(S,S)Cy, 3-F-(R,R)Cy, 3-F-diPh, 3-F-(S,S)diPh, and 3-F-(R,R)diPh are examined by circular dichroism (CD) spectra. As depicted in Fig. 9, 3-F-(S,S)diPh shows a positive cotton effect at 326 nm and further strong peaks at 270, and 249 nm. The CD spectrum of 3-F-(R,R)diPh exactly mirrors that of 3-F-(S,S)diPh, indicating that the two compounds are a pair of enantiomers. Moreover, racemic 3-F-diPh has little CD signals. Similar phenomena are observed for 3-F-Cy, 3-F-(S,S)Cy, and 3-F-(R,R)Cy as well. Moreover, 3-F-(S,S)Cy and 3-F-(S,S)diPh (or 3-F-(R,R)Cy and 3-F-(R,R)diPh) exhibit similar CD spectra, revealing that they have the same chiral configuration and energy transition.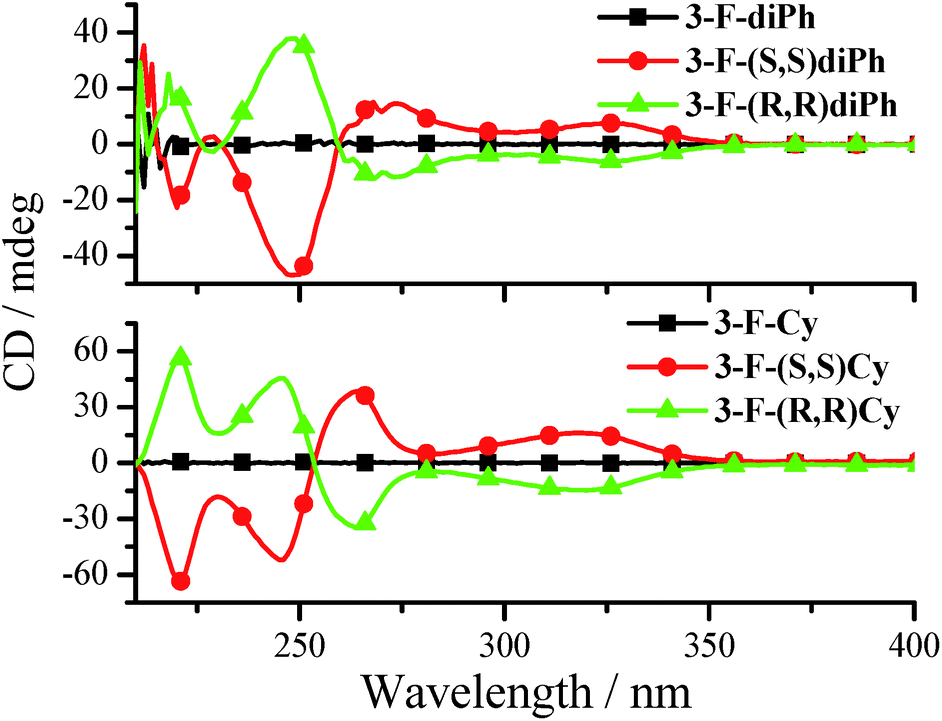 | ||
| Fig. 9 CD spectra of 3-F-Cy, 3-F-(R,R)Cy, 3-F-(S,S)Cy, 3-F-diPh, 3-F-(R,R)diPh, and 3-F-(S,S)diPh in MeCN (3.0 × 10−5 mol dm−3). | ||
The fluorescence responses towards various L- or D-amino acids were investigated under the similar experimental conditions with anion detection (Tables S4–S10, Fig. 10, and S31–S63†). Considering the problem of solubility in both water and DMSO, we chose ten amino acids including alanine (Ala), valine (Val), leucine (Leu), serine (Ser), proline (Pro), histidine (His), tryptophan (Trp), arginine (Arg), glutamate (Glu), and glutamine (Gln). Upon adding amino acids, the fluorescence enhancements of Cy and diPh (Table S4 and Fig. S31†) are lower than those of 3-F-Cy (Table S5 and Fig. S32–S37†), 3-Cl-Cy (Table S6 and Fig. S38–S43†), 3,5-Cl-Cy (Table S7 and Fig. S44–S49†), 3-F-diPh (Table S8 and Fig. S50–S55†), 3-Cl-diPh (Table S9 and Fig. S56–S61†), and 3,5-Cl-diPh (Table S10, Fig. 10, S62, and