
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly-flexible cyclic-decavanadate ligand for interconversion of dinuclear- and trinuclear-cobalt(II) and manganese(II) cores†
Tatsuya Maruyamaa,
Yuji Kikukawa ab,
Hiroshi Sakiyamac,
Misaki Katayamad,
Yasuhiro Inadad and
Yoshihito Hayashi*a
ab,
Hiroshi Sakiyamac,
Misaki Katayamad,
Yasuhiro Inadad and
Yoshihito Hayashi*a
aDepartment of Chemistry, Graduate School of Natural Science and Technology, Kanazawa University, Kakuma, Kanazawa 920-1192, Ishikawa, Japan. E-mail: hayashi@se.kanazawa-u.ac.jp
bJST, PRESTO, 4-1-8 Honcho, Kawaguchi 332-0012, Saitama, Japan
cDepartment of Science, Faculty of Science, Yamagata University, Kojirakawa, Yamagata 990-8560, Japan
dDepartment of Applied Chemistry, Graduate School of Life Sciences, Ritsumeikan University, 1-1-1 Noji-Higashi, Kusatsu 525-8577, Japan
First published on 1st August 2017
Abstract
The structure transformation of multinuclear-metal-cores can change catalytic, optical, and magnetic properties. Cyclic decavanadate ligands exhibit versatility in the number and the direction of the coordination sites by changing the conformation to stabilize various multinuclear-metal-cores, while organic multinucleating ligands require specific design for each of the multimetal complexes due to their structure directing ability. The flexibility of cyclic decavanadate ligands is demonstrated here to achieve accommodation of dinuclear or trinuclear units by using the same ligand. The reaction of a dinuclear-cobalt-core-containing decavanadate [Co2(H2O)2V10O30]6− (Co2) with 1 equiv. of Co(OAc)2 (OAc = acetate) gave a trinuclear-cobalt-core-containing decavanadate [Co3(H2O)(OAc)V10O30]5− (Co3) in high yield. The central cobalt core exhibited an incomplete-cubane-type structure. The decavanadate ring contracts to accommodate a smaller dinuclear unit by taking a wavy conformation and expands to accommodate the larger trinuclear unit. The reverse reaction quantitatively proceeded by the addition of 5 equiv. of [VO3]− with respect to Co3. Although a trinuclear-manganese-core-containing decavanadate [Mn3(H2O)(OAc)V10O30]5− (Mn3) possesses the same structure as that of Co3, the addition of 5 equiv. of [VO3]− yielded a different structure of a dinuclear-manganese-core-containing decavanadate [Mn2V10O30]6− (Mn2) with two cyclic pentavanadate ligands sandwiching the manganese core. Thus, the conformations of the cyclic decavanadates are rearranged to respond to the central metal core structures. EXAFS study suggests both manganese complexes maintain the molecular structure in solution. The simultaneous analyses of the magnetic susceptibility data and the magnetization data revealed the switch of magnetic interaction modes from ferromagnetic in dinuclear complexes to mixed ferromagnetic and antiferromagnetic interactions in trinuclear complexes: the ferromagnetic interaction in dinuclear units of Co2 (J = 8.05 cm−1) and Mn2 (J = 0.76 cm−1) (Hex = −JS1S2), and the ferromagnetic and antiferromagnetic interactions in Co3 (J = −1.59 cm−1 and J′ = 13.6 cm−1) and Mn3 (J = −2.20 cm−1 and J′ = 0.07 cm−1) (Hex = −JSA1SA2 − J′[SA1SB + SA2SB]) were studied.
