
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chloroquine exacerbates serum withdrawal-induced G1 phase arrest via an autophagy-independent mechanism†
Lei Gao a,
Hongming Zhu*ab,
Huimin Fan*ab and
Zhongmin Liu*ab
a,
Hongming Zhu*ab,
Huimin Fan*ab and
Zhongmin Liu*ab
aResearch Institute of Heart Failure, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, China. E-mail: liu.zhongmin@tongji.edu.cn; frankfan@tongji.edu.cn; zhm@tongji.edu.cn
bTranslational Medical Center for Stem Cell Therapy, Institute for Regenerative Medicine, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, China
First published on 28th September 2017
Abstract
Chloroquine (CQ) is a widely used anti-malaria or complementary drug in the clinic. However, its effect on ischemic endothelial cells remains unclear. Herein, we showed that serum withdrawal induced G1 phase arrest and autophagy in human umbilical vein endothelial cells. Moreover, CQ exacerbated serum withdrawal-induced G1 phase arrest, whereas autophagy inhibition by Atg5 knockdown did not. Pathway analyses of Akt and MEK1/2/ERK1/2 verified the cell cycle difference between CQ and Atg5 knockdown. Additionally, CQ also exacerbated serum withdrawal-induced G1 phase arrest in the cells with Atg5 knockdown. Through employing glutathione, a reactive oxygen species scavenger, we confirmed that CQ-enhanced intracellular oxidative stress during serum withdrawal aggravated G1 phase arrest. We thus demonstrate that CQ exacerbates serum withdrawal-induced G1 phase arrest via an autophagy-independent, but an oxidative stress-dependent mechanism in endothelial cells.
Introduction
Atherosclerosis is a widespread disease and the incidence rate among the population is approximately 1.7% in the USA.1 Arterial stenosis caused by atherosclerosis largely limits nutrient supply and leads to endothelium damage, which have been found to exacerbate the pathogenesis of many tissue injuries.2 Cancer patients with atherosclerosis are comparatively common,3,4 however, most of the current studies focus on the adverse effects of chemotherapy on critical organs, such as cardiotoxicity and renal toxicity.5,6 The details of chemotherapy drugs affecting diseased endothelial cells, especially those suffering atherosclerosis-induced nutrient deficiency, will thus have a significant appeal.Chloroquine (CQ) is a commonly used anti-malaria drug because of its lysosomotropic and lysosomal proteases inhibitory properties.7 Due to the immunomodulatory function, CQ has also been currently used in the clinic for many other diseases such as rheumatoid arthritis and systemic lupus erythematosus.8 Autophagy is a highly conserved pro-survival process, which cellular components are sequestered into autophagosomes and subsequently transported to lysosomes for recycling under various stress conditions.9 Since CQ also is a classic autophagy blocker, several clinical trials are thus evaluating its beneficial effects for diverse cancers treatment.10 Moreover, autophagy process also plays critical roles in the endothelial turnover and cellular adaptation against stress signals such as nutrient deficiency.11,12 What's interesting though is that CQ has been demonstrated its protective function in the ischemia or ischemia/reperfusion injury of heart and kidney.13,14 To explain these converse results, a recent study reports that CQ can prevent liver from ischemia/reperfusion injury at the early phase, whereas worsen at the late phase.15
However, up to now, few data are reported about the potential effects of CQ on nutrient deficient endothelium, in spite of the high prevalence of cancer patients with atherosclerosis. Collectively, whether and how the CQ treatment affects the nutrient-deficient endothelial cells is still yet undefined, the current study was thus designed.
Materials and methods
Reagents and antibodies
Dulbecco's Modified Eagle Medium (DMEM), trypsin EDTA, penicillin, and streptomycin were purchased from GIBCO BRL (Gaithersburg, USA). Fetal bovine serum (FBS) was purchased from Hyclone (Buckinghamshire, UK). CQ, glutathione (GSH), and N-acetyl cysteine (NAC) were purchased from Sigma-Aldrich (St. Louis, USA). Cell Counting Kit-8 (CCK-8) was procured from Dojindo (Kumamoto, Japan). Cell-Light EdU DNA cell proliferation kit and riboFECT CP Transfection Kit were obtained from RiboBio (Guangzhou, China). The Reactive Oxygen Species Assay Kit and the Cell Cycle and Apoptosis Analysis Kit were obtained from Beyotime Institute of Biotechnology (Jiangsu, China). Primary antibodies against β-actin, LC3B, Atg5, p16, p19, and p21 were purchased from Proteintech (Chicago, USA). Primary antibodies against p53, p-p53, Akt, p-Akt, MEK1/2, p-MEK1/2, ERK1/2, and p-ERK1/2 were purchased from Cell Signaling Technology (Danvers, USA). Secondary antibody was obtained from KPL (Washington, USA).Cell culture
Human umbilical vein endothelial cells (HUVECs), human cervical cancer cells (HeLa), and human embryonic kidney cells (293T) were obtained from the Shanghai Institute of Biochemistry and Cell Biology. These cell lines were maintained in high-glucose DMEM supplemented with 10% FBS, 100 units per ml penicillin and 100 μg ml−1 streptomycin. Cells were incubated in a humidified atmosphere of 5% CO2 at 37 °C.Cell viability analysis
Cell viability was measured using CCK-8 assay as previously reported.16 Briefly, cells were seeded into 96-well plates and incubated overnight. CCK-8 solutions (10 μl) were added into each well after treatment. After 4 h additional incubation, the absorbance was measured at 450 nm with a microplate reader (Dynatech, USA).EdU assay
Cell proliferation was verified by the Cell-Light EdU DNA cell proliferation kit (RiboBio, China) as reported.17 Cells were stained with 50 μM EdU reagent for 2 h at 37 °C. After being fixed with 4% paraformaldehyde for 30 min, cells were washed with 2 mg ml−1 glycine and permeabilized with 0.5% Triton X-100. Cells were reacted with Apollo reaction cocktail (1×) for 30 min, and nucleus was labeled with Hoechst 33342 reagent. Cells were visualized under a fluorescent microscope (Leica, DMI3000 B).Cell cycle analysis
After the treatments, cells were carefully collected and fixed in 70% ethanol overnight at 4 °C. According to the instruction of manufacturer, PI staining reagent (50 mg ml−1 PI and 1 mg ml−1 RNAse in 1 ml of sodium citrate buffer) was prepared and then incubated with samples in the dark at 37 °C for 30 min. Cell cycle distribution was determined by FACScan flow cytometry (BD FACSAria II; BD Co; America), and the data were analyzed using the multicycle program from Phoenix Flow Systems (San Diego, CA).Measurement of intracellular reactive oxygen species (ROS)
The intracellular ROS level was measured via using the Reactive Oxygen Species Assay Kit. Cells were cultured in 6-well plates, collected, and washed with PBS for three times after treatment. Cells were then suspended with serum-free medium containing 10 μM DCFH-DA for 30 min in dark at 37 °C. After being washed with serum-free medium for three times, samples were determined using FACScan flow cytometry (BD FACSAria II; BD Co; America).Small interfering RNA transfection
Double-stranded small interfering RNA for human Atg5 (siAtg5) and scrambled negative control siRNA (siNC) were purchased from RiboBio (Guangzhou, China) and transfected using riboFECT CP Transfection Kit as previously described.18 Cellular levels of the proteins specific for the siRNA transfection were checked by immunoblotting.Adenoviral infection and immunofluorescence analysis
The Adenoviruses harbouring tandem fluorescent mRFP-GFP-LC3 (Ad-tf-LC3) were purchased from the HANBIO (Shanghai, China). HUVECs were infected with adenoviruses at 50 multiplicities of infection for 24 h. Immunofluorescence samples were produced according to previous introduction.19 Images were then captured using the confocal laser microscope (Leica TCS SP5II STED).Western blot analysis
The exact procedures of western blot were carried out as previously described.19 Briefly, protein concentration was quantified using the BCA protein assay kit (Pierce, USA). Protein extracts were separated by 12% SDS-polyacrylamide gel and transferred to polyvinylidene difluoride (PVDF) membrane. PVDF membranes were incubated with specific primary antibodies diluted in TBST buffer at 4 °C. PVDF membranes probed with secondary antibody (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10000) diluted in TBST for 1 h at room temperature. Bands were visualized using the LI-COR system (Odyssey, USA), and quantitated using the Gel-Pro software (Media Cybernetics, USA).