S63†). Since all nature amino acids are L-type ones, firstly we used 3,5-Cl-(S,S)diPh and 3,5-Cl-(R,R)diPh to probe different L-amino acids. As shown in Fig. 10a and Table S10,† like adding anion, adding different L-amino acids to 3,5-Cl-(S,S)diPh or 3,5-Cl-(R,R)diPh DMSO solutions would turn on their fluorescence (Ф up to 0.26), indicating that they might be used as universal amino acid probes (Fig. 10c, S62, and S63†). On the whole, 3,5-Cl-(S,S)diPh and 3,5-Cl-(R,R)diPh have a similar response that L-Arg, L-Pro, and L-Ala would induce stronger fluorescence enhancements than other amino acids. Moreover, chiral 3,5-Cl-(S,S)diPh and 3,5-Cl-(R,R)diPh could also discriminate the enantiomers of some amino acids (Fig. 10b and Table S10†). For example, adding D- or L-Gln to 3,5-Cl-(S,S)diPh solution, the fluorescence of 3,5-Cl-(S,S)diPh would turn on with a 36.2- and 5.82-fold enhancement, respectively. The selective recognition of chiral molecular enantiomers is related to the enantiomeric fluorescence difference ratio, ef, according to ef = (ID − I0)/(IL − I0), in which I0 represents the fluorescence emission intensity of receptor in the absence of a chiral substrate and ID and IL are the fluorescence intensities in the presence of D- and L-substrates, respectively. 3,5-Cl-(S,S)diPh shows an good ef value of 6.22 for enantioselective recognition of D- and L-Gln. For other amino acids, His (ef = 2.71), Arg (ef = 2.27), and Trp (ef = 2.13) enantiomers could be discriminated by the 3,5-Cl-(S,S)diPh. 3,5-Cl-(R,R)diPh can identify Gln (ef = 2.93) and His (ef = 2.46) enantiomers.
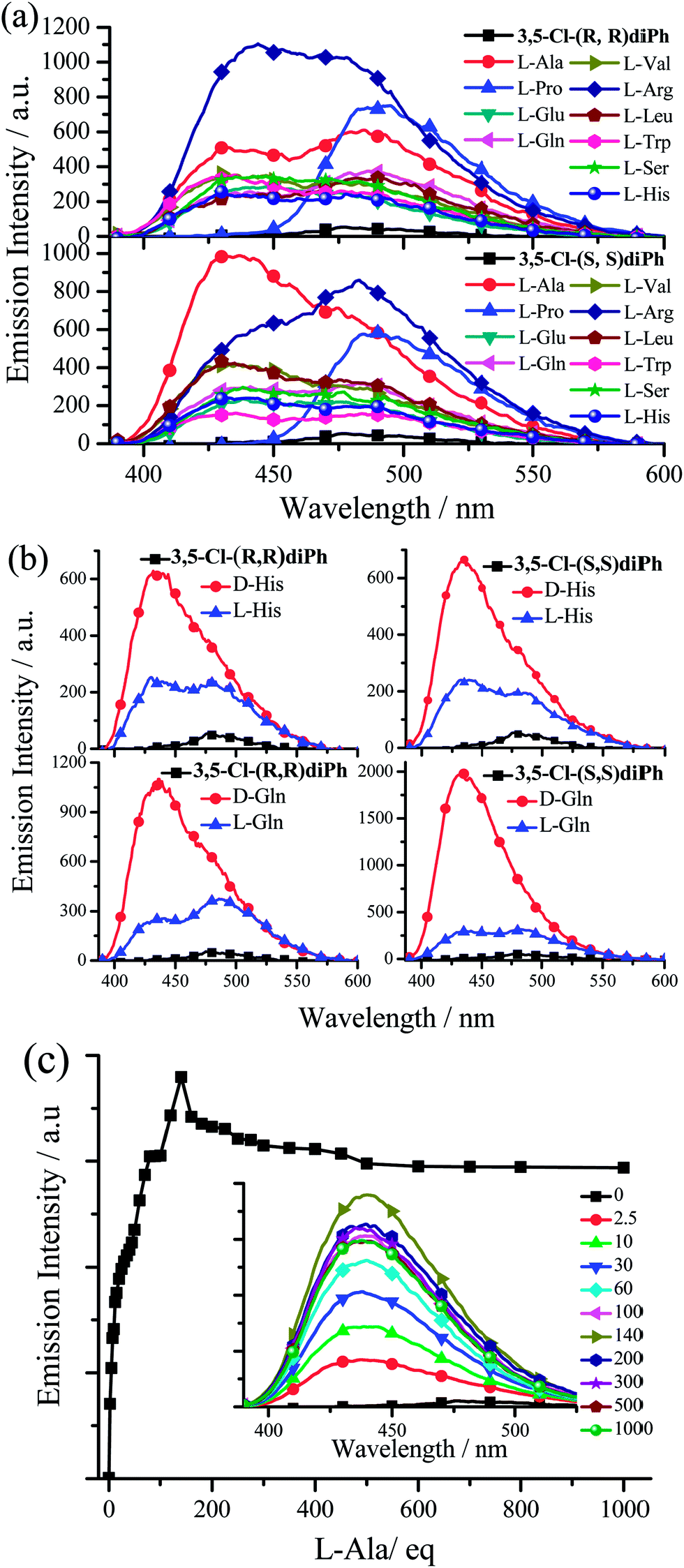 | ||