Introduction
The design and functionalization of multinuclear-metal-cores have crucial importance in many fields such as catalysis, magnetism, electrochemistry, and photochemistry.1,2 Multinuclear-metal-cores are also important in biological systems such as in metalloenzymes and utilize the protein chains as a ligand to stabilize the metal core, and these systems also require a dynamic change in the number and direction of the coordination sites of the cores during the catalytic cycle.3,4Multinuclear complexes are fine-tuned to achieve a specific property by using tailor made organic ligands in coordination chemistry. In the functional multimetal complexes, the specific ligand design is necessary to fit in metal coordination environments, for example, a dinucleating ligand for a dinuclear complex and a trinucleating ligand for a trinuclear complex.5–17 The inorganic ligands, composed of metal–oxygen clusters, polyoxometalates, also stabilize multinuclear-metal-cores.18 The advantage in the inorganic ligands is its diverse properties in redox and electron-transfer properties, acidity, and solubility by optimizing constituent elements, structures, and counter cations.19 Although the typical Keggin– and Wells–Dawson type polyoxometalates have low coordination ability, the metastable lacunary polyoxometalates, ranging from mono- to hexalacunary ones, are utilized to accommodate metal cluster cores by offering more than four oxygen atoms as donor sites. The multinuclear-metal-core-containing polyoxometalates exhibit the greatest properties in a wide field of research.20–28 In addition, the reversible structure transformation of multinuclear metal cores with using [SiW10O36]8− ligands are reported.29,30 By addition of the required components, the reversible transformations between dinuclear- and tetranuclear-copper and cobalt cores are achieved.29,30 The water- and temperature-triggered geometry control between cubane- and planer-type tetranuclear-cobalt-cores are demonstrated.31 These tailor-made organic ligands or inorganic ligands have a rigid coordination configuration defined by the arrangement of oxygen and/or nitrogen donor atoms. For the design of enzyme like flexible ligand system, the new ligand system that can meet variable coordination environments even for a different nuclearity of the metal cores has to be developed.
Vanadium-based polyoxometalates exhibit structural versatility based on three types of polyhedral units: tetrahedral VO4, square pyramidal VO5, and octahedral VO6.32–34 Among these structural units, VO4-based polyoxovanadates construct a ring structure by corner-sharings of VO4 units. The cyclic polyoxovanadates act as ligands for various metal cores to form inorganic complexes, such as [Ln(VO3)n]−n+3 (n = 8, 9, 10: Ln = lanthanides(III)), [Mn2(V5O15)2]6−, [Co2(OH2)2V10O30]6−, [Ni4(OH)2(OH2)6V10O30]4−, and [Cu2V8O24]4−.35–39 The macrocyclic [VO3]nn− has a freedom of V–O–V bridge angles and one of two oxo-groups on each vanadium atom has coordination ability to the metal center. The number of coordination donor-atoms from [VO3]nn− can be varied by either one oxo-group join to the coordination to the metal center or two oxo-groups are remain free from coordination. The maximum coordination donor atoms from [VO3]nn− ligand is n. This flexible change of the number of donor-atoms and the spatial flexibility due to its conformational change allows the ligands to form versatile inorganic complexes with a different number of central metal nuclearity. The coordination sites on the metal core which is not occupied from the cyclic ligands is available to other ancillary ligands such as water ligands, and it provide the active site for exchange reaction to the other types of ligands.39–41 The inorganic complex may be a scaffold to construct a unique polyoxovanadate in a bowl-shape by removing the metal cores from [Cu2V8O24]4− to give dodecavanadate [V12O32(Cl)]5−.42 The inorganic complexes by [VO3]nn− ligand have been investigated in a solid state with crystal structures, but this excellent flexibility may allow a dynamic transformation of the nuclearity of the metal cores even in solution state within the same ligand system. Here, we demonstrate the [VO3]nn− (n = 5 or 10) ligands respond to the change of central metal cores between dinuclear- and trinuclear-metal-cores and it adopts two types of completely different coordination sites that are different in coordination site number and the arrangements through the variable coordination modes of the same ligands (Fig. 1). We focused on the cobalt-core-containing polyoxovanadates because of the structural variety and their unique properties, such as magnetism, photochemistry, electrochemistry, and catalysis.43–49 We choose dinuclear-cobalt-core-containing polyoxometalate (Co2) for the first precursor, because Co2 possess the disk-type structure to which an additional cobalt cation can access to form a trinuclear-core, and two non-coordinating VO4 units which can work as the additional coordination donor groups.36 We successfully synthesize a novel trinuclear-cobalt-core-containing polyoxovanadate, [Co3(H2O)(OAc)V10O30]5− (Co3). Their reversibility and magnetic properties are also investigated.