:10000) diluted in TBST for 1 h at room temperature. Bands were visualized using the LI-COR system (Odyssey, USA), and quantitated using the Gel-Pro software (Media Cybernetics, USA).
Statistical analysis
Data are expressed as mean ± standard deviation (SD) of three replicates. Two treatment groups were compared using the two-tailed Student's t-test. Multiple group comparisons were assessed with the One-way ANOVA test and Tukey's multiple comparison test. SPSS 17.0 software was used to analyze all the data. P values < 0.05 were regarded as significant.Results
Non-lethal serum withdrawal induces G1 phase arrest and autophagy in endothelial cells
In the current study, we deprived serum to simulate non-lethal nutrient deficiency condition in vitro, which can also induce autophagy.20 Consistent with previous study,21 serum deprivation significantly decreased cell viability of HUVECs compared with control (Fig. 1A). We then analyzed cell cycle distribution after 24 h serum deprivation, and observed that cells were accumulated at G1 phase (Fig. 1B). A previous study reports that serum deprivation is insufficient to induce autophagy in cardiac myocytes.22 To determine whether serum withdrawal can induce autophagy in endothelial cells, we deprived serum for 24 h, and then evaluated the level of LC3B, an autophagy indicator, and Atg5, an essential protein required for autophagy initiation.7 As shown in Fig. 1C, the expression of Atg5 and LC3B-II were significantly increased when compared with control. The Ad-tf-LC3 can be used to distinguish autophagosomes and autolysosomes, which yellow dots in merged images indicate autophagosomes and red dots indicate autolysosomes, and thus monitor autophagy.7 As shown in Fig. 1D, the Ad-tf-LC3 was overexpressed in control that diffuse cytoplasmic staining was observed, and Ad-tf-LC3 dots were observed after serum deprivation. These results suggest that non-lethal serum withdrawal induces G1 phase arrest and autophagy in endothelial cells. | ||
| Fig. 1 Serum withdrawal decreases cell viability, causes G1 phase arrest, and induces autophagy in HUVECs. (A) Cells were exposed to normal or serum-deprived medium for 24 h. Cell viability was measured by CCK-8 assay. (B) Analysis of cell cycle distribution. Cells were subjected to normal or serum-deprived medium, and cell cycle distribution was then analyzed by flow cytometry. (C) Western blot analysis showed that 24 h serum withdrawal increased the expression of Atg5 and LC3B-II. (D) Cells were transduced with Ad-tf-LC3 for 24 h before serum deprivation, and then exposed to normal or serum-deprived medium for 24 h. Representative images of Ad-tf-LC3 staining (scale bar, 10 μm) and quantitative analysis of autophagosomes (black bars) and autolysosomes (gray bars) were shown (arrows indicate autophagosomes). The values are expressed as mean ± SD of three replicates. *Significantly different from control, *p < 0.05, **p < 0.01. | ||
CQ exacerbates the G1 phase arrest caused by serum withdrawal in endothelial cells
To determine the effect of CQ on G1 phase arrest caused by serum withdrawal, cells were pre-treated with CQ for 2 h, and then subjected to serum-deprived medium, in the absence or presence of CQ. To verify autophagy inhibition effect of CQ, we then performed the LC3 turnover assay.7 As shown in Fig. 2A, CQ could increase the LC3B-II level, and LC3B-II net flux was also significantly increased by serum deprivation compared with control, confirming that serum withdrawal induced autophagy and the autolysosomal degradation was inhibited by CQ during serum withdrawal. Through employing the Ad-tf-LC3, we verified that CQ significantly enhanced the accumulation of autolysosomes and autophagosomes compared with serum deprivation-only group (Fig. 2B). We then performed EdU and CCK-8 assays to study the effect of CQ on the impaired cell proliferation caused by serum withdrawal. As shown in Fig. 2C, serum deprivation significantly decreased the ratio of EdU positive cells comparing with control, and the ratio of EdU positive cells was further decreased after CQ treatment. Consistent with EdU results, the number of cells was reduced by CQ treatment during serum withdrawal (Fig. 2D), which was further confirmed by CCK-8 results (Fig. 2E). In term of cell cycle distribution, the percentage of G1 phase was significantly increased from 66.33% to 74.30% by CQ during serum withdrawal as compared with serum deprivation group (Fig. 2F). p16, p19, and p21 are three critical cyclin dependent kinases inhibitors, which function as negative regulators of cell cycle progression from G1 to S phase.23 As shown in Fig. 2G, only the p19 level was increased by serum deprivation, and further significantly up-regulated in the presence of CQ. Although the expression of p16 and p21 was not altered by serum deprivation, the level of p16 and p21 was markedly increased by CQ during serum deprivation when compared with control. Our results thus suggest that CQ exacerbates the G1 phase arrest caused by serum withdrawal in endothelial cells. | ||