| Fig. 10 (a) Emission spectra of 3,5-Cl-(S,S)diPh and 3,5-Cl-(R,R)diPh upon the addition of 100 equivalent of different L-amino acids. (b) Emission spectra of 3,5-Cl-(S,S)diPh and 3,5-Cl-(R,R)diPh upon the addition of 100 equivalent of different L-and D-amino acids. (c) Plot of emission intensity of 3,5-Cl-(R,R)diPh at 440 nm as a function of L-Arg concentration. Insert: emission spectra of 3,5-Cl-(R,R)diPh upon the addition of different equivalent of L-Arg. In all cases, the probe is 1.0 × 10−5 mol dm−3 in DMSO and is excited at 380 nm. | ||
In addition, 3-F-(S,S)diPh has an excellent ef value of 0.11 for enantioselective recognition of D- and L-Val (Table S8 and Fig. S54†). 3-F-(R,R)diPh can discriminate D- and L-Arg (ef = 4.89) (Table S9†). 3-Cl-(S,S)diPh can efficiently discriminate Arg (ef = 5.48) and Gln (ef = 0.21) enantiomers, on the other hand, Arg (ef = 4.43) and Trp (ef = 0.18) enantiomers can be can be distinguished by 3-Cl-(R,R)diPh (Table S9†). 3-F-Cy (Table S5†), 3-Cl-Cy (Table S6†), and 3,5-Cl-Cy (Table S7†) generally exhibit worse ef values than 3-F-diPh (Table S8†), 3-Cl-diPh (Table S9†), and 3,5-Cl-diPh (Table S10†). However, since it is too difficult to grow good-quality single crystals from the mixture of the dye and amino acid in DMSO solution, we failed to get a clear insight into the chiral discrimination ability (see the later discussion). We just guess that diPhs have a 1,2-diphenylethane bridge but not a circular bridge of cyclohexane, hence, compared with Cys, they would be more flexible and consequently might have a better ability to form a cavity4a for the recognition of a tiny chiral structure difference between D- and L-amino acid.