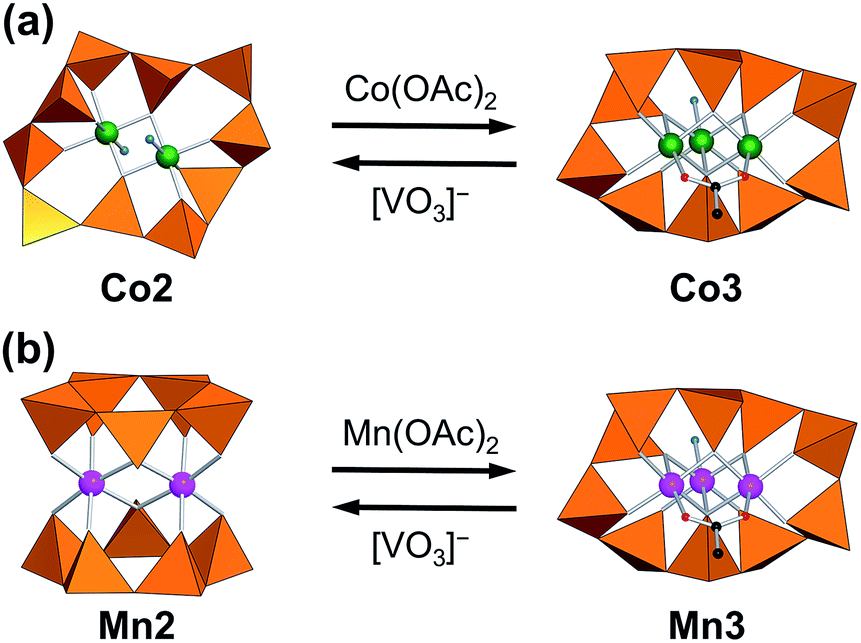 | ||
| Fig. 1 Interconversion of decavanadates with dinuclear- and trinuclear- (a) cobalt and (b) manganese cores. Orange tetrahedra represent VO4 units. Green, pink, black, red and blue spheres represent cobalt atoms, manganese atoms, carbon atoms, oxygen atoms of acetate, and water oxygen atoms, respectively. | ||
Experimental section
Instruments
IR spectra were measured on a JASCO FT/IR-4100 using attenuated total reflection (ATR) method. Elemental analyses of C, H and N were performed by the Research Institute for Instrumental Analysis at Kanazawa University. The ICP-AES analyses were performed with iCAP 6300 instrument from Thermo Fisher Scientific. Magnetic susceptibilities of polycrystalline samples were measured on Quantum Design SQUID magnetometer operating between 2 K and 300 K under 1000 Oe magnetic field. The magnetic analyses, including susceptibility and magnetization simulation, and simultaneous fitting, were conducted by MagSaki(A), Magsaki(Tri), and MagSaki(Basic) software of MagSaki series.50–52Materials
Mn(OAc)2, Co(OAc)2, acetonitrile, and diethyl ether were obtained from Wako Chemical and used as received. {Et4N}[VO3],53 {Et4N}6[Mn2(V5O15)2],36 {Et4N}6[Co2(H2O)2V10O30],36 and {Et4N}5[Mn3(OAc)(H2O)V10O30]39 were synthesized according to the reported procedures.EXAFS studies
The extended X-ray analysis fine structure (EXAFS) measurements for solid samples and propylene carbonate solution samples were performed at 295 K using the BL-3 at the SR Center of Ritsumeikan University (Kusatsu, Japan). All samples were prepared in an anaerobic environment using a polyethylene glove bag filled with dry nitrogen because of the hygroscopic nature of the samples. Solid samples were diluted with boron nitride and pressed into pellets of 20 mm diameter. The pellets were sealed in a polyethylene bag in order to avoid moisture. Sample solutions with manganese concentrations of 0.1 mM were prepared by dissolving the powder of each complex in propylene carbonate at 353 K. The solution samples were sealed in a transparent plastic bag with approximate thickness of 1.5 mm through a handmade cell. The white synchrotron radiations were monochromatized by a Si(220) double-crystal monochromator. Data collections of the Mn and Co K-edge absorption edges were performed in transmission mode. EXAFS data were analyzed with the ATHENA and ARTEMIS programs in the Demeter package.54X-ray crystallographic analysis
Diffraction measurements of Co3 were made on a Rigaku/MSC Mercury diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71069 Å) at 83 K. The data of Co3 were collected and processed using CrystalClear program.55 Numerical absorption corrections were applied by using CrystalClear and corrections for Lorenz and polarization effects were performed. The structural analysis was performed using CrystalStructure56 and ShelXle57 for Windows software. The structure was solved by SHELXS-2014 58 (direct methods) and refined by SHELXL-2014.58 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms are positioned geometrically and refined using a riding model. CCDC 1546869 (for Co3) contain the supplementary crystallographic data.†Synthesis of Co3