| Fig. 2 CQ exacerbates the G1 phase arrest caused by serum withdrawal in HUVECs. (A) Western blot analysis of LC3B. Cells were subjected to normal or serum-deprived medium for 24 h, in the absence or presence of CQ. LC3B-II net flux was determined by subtracting the densitometric value of LC3B-II in samples non-treated with CQ (LC3B-II − CQ) from the sample treated with CQ (LC3B-II + CQ). (B) Cells were transduced with Ad-tf-LC3 for 24 h prior to treatment. Cells were then exposed to normal or serum-deprived medium for further 24 h, in the absence or presence of 20 μM CQ. Representative images of Ad-tf-LC3 staining (scale bar, 10 μm; arrows indicate autophagosomes, arrowheads indicate autophagosomes) and quantitative analysis of autophagosomes (black bars) and autolysosomes (gray bars) were shown. (C) EdU incorporation assay was analyzed by fluorescent microscope (scale bar, 50 μm). Cells were exposed to normal or serum-deprived medium for 24 h, in the absence or presence of CQ. The EdU incorporation ratio was expressed as the ratio of EdU positive cells to total Hoechst 33342 positive cells. Cells were exposed to normal or serum-deprived medium for 24 h, in the absence or presence of CQ. Next, the morphological images were taken by the inverted contrast phase microscope (D) and cell viability was measured by CCK-8 assay (E). (F) Analysis of cell cycle distribution. Cells were subjected to normal culture medium or serum-deprived medium for 24 h, in the absence or presence of CQ. Cell cycle distribution was analyzed by flow cytometry. (G) Western blot analysis of p16, p19, and p21. Cells were subjected to normal or serum-deprived medium for 24 h, in the absence or presence of CQ. The values are expressed as mean ± SD of three replicates. *Significantly different from control, *p < 0.05, **p < 0.01. #Significantly different from serum deprivation group, #p < 0.05. | ||
Knockdown of Atg5 does not influence the G1 phase arrest caused by serum withdrawal in endothelial cells
To determine whether the effect of CQ is due to autophagy inhibition function, we aimed to search for a genetic method suppressing autophagy to reproduce the phenomenon caused by CQ. To avoid phased influence, mechanistic studies usually inhibit autophagy at the initial phase via using genetic method, such as Atg5 silencing,7 and no genetic methods are currently available to inhibit autophagy similar to CQ. We then suppressed autophagy by employing siRNA to knock down the Atg5, an autophagy-specific gene involved in autophagy initiation stage.24,25 As shown in Fig. 3A, under normal condition, knockdown of Atg5 suppressed the Atg5 expression, whereas did not alter the LC3B-II level. Nevertheless, it is necessary to evaluate autophagy under serum withdrawal condition after genetically inhibiting autophagy.7 Knockdown of Atg5 not only significantly suppressed the expression of Atg5, but also the expression of LC3B-II during serum deprivation compared with serum deprivation-only group, suggesting that autophagy was indeed inhibited (Fig. 3B). We next found that the number of autophagosomes and autolysosomes were decreased after Atg5 knockdown during serum withdrawal (Fig. 3C). However, interestingly, the ratio of EdU positive cells was not altered after genetically inhibiting autophagy during serum withdrawal (Fig. 3D). Additionally, knockdown of Atg5 also did not affect cells morphology and viability (Fig. 3E and F). We then analyzed cell cycle distribution, and observed that 67.5% of cells were arrested at G1 phase after serum deprivation, while 66.46% of cells were arrested at G1 phase by Atg5 knockdown during serum withdrawal (Fig. 3G). As shown in Fig. 3H, knocking down the Atg5 level also did not influence the level of three cyclin dependent kinases inhibitors including p16, p19, and p21. Taken together, our data suggest that knockdown of Atg5 inhibiting autophagy does not influence the G1 phase arrest caused by serum withdrawal in endothelial cells.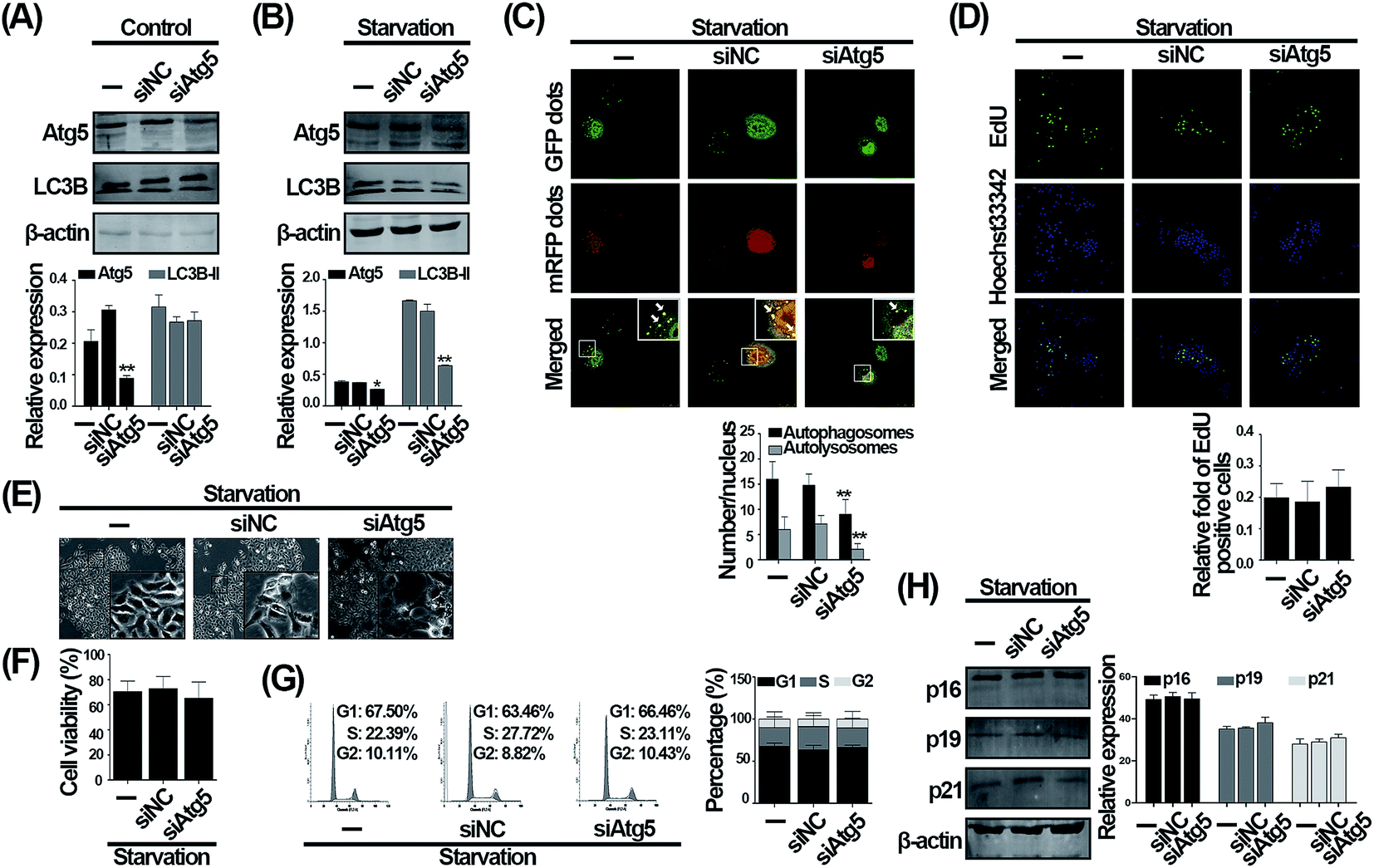 | ||
| Fig. 3 Knockdown of Atg5 does not affect the cell cycle distribution induced by serum withdrawal in HUVECs. Western blot analysis of Atg5 and LC3B. Cells were transfected with negative control (siNC) or Atg5 siRNA (siAtg5) for 48 h. Before cells were harvested, cells were exposed to normal (A) or serum-deprived medium (B) for 24 h. (C) Cells were transduced with Ad-tf-LC3 for 24 h prior to transfection with siNC or siAtg5. After another 48 h, cells were exposed to serum-deprived medium for 24 h. Representative images of Ad-tf-LC3 staining (scale bar, 25 μm; arrows indicate autophagosomes) and quantitative analysis of autophagosomes (black bars) and autolysosomes (gray bars) were shown. The values are expressed as mean ± SD of three replicates. *Significantly different from control, *p < 0.05, **p < 0.01. (D) EdU