Mechanism of anion and amino acid probes
In order to investigate the possible interaction mechanism between the dye and anion/amino acid, 1H nuclear magnetic resonance (1H NMR) analysis was done. As shown in Fig. 11a, the 1H NMR signal of H1 atoms in 3,5-Cl-diPh appears a small broad peak at downfield (δ = 14.5 ppm) due to the existence of the intramolecular hydrogen bonds (–OH⋯N, Fig. 5).15,27 If adding 100 equivalent of OH− (Fig. 11b), F− (Fig. 11c), or L-Arg (Fig. 11d) (in D2O), this downfield peak would disappear and reform a new small sharp peak at upfield (δ = 10.1–10.2 ppm), which might be assigned to the ArOH without intramolecular hydrogen bonds.27,28 This upfield shift indicates that the strong intramolecular hydrogen bonds are destroyed by adding anion/amino acid. At the same time, the reduction (about 1/2–2/3) of peak area in new peak reveals the formation of new intermolecular hydrogen bonds. These intermolecular hydrogen bonds would increase the molecular electron density through-bond effect and lead to upfield shifts of other H atoms consequently.29 Moreover, the 1H NMR signal of H3 at chiral carbon atoms also shows an obvious upfield shift from δ = 5.2 ppm into δ = 4.7, (5.0 and 4.2), and 3.9 ppm upon adding OH−, F−, and L-Arg, respectively, which might indicate that the neighboring nitrogen atoms have strong intermolecular interactions with anion and amino acid through hydrogen and halogen bonds. Furthermore, the blank experiments (Fig. S64 and S65†) revealed that the 1H NMR peak at 10.1–10.2 was not caused by an impurity. | ||
| Fig. 11 1H NMR spectra of 3,5-Cl-diPh (a), 3,5-Cl-diPh + 100 equivalent of OH− (b), 3,5-Cl-diPh + 100 equivalent of F− (c), and 3,5-Cl-(R,R)diPh + 100 equivalent of L-Arg (d) in DMSO-d6. | ||
The mechanisms of the aggregation- and anion/amino acid-induced fluorescence enhancement are totally different. For example, the green AIE of 3-F-Cy is originated from the aggregated state (in water or solid) but its blue anion/amino acid-induced fluorescence is originated from the monomer (1.0 × 10−5 mol dm−3 in DMSO). To gain insight into the nature of the excited states and transitions, gas-phase density functional theory (DFT) and time-dependent-DFT (TD-DFT) were also carried out for 3-F-Cy with the Gaussian 09 program package (B3LYP 6-31G(d,p)). The energy absorption bands of 3-F-Cy (λabs = 316, 253, and 211 nm in MeCN) are reproduced well by the computation, which predict three absorption peaks at 318, 248, and 213 nm (Fig. 12). The lower energy absorption is mainly contributed to the highest occupied molecular orbital (HOMO) → lowest unoccupied molecular orbital (LUMO) (318 nm, oscillator strength, fosc = 0.0814). The energy level and orbital isosurfaces diagrams of 3-F-Cy (Fig. 12) reveal that its HOMO and LUMO are mainly made up of the π-functions of iminomethylphenol units rather than the cyclohexane bridge. Therefore, the lower energy absorption can mainly be assigned to the π → π* transition involving molecular orbitals essentially localized on the iminomethylphenol units with little contribution from the non-conjugated cyclohexane bridge. As mentioned above, F−, OH−, or L-Arg would lead to changing –OH into O−, and thus 3-F-Cy in form of O− is used to evaluate the nature of the anion/amino acid-induced excited states and transitions by calculation. For optimized structure, 3-F-Cy (in form of O−, Fig. 12) has a much bigger dihedral angle (79.0°) between two phenol rings than 3-F-Cy (in form of OH, 51.5°). The lower energy absorption band of 3-F-Cy (in form of O−, λabs = 365 and 350 nm for experiment and computation, respectively) are mainly composed of three transitions, including HOMO → LUMO (360 nm, fosc = 0.150), HOMO−1 → LUMO+1 (344 nm, fosc = 0.175), and HOMO → LUMO+1 (338 nm, fosc = 0.127). The π-functions of HOMO, HOMO−1, LUMO and LUMO+1 of 3-F-Cy in form of OH and O− are similar, which reveals that anion/amino acid would not induce to change of the way of transition (π → π* transition). This is much different from our previous work15 that anion would induce to change of the way of transition from n → π* into π → π* for TSBs. Moreover, anion/amino acid would red shift the lower energy absorption band with an increment of molar extinction co-efficient (Fig. 12 and S66†). This phenomenon can be reasonably explained by the fact that electron-rich ArO− anions have resonance structures of benzoquinone.28 These resonance structures lead the π-function Frontier molecular orbitals to a uniform distribution in the molecule skeleton, and consequently 3-F-Cy (in form of O−) has not only a smaller energy gap of transition but also an increment of molar extinction co-efficient and oscillator strength. This also might be contributed to its high fluorescence quantum yield in dilute DMSO solution. It is strange that dilute 3-F-Cy (in form of O−) DMSO solution having a red-shifted absorption spectrum shows a blue-shifted fluorescence spectrum, but AIE of 3-F-Cy (in form of OH, in solid or water) is green-yellow. There is no anymore ESIPT for 3-F-Cy (in form of O−), which might be contributed to its blue fluorescence.