{Et4N}[VO3] (230 mg, 1.0 mmol) was dissolved in acetonitrile (3 mL), and Co(OAc)2·4H2O (125 mg, 0.5 mmol) was added. The resulting solution was stirred for 3 h at room temperature. The precipitates formed were collected, washed with acetonitrile, and dried to afford 85 mg of Co3 (45% yield based on V). Recrystallization of Co3 from an acetonitrile gave rod shaped crystals suitable for X-ray crystallographic structure analysis. Anal. Calcd (%) for {Et4N}5[Co3(OAc)(H2O)V10O30]: C, 26.63; H, 5.59; N, 3.70. Found: C, 26.51; H, 5.67; N, 3.95. IR (ATR, cm−1): 3465, 3297, 2988, 2950, 1650, 1573, 1484, 1462, 1440, 1418, 1395, 1307, 1174, 1003, 979, 959, 944, 903, 849, 826, 784, 765, 710, 693, 668, 612, 552.Transformation of Co2 into Co3
Compound Co2 (190 mg, 100 μmol) was immersed in acetonitrile (5 mL), followed by the addition of Co(OAc)2·4H2O (25 mg, 100 μmol), giving a green suspension solution. The solution was stirred for 2 h at room temperature. Then, the yellow-green precipitates formed were collected, washed with acetonitrile, and dried to afford 166 mg of Co3 (87% yield based on Co2). The formation of Co3 was confirmed by comparing IR with authentic samples.Transformation of Co3 into Co2
Compound Co3 (38 mg, 20 μmol) was dissolved in acetonitrile (10 mL), followed by the addition of {Et4N}[VO3] (23 mg, 100 μmol), giving a green solution. The solution was stirred for 2 h at room temperature. To the solution was added diethyl ether, and the resulting solution was stood for 1 day at room temperature. Then, the precipitates formed were collected, washed with acetonitrile, and dried to afford 51 mg of Co2 (87% yield based on Co3). The formation of Co3 was confirmed by comparing IR with authentic samples.Transformation of Mn2 into Mn3
Compound Mn2 (94 mg, 50 μmol) was immersed in acetonitrile (10 mL), followed by the addition of Mn(OAc)2·4H2O (12 mg, 50 μmol), giving an orange solution. The solution was stirred for 2 h at room temperature, and a red-orange suspension was obtained. The resulting solution was concentrated to ca. 1 mL. Then, the orange precipitates formed were collected, washed with acetonitrile, and dried to afford 91 mg of Mn3 (96% yield based on Mn2). The formation of Mn3 was confirmed by comparing IR with authentic samples.Transformation of Mn3 into Mn2
Compound Mn3 (94 mg, 50 μmol) was immersed in acetonitrile (10 mL), followed by the addition of {Et4N}[VO3] (57 mg, 250 μmol), giving an orange solution. The solution was stirred for 2 h at room temperature, and a red-orange suspension was obtained. The resulting solution was concentrated to ca. 1 mL. Then, the orange precipitates formed were collected, washed with acetonitrile, and dried to afford 125 mg of Mn2 (88% yield based on Mn3). The formation of Mn2 was confirmed by comparing IR with authentic samples.Results and discussion
Synthesis of Co3 and reversible transformation between Co2 and Co3