incorporation assay was analyzed by fluorescent microscope (scale bar, 50 μm). Cells transfected with siNC or siAtg5 for 48 h were exposed to serum-deprived medium for 24 h. The EdU incorporation ratio was expressed as the ratio of EdU positive cells to total Hoechst 33342 positive cells. Cells transfected with siNC or siAtg5 for 48 h were then exposed to serum-deprived medium for 24 h. The morphological images were taken by inverted contrast phase microscope (E) and cell viability was measured by CCK-8 assay (F). (G) Analysis of cell cycle distribution. After transfection with siNC or siAtg5, cells were treated with serum-deprived medium for 24 h. Cell cycle distribution was then analyzed by flow cytometry. (H) Western blot analysis of p16, p19, and p21. Cells transfected with siNC or siAtg5 were treated with serum-deprived medium for 24 h. | ||
CQ-exacerbated G1 phase arrest during serum withdrawal is independent of autophagy status in endothelial cells
The Akt and MEK1/2/ERK1/2 signaling are two critical pathways for cell proliferation. In detail, Akt is phosphorylated by multiple stimuli, while ERK1/2 is phosphorylated by MEK1/2. Subsequently, activated p-Akt and p-ERK1/2 promote the transition from G1 to S phase via activating many downstream molecules.26–28 Besides, nutrient deficiency suppresses the sustained phosphorylation of Akt and MEK1/2/ERK1/2 and thereby accumulates cells at G1 phase,29 suggesting that the accumulation of cells in G1 phase can be mechanistically verified by the activation of Akt, MEK1/2, and ERK1/2. Therefore, to verify that CQ and Atg5 knockdown differently affect serum withdrawal-induced G1 phase arrest, we subsequently evaluated the phosphorylation of these proteins. As shown in Fig. 4A, serum deprivation significantly decreased the ratio of p-Akt/Akt when compared with control, while the ratio of p-Akt/Akt was further decreased by CQ. Similarly, the ratio of p-ERK1/2/ERK1/2 was also significantly decreased by CQ during serum withdrawal, whereas the ratio of p-MEK1/2/MEK1/2 was not altered. These results suggest that CQ exacerbates the suppression of Akt and MEK1/2/ERK1/2 signaling pathways caused by serum withdrawal. However, inhibition of autophagy by Atg5 knockdown did not alter the signaling pathways during serum withdrawal. As shown in Fig. 4B, it seemed that Atg5 knockdown might even increase the ratio of p-Akt/Akt compared with negative control. However, both the ratios of p-MEK1/2/MEK1/2 and p-ERK1/2/ERK1/2 were not altered after Atg5 knockdown. These results thus suggest that inhibition of autophagy by Atg5 knockdown cannot phenocopy the exacerbated effect of CQ on the Akt and MEK1/2/ERK1/2 signaling pathways during serum withdrawal in endothelial cells. To explore CQ's effect on cell cycle distribution of endothelial cells that serum withdrawal-induced autophagy is suppressed, CQ was added into cells with Atg5 knockdown during serum withdrawal. As shown in Fig. 4C, CQ increased the percentage of G1 phase from 62.20% to 71.14% compared with starvation-only group, and the percentage also was not significantly altered when starvation-induced autophagy was suppressed, suggesting that CQ-exacerbated G1 phase arrest during serum withdrawal is independent of cellular autophagy status. Collectively, our results suggest that CQ exacerbates serum withdrawal-induced G1 phase arrest via an autophagy-independent manner in endothelial cells. | ||
| Fig. 4 CQ and knockdown of Atg5 differently affect the Akt and MEK1/2/ERK1/2 signaling pathways during serum withdrawal in HUVECs. (A) Western blot analysis of the Akt and MEK1/2/ERK1/2 signaling pathways. After pretreatment with 20 μM CQ for 2 h, cells were subjected to normal or serum-deprived medium for 24 h, in the absence or presence of CQ. The values are expressed as mean ± SD of three replicates. *Significantly different from control, *p < 0.05, **p < 0.01. #Significantly different from serum deprivation group, #p < 0.05. (B) Western blot analysis of the Akt and MEK/ERK signaling pathways. After transfection with siNC or siAtg5, cells were exposed to serum-deprived medium for 24 h. *Significantly different from starvation-only group, *p < 0.05. #Significantly different from negative control group, #p < 0.05. (C) After transfection with siNC or siAtg5, cells were exposed to serum-deprived medium for 24 h, in the absence or presence of CQ. Flow cytometry was used to analysis cell cycle distribution. | ||
The effect of CQ with different concentrations on cell cycle distribution during serum withdrawal in endothelial cells