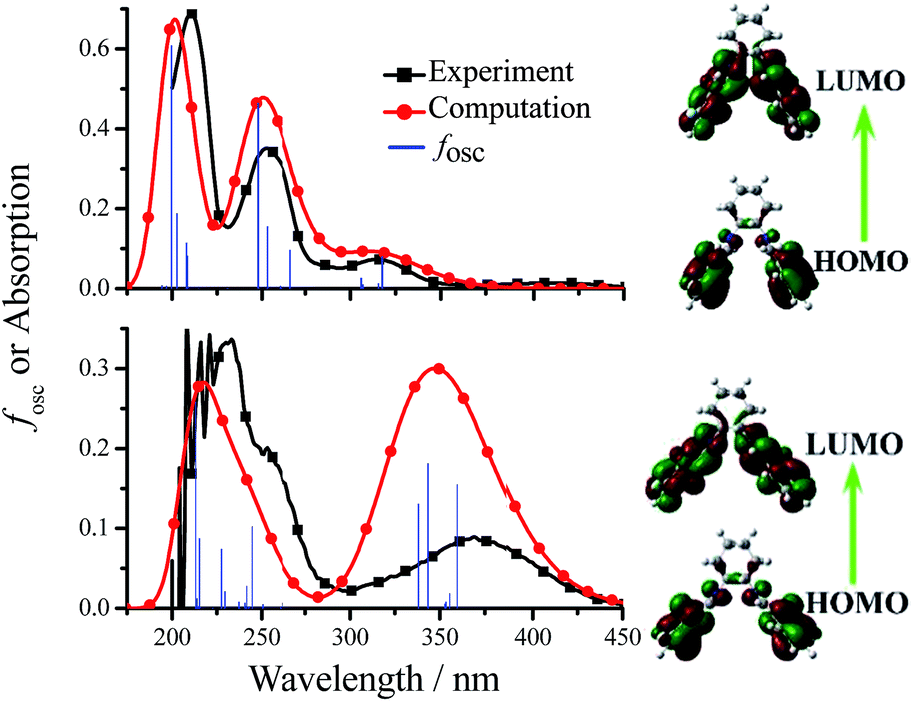 | ||
| Fig. 12 Computational (gas-phase) and experimental absorption spectra and Frontier molecular orbitals of 3-F-Cy (top, in form of OH) and 3-F-Cy + 100 equivalent of OH− (bottom, in form of O−) MeCN solution. | ||
Living cell imaging
These non-conjugated AIE-active materials might have a lower cytotoxicity and better biocompatibility, and thus green-light-emitting 3,5-Cl-Cy and red-light-emitting 3-NO2-Cy were used in cell imaging applications. The HeLa cells were imaged by the dye using a standard cell-staining protocol. As can be seen from Fig. 13, incubated with 3,5-Cl-Cy or 3-NO2-Cy, the HeLa cells grow similar as in the control experiments in the absence of the dyes, indicating that both dyes have little toxicity on the living cells. HeLa cells show negligible background fluorescence. However, intense intracellular green and red AIE is observed after HeLa cells are incubated with 3,5-Cl-Cy or 3-NO2-Cy, respectively. Therefore, these AIE-active materials may have potentially applications in probing or monitoring biologically important processes in vitro as well as in vivo.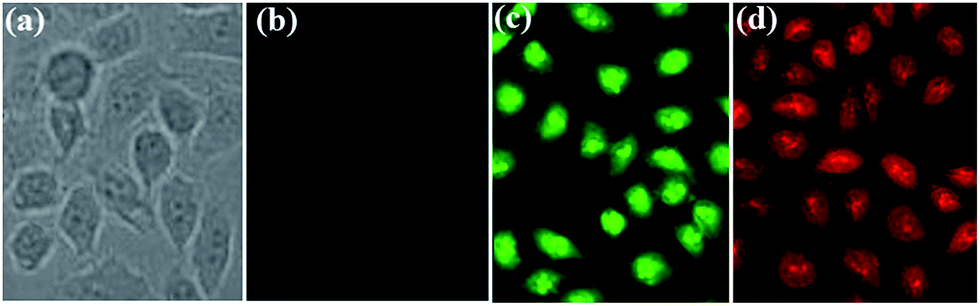 | ||
| Fig. 13 Brightfield (a) incubated without dye and fluorescence images (b) incubated without dye; (c) incubated with 3,5-Cl-Cy; (d) incubated with 3-NO2-Cy of HeLa cells. | ||
Conclusion
In the present work, we demonstrate the unique AIE, chirality, anion/amino acid probe, and cell imaging properties a series of non-conjugated and cyclohexane/1,2-diphenylethane-linked salen ligands. These V- or propeller-type salen ligands with small π-conjugated systems emit strong blue, green, and red AIE through little π–π interactions and strong non-covalent intermolecular interactions, such as O⋯H, H⋯H, C⋯H, F⋯H, F⋯F, Cl⋯H, Cl⋯O, and Cl⋯Cl. Moreover, combine with the advantages of non-conjugation and chirality, these soft materials have multiple and strong interactions with anions and amino acids by hydrogen and halogen bonds, and thus they might be