By the reaction of Co2 with 1 equiv. of Co(OAc)2 with respect to Co2 in acetonitrile, the precipitates of Co3 were obtained in 87% yield based on Co2 (Fig. 1a). The IR spectrum of Co3 showed the characteristic bands due to asymmetrical and symmetrical stretching vibration of acetate at 1573 cm−1 and 1418 cm−1 (Fig. 2A), respectively. The peak separation value of these two peaks (Δ = 155 cm−1) was in the range of those of bridging acetate.59,60 In the fingerprint region of the polyoxovanadate frameworks (1100–500 cm−1), the spectrum was different from that of Co2. The weak peak of terminal V–O stretching vibration around 1000 cm−1 that was the characteristic feature of the VO4-based circular polyoxovanadates, was observed.35–39 Single crystals suitable for X-ray crystallographic analysis were successfully obtained from the saturated acetonitrile solution. The bond valence sum values of cobalt (1.99–2.07) and vanadium (4.98–5.06) indicate that respective valences are +2 and +5. The anion structure of Co3 was composed of a VO4-based cyclic decavanadate ligand and a trinuclear-cobalt-core. The coordination sites of trinuclear-cobalt units are occupied by thirteen oxygen atoms: one oxygen atom from aquo-ligand, two oxygen atoms from acetate group and ten oxygen atoms from VO4 units (Table S1†). The X-ray data and elemental analyses show that the formula of Co3 is {Et4N}5[Co3(H2O)(OAc)V10O30]. The formation of Co3 from Co2 is expressed by eqn (1).| {Et4N}6[Co2(H2O)2V10O30] + Co(OAc)2 → {Et4N}5[Co3(H2O)(OAc)V10O30] + {Et4N}OAc + H2O | (1) |
 | ||
| Fig. 2 (A) IR spectra of (a) Co2, (b) Co2 treated with 1.0 equiv. of Co(OAc)2, and (c) Co3. (B) IR spectra of (a) Co3, (b) Co3 treated with 5.0 equiv. of {Et4N}[VO3], and (c) Co2. | ||
The trinuclear cobalt core was the incomplete-cubane type structure with Co3O4 fragment. The value of the bond lengths and bond angles are similar to those of the previously reported compounds with the incomplete-cubane-type Co3O4 cores.61–63 The distribution of the Co–O bond lengths in Co3 was small in the range. Such an uniform incomplete-cubane framework was only observed in the compound with each of metal coordinated by the same small ligands without the structural bulkiness,61–63 indicating that the cyclic decavanadate ligand comfortably offers an ideal donor-sites arrangement by adjusting their conformation to the central Co3O4 core. The bridging acetate ligand restricts the Co⋯Co distance in a shorter length of 3.00 Å leaving other inter-metallic distances at 3.30 Å. Even Co2 and Co3 are accommodated by the same decavanadate ligands, their conformations were different each other. The decavanadate ligand in Co2 provides eight coordination donors to the central cobalt core. The V10O10 ring in Co2 contracts by taking the wavy conformation composed from the five zig-zag chain-blocks (V1–V2–V3, V3–V4–V5, V5–V1*–V2*, V2*–V3*–V4*, and V4*–V5*–V1) and two of the VO4 units are not involved in the coordination (Fig. 3a). On the other hand, decavanadate ligand in Co3, expands that all VO4 units were coordinated to the central cobalt core. The V10O10 ring structure in Co3 is considered to be constructing from two kinds of zig-zag chain-blocks: (V3–V4–V5 and V5–V6–V7) followed by two relatively longer waves (V7–V8–V9–V10 and V10–V1–V2–V3), to cover the three-dimensional trinuclear-cobalt-core (Fig. 3b).