To explore whether the effect of CQ on G1 phase arrest is a dose-dependent manner, we treated cells with 10 and 50 μM CQ during serum starved condition. Cells exposed to 50 μM CQ during serum withdrawal showed typical morphological changes of apoptosis, such as cell shrinkage and vacuolation formation,19 while 10 μM CQ did not (Fig. 5A). Moreover, as shown in Fig. 5B, 10 μM CQ did not affect cell cycle distribution in both normal and starved conditions, whereas 50 μM CQ increased the percentage of G1 phase from 47.89% to 61.13% during normal condition, and further increased the percentage from 58.15% to 73.93% during starved condition. Therefore, our results indicate that 20 μM CQ is sufficient to exacerbate serum withdrawal-induced G1 phase arrest in endothelial cells, and a higher CQ concentration possesses the potential cytotoxicity. To examine whether the effect of CQ is specifically occurred in endothelial cells, HeLa and 293T cell lines were employed. As shown in Fig. S1,† the percentage of G1 phase was increased from 75.68% to 85.70% after CQ treatment during serum withdrawal in HeLa cells. Moreover, serum withdrawal-induced G1 phase arrest was not altered after autophagy inhibition, whereas increased from 77.32% to 84.52% after CQ treatment. Conversely, CQ even decreased the percentage from 87.66% to 77.93% during serum withdrawal in 293T cells, and when compared with Atg5 knockdown, CQ treatment did not affect cell cycle distribution. These data suggest that CQ can exacerbate serum withdrawal-induced G1 phase arrest via an autophagy-independent manner in HeLa cells, rather than 293T cells. | ||
| Fig. 5 The effect of CQ with different concentrations on cell cycle distribution in HUVECs. Cells were subjected to normal culture medium or serum-deprived medium for 24 h, in the absence or presence of CQ (10 and 50 μM, respectively). Cell morphology was captured using inverted contrast phase microscope (A). Cell cycle distribution was analyzed by using flow cytometry (B). | ||
CQ and knockdown of Atg5 differently regulate the intracellular oxidative stress status during serum withdrawal in endothelial cells
To investigate the underlying mechanism that CQ-exacerbated G1 phase arrest during serum withdrawal, we evaluated the intracellular oxidative stress status, a critical mechanism causing cell proliferation inhibition in response to extracellular stress.30,31 As compared with control, serum deprivation significantly increased the intracellular ROS generation, and a tendency towards higher ROS generation was seen after CQ treatment during serum withdrawal (Fig. 6A). We next evaluated the ratio of p-p53/p53, which can indicate the intracellular oxidative stress status.32 As shown in Fig. 6B, the ratio of p-p53/p53 was significantly increased by serum deprivation compared with control, and CQ treatment further increased the ratio of p-p53/p53 during serum withdrawal. However, after inhibiting autophagy induced by serum withdrawal via knocking down Atg5, the excessive intracellular ROS level caused by serum deprivation was even reduced (Fig. 6C). Moreover, knockdown of Atg5 to inhibit autophagy also did not alter the ratio of p-p53/p53 during serum withdrawal (Fig. 6D). Our results thus indicate that CQ can exacerbate the intracellular oxidative stress status caused by serum withdrawal in endothelial cells, whereas inhibition of autophagy by Atg5 knockdown does not. | ||
| Fig. 6 CQ and knockdown of Atg5 differently affect the oxidative stress during serum withdrawal in HUVECs. (A) After pretreatment with CQ, cells were exposed to normal or serum-deprived medium for 24 h, in the absence or presence of CQ. The level of intracellular ROS was detected by flow cytometry. (B) Western blot analysis of p53 and p-p53. After pretreatment with CQ, cells were subjected to normal culture medium or serum-deprived medium for 24 h, in the absence or presence of CQ. The values are expressed as mean ± SD of three replicates. *Significantly different from control, *p < 0.05, **p < 0.01. #Significantly different from serum deprivation group, #p < 0.05. (C) Cells transfected with siNC or siAtg5 were exposed to serum-deprived medium for 24 h. The level of intracellular ROS was then detected by flow cytometry. (D) Western blot analysis of p53 and p-p53. Cells were transfected with siNC or siAtg5 and then exposed to serum-deprived medium for 24 h. The values are expressed as mean ± SD of three replicates. The values are expressed as mean ± SD of three replicates. *Significantly different from control, **p < 0.01. #Significantly different from serum deprivation group, #p < 0.05. | ||
GSH attenuates the exacerbated G1 phase arrest caused by CQ during serum withdrawal in endothelial cells