used as universal anion probes and chiral receptors of unprotected amino acids with a good enantiomeric selectivity. Therefore, we believe that these simple salen ligands provide a new paradigm in the design of chiral and non-conjugated fluorescent materials for developing advanced organic optoelectronic devices, florescent bio-probes, and cell imaging, and so on. Further studies on circularly polarized luminescence30 of these salen ligands are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21372169). We acknowledge the comprehensive training platform of the specialized laboratory of the College of Chemistry, Sichuan University, for material analysis.Notes and references
- (a) H. C. Aspinall, Chem. Rev., 2002, 102, 1807 CrossRef CAS; (b) J. Crassous, Chem. Soc. Rev., 2009, 38, 830 RSC; (c) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304 CrossRef CAS.
- (a) L. Pu, Chem. Rev., 2004, 104, 1687 CrossRef CAS PubMed; (b) G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1 CrossRef CAS; (c) A. Zehnacker and M. A. Suhm, Angew. Chem., Int. Ed., 2008, 47, 6970 CrossRef CAS; (d) A. Accetta, R. Corradini and R. Marchelli, Top. Curr. Chem., 2011, 300, 175 CrossRef CAS; (e) L. Pu, Acc. Chem. Res., 2012, 45, 150 CrossRef CAS; (f) D. Leung, S. O. Kang and E. V. Anslyn, Chem. Soc. Rev., 2012, 41, 448 RSC; (g) X. Zhang, J. Yin and J. Yoon, Chem. Rev., 2014, 114, 4918 CrossRef CAS; (h) L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840 CrossRef CAS PubMed.
- (a) X. Yang, X. Liu, K. Shen, C. Zhu and Y. Cheng, Org. Lett., 2011, 13, 3510 CrossRef CAS PubMed; (b) F. Li, L. Li, W. Yang, L. S. Zheng, Z. J. Zheng, K. Jiang, Y. Lu and L. W. Xu, Tetrahedron Lett., 2013, 54, 1584 CrossRef CAS; (c) Z. Huang, S. Yu, K. Wen, X. Yu and L. Pu, Chem. Sci., 2014, 5, 3457 RSC.
- (a) H. T. Feng, X. Zhang and Y. S. Zheng, J. Org. Chem., 2015, 80, 8096 CrossRef CAS; (b) J. B. Xiong, W. Z. Xie, J. P. Sun, J. H. Wang, Z. H. Zhu, H. T. Feng, D. Guo, H. Zhang and Y. S. Zheng, J. Org. Chem., 2016, 81, 3720 CrossRef CAS.