To further confirm that the exacerbated G1 arrest caused by CQ is mediated by the excessive ROS generation, we firstly used GSH to scavenge ROS.33 As shown in Fig. 7A, we found that the increased ROS caused by CQ during serum withdrawal, was significantly reduced after GSH treatment. However, since GSH can be broken down into amino acids, which may indirectly resupply nutrients to cells, we then used NAC to scavenge intracellular ROS to ensure that the effect is not due to amino acids resupply.34 As shown in Fig. 7B, we found that NAC also significantly decreased the excessive ROS caused by CQ during serum withdrawal. Additionally, we found that the reduced cell viability caused by serum deprivation, in the absence or presence of CQ, were significantly increased by GSH, while CQ treatment did not significantly alter cell viability after GSH treatment during serum withdrawal (Fig. 7C). Furthermore, GSH reduced the percentage of G1 phase arrest caused by serum withdrawal, especially in the presence of CQ. As shown in Fig. 7D, the percentage of G1 phase was 61.61% for serum deprivation group, while only 56.07% of cells were arrested at G1 phase after GSH treatment. Additionally, the percentage of G1 phase was 70.19% for CQ-treated serum deprivation group, while only 51.06% of cells were arrested at G1 phase after GSH treatment. Taken together, these results suggest that the exacerbated G1 phase arrest caused by CQ during serum withdrawal was mediated by the intracellular ROS generation in endothelial cells.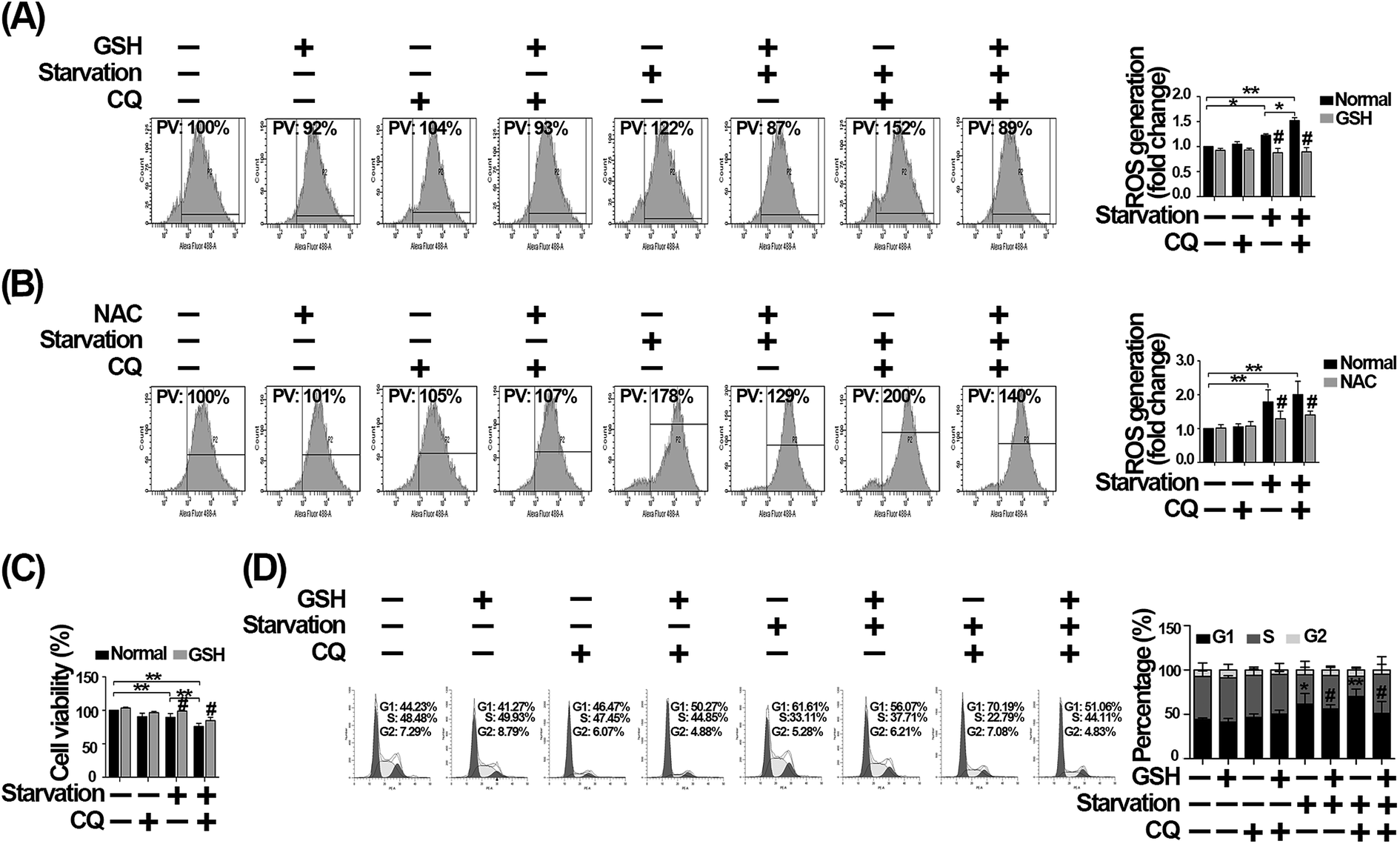 | ||
| Fig. 7 GSH attenuates G1 phase arrest caused by CQ during serum withdrawal in HUVECs. (A) Cells were treated with 10 mM GSH and CQ for 2 h, and then exposed to normal or serum-deprived medium for 24 h, in the absence or presence of GSH and CQ. PV: percentage values. (B) After pretreatment with 10 μg ml−1 NAC and CQ for 2 h, cells were exposed to normal or serum-deprived medium for 24 h, in the absence or presence of NAC and CQ. Cells were then collected, and the intracellular ROS level was detected by flow cytometry. (C) Cell viability was measured by CCK-8 assay. After pretreatment with GSH and CQ, cells were exposed to normal or serum-deprived medium for 24 h, in the absence or presence of GSH and CQ. (D) Analysis of cell cycle distribution. Cells were subjected to normal culture medium or serum-deprived medium for 24 h after pretreatment with GSH and CQ for 2 h. Cell cycle distribution was analyzed by flow cytometry. The values are expressed as mean ± SD of three replicates. *Denotes a statistical significance, *p < 0.05, **p < 0.01. #Significantly different from treatment without GSH or NAC, #p < 0.05. | ||
Discussion
In the present study, we demonstrate that CQ exacerbates serum withdrawal-induced G1 phase arrest via an autophagy-independent mechanism in endothelial cells, which the underlying mechanism is largely ascribed to the CQ-caused increased intracellular oxidative stress during serum withdrawal.Maintenance of adequate nutrient supply in endothelial cells is vital for their functions, and ischemia-induced nutrient shortages can lead to the cell cycle arrest via altering the intracellular homeostasis.35 Although several recent studies have reported the promising effect of CQ on the endothelial-dependent vascular relaxation via increasing the release of nitric oxide in endothelial cells,21,36 the efficiency of CQ application over the ischemic diseases is still largely controversial.13–15 The effect of CQ on the cell cycle arrest caused by nutrient deficiency in endothelial cells and the underlying mechanisms have not been fully characterized. Herein, we demonstrate that serum withdrawal can induce G1 phase arrest in endothelial cells, while CQ application even exacerbates the cell cycle arrest during serum withdrawal. Considering the fact that CQ also functions as an autophagy blocker,7 whereas autophagy usually is a protective mechanism for cells during nutrient deficiency.37 We thus question that whether the exacerbated G1 phase arrest caused by CQ during serum deprivation is due to its autophagy inhibitory mechanism. However, to our knowledge, there are currently no suitable genetic methods to inhibit autophagy at a late stage like CQ.38 Considering the fact that regardless of autophagy inhibition at early or late stage, autophagy-dependent small molecules recycling effect is invariably inhibited,7 we thus genetically suppressed autophagy at an early stage via employing siRNA against Atg5, an autophagy-specific gene involved in autophagy initiation stage.7 Through employing Atg5 siRNA to inhibit autophagy, although serum withdrawal-induced autophagy was suppressed, the cell cycle distribution was not significantly altered. Interestingly, a recent study has reported that the chemotherapy sensitizing effect of CQ can be independent of its autophagy inhibition function,11,39 while one another study also shows that CQ-reduced vascular sprouting is also autophagy independent via using the spheroid sprouting model.25 Our study thus demonstrate that CQ can exacerbate serum withdrawal-induced G1 arrest in endothelial cells via an autophagy-independent mechanism, although it is a classic autophagy inhibitor. Additionally, we also observed that the similar phenomenon could occur in HeLa cells, but not in 293T cells. Collectively, these evidences thus remind us that autophagy-independent G1 phase arrest effect of CQ should be considered in future studies due to the high prevalence of CQ-based application and study.
It has been well established that nutrient deprivation can arouse the intracellular ROS generation40 and the ratio of p-p53/p53, which in turn will lead to the cell cycle arrest.41 Consistent with these reports, we found that serum withdrawal could arouse an increase in intracellular oxidative stress evidenced by an up-regulated intracellular ROS generation and the ratio of p-p53/p53.42 In addition, we found that CQ could further promote a dramatic increase in intracellular oxidative stress during serum withdrawal, whereas Atg5 knockdown did not (Fig. 6). To confirm that the CQ-caused cell cycle arrest is triggered by oxidative stress, GSH, a ROS scavenger, was next employed.33 After pretreatment with 10 mM GSH for 2 h, cells were subjected to CQ treatment in the presence of GSH during serum withdrawal. As result, ROS removal by GSH significantly appeased CQ-exacerbated G1 phase arrest, suggesting that excessive ROS generation by CQ is the critical trigger exacerbating G1 phase arrest (Fig. 7). Therefore, our data suggest that the exacerbated G1 phase arrest caused by CQ during serum withdrawal is largely attributed to the enhanced intracellular oxidative stress in endothelial cells. In contrast to the augmentation effect of CQ on starvation-induced excessive ROS generation, autophagy inhibition by Atg5 knockdown could even reduce the excessive ROS level in endothelial cells (Fig. 6), which is consistent with previous report.43 It has been established that extracellular stressors can increase the intracellular ROS generation via promoting the degradation of catalase, an important endogenous ROS scavenger, whereas autophagy inhibition by Atg5 knockdown can prevent the catalase degradation, and thus decrease ROS generation.43,44 Similarly, several studies have also reported that serum deprivation can induce excessive ROS generation via promoting the degradation of many molecules, such as GSH and mitochondrial ROS modulator 1.45,46 Altogether, we thus speculate that siRNA against Atg5 may reduce ROS level via preventing the degradation of intracellular antioxidants during nutrient deficiency. Nevertheless, additional in vivo studies are welcome to define the autophagy-independent effects of CQ application on ischemic endothelium.
In summary, our results demonstrate that CQ exacerbates the G1 phase arrest caused by serum withdrawal via an autophagy-independent, but an intracellular oxidative stress-mediated mechanism in endothelial cells.
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by National Basic Research Program of China (973 Program, 2014CB965103), Shanghai Sailing Program (16YF1409500), National Natural Science Foundation of China (81600282), Health industry Project of Pudong Health Bureau of Shanghai (PW2013E-1), and the Shanghai Municipal Health and Family Planning Commission (ZY3-LCPT-2-1003).References
- J. C. Wang and M. Bennett, Circ. Res., 2012, 111, 245–259 CrossRef CAS PubMed.
- N. Jackson, D. Atar, M. Borentain, G. Breithardt, M. van Eickels, M. Endres, U. Fraass, T. Friede, H. Hannachi, S. Janmohamed, J. Kreuzer, M. Landray, D. Lautsch, C. Le Floch, P. Mol, H. Naci, N. J. Samani, A. Svensson, C. Thorstensen, J. Tijssen, V. Vandzhura, A. Zalewski and P. Kirchhof, Eur. Heart J., 2016, 37, 747–754 CrossRef PubMed.
- A. Matyszewski, A. M. Czarnecka, P. Stachowiak, M. Nowakowska, Z. Kornacewicz-Jach, J. D. Kasprzak and C. Szczylik, Future Oncol., 2017, 13, 593–602 CrossRef CAS PubMed.
- B. Melichar, H. Kalabova, L. Ungermann, L. Krcmova, R. Hyspler, M. Kasparova, M. Pecka, V. Sramek, H. Prochazkova-Studentova, A. Svobodnik, L. Pecen and D. Solichova, Anticancer Res., 2012, 32, 4077–4084 CAS.
- M. de Forni and J. P. Armand, Curr. Opin. Oncol., 1994, 6, 340–344 CrossRef CAS PubMed.
- I. Sekine, K. Yamada, H. Nokihara, N. Yamamoto, H. Kunitoh, Y. Ohe and T. Tamura, Cancer Sci., 2007, 98, 1408–1412 CrossRef CAS PubMed.
- N. Mizushima, T. Yoshimori and B. Levine, Cell, 2010, 140, 313–326 CrossRef CAS PubMed.
- A. Lesiak, J. Narbutt, A. Sysa-Jedrzejowska, J. Lukamowicz, D. P. McCauliffe and A. Wozniacka, Lupus, 2010, 19, 683–688 CrossRef CAS PubMed.
- Y. Ohsumi, Cell Res., 2014, 24, 9–23 CrossRef CAS PubMed.
- J. Sotelo, E. Briceno and M. A. Lopez-Gonzalez, Ann. Intern. Med., 2006, 144, 337–343 CrossRef CAS PubMed.
- P. Maycotte, S. Aryal, C. T. Cummings, J. Thorburn, M. J. Morgan and A. Thorburn, Autophagy, 2012, 8, 200–212 CrossRef CAS PubMed.
- G. R. De Meyer, M. O. Grootaert, C. F. Michiels, A. Kurdi, D. M. Schrijvers and W. Martinet, Circ. Res., 2015, 116, 468–479 CrossRef CAS PubMed.
- T. Fazekas and L. Szekeres, Acta Physiol. Hung., 1988, 72, 191–199 CAS.
- Z. Todorovic, B. Medic, G. Basta-Jovanovic, S. Radojevic Skodric, R. Stojanovic, B. Rovcanin and M. Prostran, PLoS One, 2014, 9, e92673 Search PubMed.
- H. Fang, A. Liu, U. Dahmen and O. Dirsch, Cell Death Dis., 2013, 4, e694 CrossRef CAS PubMed.
- L. Cao, C. Chen, H. Zhu, X. Gu, D. Deng, X. Tian, J. Liu and Q. Xiao, Oncotarget, 2016, 7(32), 51865–51874 Search PubMed.
- T. Guo, W. Wang, H. Zhang, Y. Liu, P. Chen, K. Ma and C. Zhou, PLoS One, 2011, 6, e22387 CAS.
- C. Ye, N. X. Sun, Y. Ma, Q. Zhao, Q. Zhang, C. Xu, S. B. Wang, S. H. Sun, F. Wang and W. Li, FEBS Lett., 2015, 589, 702–709 CrossRef CAS PubMed.
- L. Gao, Y. Wang, Z. Xu, X. Li, J. Wu, S. Liu, P. Chu, Z. Sun, B. Sun, Y. Lin, J. Peng, G. Han, S. Wang and Z. Tang, Apoptosis, 2015, 20, 1636–1650 CrossRef CAS PubMed.
- S. Steiger-Barraissoul and A. Rami, Apoptosis, 2009, 14, 1274–1288 CrossRef CAS PubMed.
- C. R. Pestana, J. C. Oishi, H. S. Salistre-Araujo and G. J. Rodrigues, Cell. Physiol. Biochem., 2015, 37, 1168–1177 CrossRef CAS PubMed.