- (a) H. L. Liu, H. P. Zhu, X. L. Hou and L. Pu, Org. Lett., 2010, 12, 4172 CrossRef CAS; (b) H. Jintoku, M. Takafuji, R. Odac and H. Ihara, Chem. Commun., 2012, 48, 4881 RSC; (c) T. Liu, Y. Su, H. Song and Y. Lv, Analyst, 2013, 138, 6558 RSC; (d) B. Aswathy and G. Sony, J. Lumin., 2014, 154, 541 CrossRef CAS.
- (a) U. Mitschke and P. Bauerle, J. Mater. Chem., 2000, 10, 1471 RSC; (b) L. S. Hung and C. H. Chen, Mater. Sci. Eng., A, 2002, 39, 143 CrossRef; (c) J. D. Slinker, J. Rivnay, J. S. Moskowitz, J. B. Parker, S. Bernhard, H. D. Abruoac and G. Malliaras, J. Mater. Chem., 2007, 17, 2976 RSC; (d) Q. Zhao, C. H. Huang and F. Y. Li, Chem. Soc. Rev., 2011, 40, 2508 RSC; (e) J. F. Zhang, Y. Zhou, J. Yoon and J. S. Kim, Chem. Soc. Rev., 2011, 40, 3416 RSC; (f) Y. Feng, J. H. Cheng, L. Zhou, X. G. Zhou and H. F. Xiang, Analyst, 2012, 137, 4885 RSC; (g) H. F. Xiang, J. H. Cheng, X. F. Ma, X. G. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC; (h) Y. M. Yang, Q. Zhao, W. Feng and F. Y. Li, Chem. Rev., 2013, 113, 192 CrossRef CAS; (i) J. H. Cheng, X. G. Zhou and H. F. Xiang, Analyst, 2015, 140, 7082 RSC; (j) J. Liang, B. Z. Tang and B. Liu, Chem. Soc. Rev., 2015, 44, 2798 RSC.
- (a) J. B. Birks, Photophysics of Aromatic Molecules, Wiley, New York, 1970 Search PubMed; (b) X. F. Ma, R. Sun, J. H. Cheng, J. Y. Liu, F. Gou, H. F. Xiang and X. G. Zhou, J. Chem. Educ., 2016, 93, 345 CrossRef CAS.
- B. Z. Tang, New Structural Motifs for Solid Light Emitters, Cutting-Edge Chemistry, ACS, 2016, https://www.acs.org/content/acs/en/pressroom/cutting-edge-chemistry/new-structural-motifs-for-solid-light-emitters.html?cid=home_promo Search PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- C. M. Xing, J. W. Y. Lam, A. Qin, Y. Dong, M. Haussler, W. T. Yang and B. Z. Tang, Polym. Mater. Sci. Eng., 2007, 96, 418 CAS.
- A. Pucci, R. Rausa and F. Ciardelli, Macromol. Chem. Phys., 2008, 209, 900 CrossRef CAS.
- M. S. Mathew, K. Sreenivasan and K. Joseph, RSC Adv., 2015, 5, 100176 RSC.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC; (b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS.
- (a) L. Zhou, P. Y. Cai, Y. Feng, J. H. Cheng, H. F. Xiang, J. Liu, D. Wu and X. G. Zhou, Anal. Chim. Acta, 2012, 735, 96 CrossRef CAS; (b) L. Zhou, Y. Feng, J. H. Cheng, N. Sun, X. G. Zhou and H. F. Xiang, RSC Adv., 2012, 2, 10529 RSC; (c) J. H. Cheng, K. Y. Wei, X. F. Ma, X. G. Zhou and H. F. Xiang, J. Phys. Chem. C, 2013, 117, 16552 CrossRef CAS; (d) J. H. Cheng, Y. H. Zhang, X. F. Ma, X. G. Zhou and H. F. Xiang, Chem. Commun., 2013, 49, 11791 RSC; (e) J. H. Cheng, X. F. Ma, Y. H. Zhang, J. Y. Liu, X. G. Zhou and H. F. Xiang, Inorg. Chem., 2014, 53, 3210 CrossRef CAS; (f) X. F. Ma, J. H. Cheng, J. Y. Liu, X. G. Zhou and H. F. Xiang, New J. Chem., 2015, 39, 492 RSC; (g) J. H. Cheng, Y. X. Li, R. Sun, J. Y. Liu, F. Gou, X. G. Zhou, H. F. Xiang and J. Liu, J. Mater. Chem. C, 2015, 3, 11099 RSC.