- Y. Matsui, H. Takagi, X. Qu, M. Abdellatif, H. Sakoda, T. Asano, B. Levine and J. Sadoshima, Circ. Res., 2007, 100, 914–922 CrossRef CAS PubMed.
- S. Gogolin, V. Ehemann, G. Becker, L. M. Brueckner, D. Dreidax, S. Bannert, I. Nolte, L. Savelyeva, E. Bell and F. Westermann, Cell Cycle, 2013, 12, 1091–1104 CrossRef CAS PubMed.
- S. Luo and D. C. Rubinsztein, Cell Death Differ., 2007, 14, 1247–1250 CrossRef CAS PubMed.
- H. Maes, A. Kuchnio, A. Peric, S. Moens, K. Nys, K. De Bock, A. Quaegebeur, S. Schoors, M. Georgiadou, J. Wouters, S. Vinckier, H. Vankelecom, M. Garmyn, A. C. Vion, F. Radtke, C. Boulanger, H. Gerhardt, E. Dejana, M. Dewerchin, B. Ghesquiere, W. Annaert, P. Agostinis and P. Carmeliet, Cancer Cell, 2014, 26, 190–206 CrossRef CAS PubMed.
- B. D. Manning and L. C. Cantley, Cell, 2007, 129, 1261–1274 CrossRef CAS PubMed.
- T. Yamamoto, M. Ebisuya, F. Ashida, K. Okamoto, S. Yonehara and E. Nishida, Curr. Biol., 2006, 16, 1171–1182 CrossRef CAS PubMed.
- C. Fremin, A. Bessard, F. Ezan, L. Gailhouste, M. Regeard, J. Le Seyec, D. Gilot, G. Pages, J. Pouyssegur, S. Langouet and G. Baffet, Hepatology, 2009, 49, 930–939 CrossRef CAS PubMed.
- E. Stabile, Y. F. Zhou, M. Saji, M. Castagna, M. Shou, T. D. Kinnaird, R. Baffour, M. D. Ringel, S. E. Epstein and S. Fuchs, Circ. Res., 2003, 93, 1059–1065 CrossRef CAS PubMed.
- G. Kroemer, L. Galluzzi, P. Vandenabeele, J. Abrams, E. S. Alnemri, E. H. Baehrecke, M. V. Blagosklonny, W. S. El-Deiry, P. Golstein, D. R. Green, M. Hengartner, R. A. Knight, S. Kumar, S. A. Lipton, W. Malorni, G. Nunez, M. E. Peter, J. Tschopp, J. Yuan, M. Piacentini, B. Zhivotovsky and G. Melino, Cell Death Differ., 2009, 16, 3–11 CrossRef CAS PubMed.
- G. Kumari, T. Ulrich, M. Krause, F. Finkernagel and S. Gaubatz, J. Biol. Chem., 2014, 289, 16072–16084 CrossRef CAS PubMed.
- H. Zhu, A. Sun, H. Zhu, Z. Li, Z. Huang, S. Zhang, X. Ma, Y. Zou, K. Hu and J. Ge, Arterioscler., Thromb., Vasc. Biol., 2014, 34, 894–901 CrossRef CAS PubMed.
- T. L. Roth, D. Nayak, T. Atanasijevic, A. P. Koretsky, L. L. Latour and D. B. McGavern, Nature, 2014, 505, 223–228 CrossRef CAS PubMed.
- S. D. Copley and J. K. Dhillon, Genome Biol., 2002, 3(5), research0025 CrossRef PubMed.
- R. Martin, J. J. Berlanga and C. de Haro, J. Cell Sci., 2013, 126, 3010–3020 CrossRef CAS PubMed.
- D. Ghigo, E. Aldieri, R. Todde, C. Costamagna, G. Garbarino, G. Pescarmona and A. Bosia, J. Clin. Invest., 1998, 102, 595–605 CrossRef CAS PubMed.
- N. Mizushima, B. Levine, A. M. Cuervo and D. J. Klionsky, Nature, 2008, 451, 1069–1075 CrossRef CAS PubMed.
- B. Pasquier, Cell. Mol. Life Sci., 2016, 73, 985–1001 CrossRef CAS PubMed.
- C. H. Eng, Z. Wang, D. Tkach, L. Toral-Barza, S. Ugwonali, S. Liu, S. L. Fitzgerald, E. George, E. Frias, N. Cochran, R. De Jesus, G. McAllister, G. R. Hoffman, K. Bray, L. Lemon, J. Lucas, V. R. Fantin, R. T. Abraham, L. O. Murphy and B. Nyfeler, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 182–187 CrossRef CAS PubMed.
- E. Desideri, R. Vegliante, S. Cardaci, R. Nepravishta, M. Paci and M. R. Ciriolo, Autophagy, 2014, 10, 1652–1665 CrossRef CAS PubMed.
- L. Jiang, N. Kon, T. Li, S. J. Wang, T. Su, H. Hibshoosh, R. Baer and W. Gu, Nature, 2015, 520, 57–62 CrossRef CAS PubMed.
- H. Jung, M. J. Kim, D. O. Kim, W. S. Kim, S. J. Yoon, Y. J. Park, S. R. Yoon, T. D. Kim, H. W. Suh, S. Yun, J. K. Min, H. G. Lee, Y. H. Lee, H. J. Na, D. C. Lee, H. C. Kim and I. Choi, Cell Metab., 2013, 18, 75–85 CrossRef CAS PubMed.
- L. Zheng, A. Terman, M. Hallbeck, N. Dehvari, R. F. Cowburn, E. Benedikz, K. Kagedal, A. Cedazo-Minguez and J. Marcusson, Autophagy, 2011, 7, 1528–1545 CrossRef CAS PubMed.
- V. Ginet, J. Puyal, C. Rummel, D. Aubry, C. Breton, A. J. Cloux, S. R. Majjigapu, B. Sordat, P. Vogel, S. Bruzzone, A. Nencioni, M. A. Duchosal and A. Nahimana, Autophagy, 2014, 10, 603–617 CrossRef CAS PubMed.
- S. B. Lee, J. J. Kim, T. W. Kim, B. S. Kim, M. S. Lee and Y. D. Yoo, Apoptosis, 2010, 15, 204–218 CrossRef CAS PubMed.
- F. Bellomo, C. Piccoli, T. Cocco, S. Scacco, F. Papa, A. Gaballo, D. Boffoli, A. Signorile, A. D'Aprile, R. Scrima, A. M. Sardanelli, N. Capitanio and S. Papa, Antioxid. Redox Signaling, 2006, 8, 495–502 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06737b |
| This journal is © The Royal Society of Chemistry 2017 |