- X. H. Zhang, G. Y. Shen, F. Gou, J. H. Cheng, X. G. Zhou and H. F. Xiang, Mater. Chem. Front., 2017, 1, 1041 RSC.
- (a) L. Canali and D. C. Sherrington, Chem. Soc. Rev., 1999, 28, 85 RSC; (b) P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410 RSC; (c) C. Baleizao and H. Garcia, Chem. Rev., 2006, 106, 3987 CrossRef CAS.
- P. S. Hariharan and S. P. Anthony, Spectrochim. Acta, Part A, 2015, 136, 1658 CrossRef CAS.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
- (a) Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951 CrossRef CAS; (b) Y. Yuan, R. T. K. Kwok, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2014, 136, 2546 CrossRef CAS PubMed.
- (a) J. Yang, L. Li, Y. Yu, Z. C. Ren, Q. Peng, S. H. Ye, Q. Q. Li and Z. Li, Mater. Chem. Front., 2017, 1, 91 RSC; (b) C. W. T. Leung, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 62 CrossRef CAS; (c) S. Chen, Y. Hong, Y. Liu, J. Liu, C. W. T. Leung, M. Li, R. T. K. Kwok, E. Zhao, J. W. Y. Lam, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 4926 CrossRef CAS; (d) L. Chen, G. W. Lin, H. R. Peng, S. Y. Ding, W. W. Luo, R. R. Hu, S. M. Chen, F. Huang, A. J. Qin, Z. J. Zhao and B. Z. Tang, Mater. Chem. Front., 2017, 1, 176 RSC; (e) J. Yang, J. Huang, Q. Q. Li and Z. Li, J. Mater. Chem. C, 2016, 4, 2663 RSC.
- J. C. Cannadine, J. P. Corden, W. Errington, P. Moore and M. G. H. Wallbridge, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1014 CrossRef.
- E. Hadjoudis, A. Rontoyianni, K. Ambroziak, T. Dziembowska and I. M. Mavridis, J. Photochem. Photobiol., A, 2004, 162, 521 CrossRef CAS.
- E. Rafii, M. Giorgi, N. Vanthuyne and C. Roussel, ARKIVOC, 2005, 10, 86 Search PubMed.
- (a) P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386 CrossRef CAS; (b) G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772 RSC.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988 RSC.
- V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169 RSC.
- J. H. Cheng, F. Gou, X. H. Zhang, G. Y. Shen, X. G. Zhou and H. F. Xiang, Inorg. Chem., 2016, 55, 9221 CrossRef CAS.
- J. Y. Liu, J. H. Cheng, X. F. Ma, X. G. Zhou and H. F. Xiang, Res. Chem. Intermed., 2016, 42, 5027 CrossRef CAS.
- (a) M. Boiocchi, L. Del Boca, D. E. Gomez, L. Fabbrizzi, M. Licchelli and E. Monzani, J. Am. Chem. Soc., 2004, 126, 16507 CrossRef CAS; (b) Q. Li, Y. Guo, J. Xu and S. J. Shao, Sens. Actuators, B, 2011, 158, 427 CrossRef CAS.
- (a) E. M. Sanchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem.–Eur. J., 2015, 21, 13488 CrossRef CAS PubMed; (b) J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General, materials, computational details, crystallographic information files (CIF), crystallographic data, absorption, and fluorescence emission data. CCDC 3-F-Cy (1551151), 3-F-(S,S)Cy (1496280), 3-F-(R,R)Cy (1496279), 3-F-diPh (1551150), and 3-Cl-diPh (1551147). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08267c |
| This journal is © The Royal Society of Chemistry 2017 |