 | ||
| Fig. 3 Comparison of the V10O10 rings in (a) Co2 and (b) Co3. | ||
The one cobalt unit in Co3 can be removed by the reaction with [VO3]−. The reaction of Co3 with 5 equiv. of {Et4N}[VO3] in acetonitrile produces Co2 exclusively (87% yield based on Co3, Fig. 1a). The IR spectrum of the precipitates produced by adding ether to the above mixture solution was identical with those of Co2 (Fig. 2B), showing that Co3 is reversibly transformed into the starting Co2. The transformation reaction was expressed by eqn (2). Thus, the nucleation number control was achieved from dinuclear to trinuclear or from trinuclear to dinuclear cobalt complexes by the stoichiometric control.
| 2{Et4N}5[Co3(H2O)(OAc)V10O30] + 10{Et4N}[VO3] + 4H2O → 3{Et4N}6[Co2(H2O)2V10O30] + 2{Et4N}OAc | (2) |
The examples of cobalt-core-containing polyoxovanadates was reported with several kinds of coordinative ligands, [Co(L)V4O12]3− (L = Cl−, OAc−, PhCOO−, OCN−, N3−).40,41,64 It is noted that Co2 and Co3 were not obtained by the reaction of [Co(OAc)V4O12]3− with {Et4N}[VO3], and Co(OAc)2, respectively.
Reversible transformation between dinuclear- and trinuclear-manganese-containing decavanadates
Recently, we reported the synthesis of a trinuclear-manganese-core-containing polyoxovanadate, [Mn3(OAc)(H2O)V10O30]5− (Mn3). Compound Mn3 was isomorphous to Co3 in crystallographic data (Table S2†).39 Because of the different ionic radii of Co2+ (0.745 Å) and Mn2+ (0.830 Å), the average Mn–O distances of Mn3 (2.177 Å) were longer than the Co–O distances of Co3 (2.097 Å). The cyclic decavanadate ligand accepted these differences by adjusting their conformation. The V–O distances and O–V–O bond angles of Mn3 and Co3 were similar to each other, and each V–O–V bond angle of Mn3 was 2.0° larger in average than that of Co3. The larger ionic radius caused the extension of the V10O10 ring size, for example, the distances between V2 and V8 in Mn3 and Co3 were 8.90 Å and 8.75 Å, respectively.Since Mn3 has the same structure of Co3, we also investigate the transformation reaction of Mn3 to a dinuclear complex with the reaction by [VO3]− ion. The IR spectrum of Mn3 as a starting material was almost identical to that of Co3. By the reaction of Mn3 with 5 equiv. of {Et4N}[VO3] with respect to Mn3 in acetonitrile, precipitates were obtained (yield: 88% based on Mn3, Fig. 1b). The IR spectrum of the precipitate was matched the same as that of the previously reported dinuclear-core-containing decavanadate (Mn2) (Fig. 4B). Although the formulas of Mn2 and Co2 were the same except for the presence of H2O, the cyclic ligand configurations are totally different. While Co2 possessed a single cyclic V10O10 ring, Mn2 possessed two V5O5 rings sandwiching a dinuclear-manganese-core. The formation of Mn2 from Mn3 were expressed by eqn (3). In addition, the reverse reaction proceeded by addition of 1 equiv. of Mn(OAc)2 (eqn (4), Fig. 1b).
| 2{Et4N}5[Mn3(OAc)(H2O)V10O30] + 10{Et4N}[VO3] → 3{Et4N}6[Mn2(V5O15)2] + 2{Et4N}OAc | (3) |
| {Et4N}6[Mn2(V5O15)2] + Mn(OAc)2 → {Et4N}5[Mn3(OAc)(H2O)V10O30] + {Et4N}OAc | (4) |
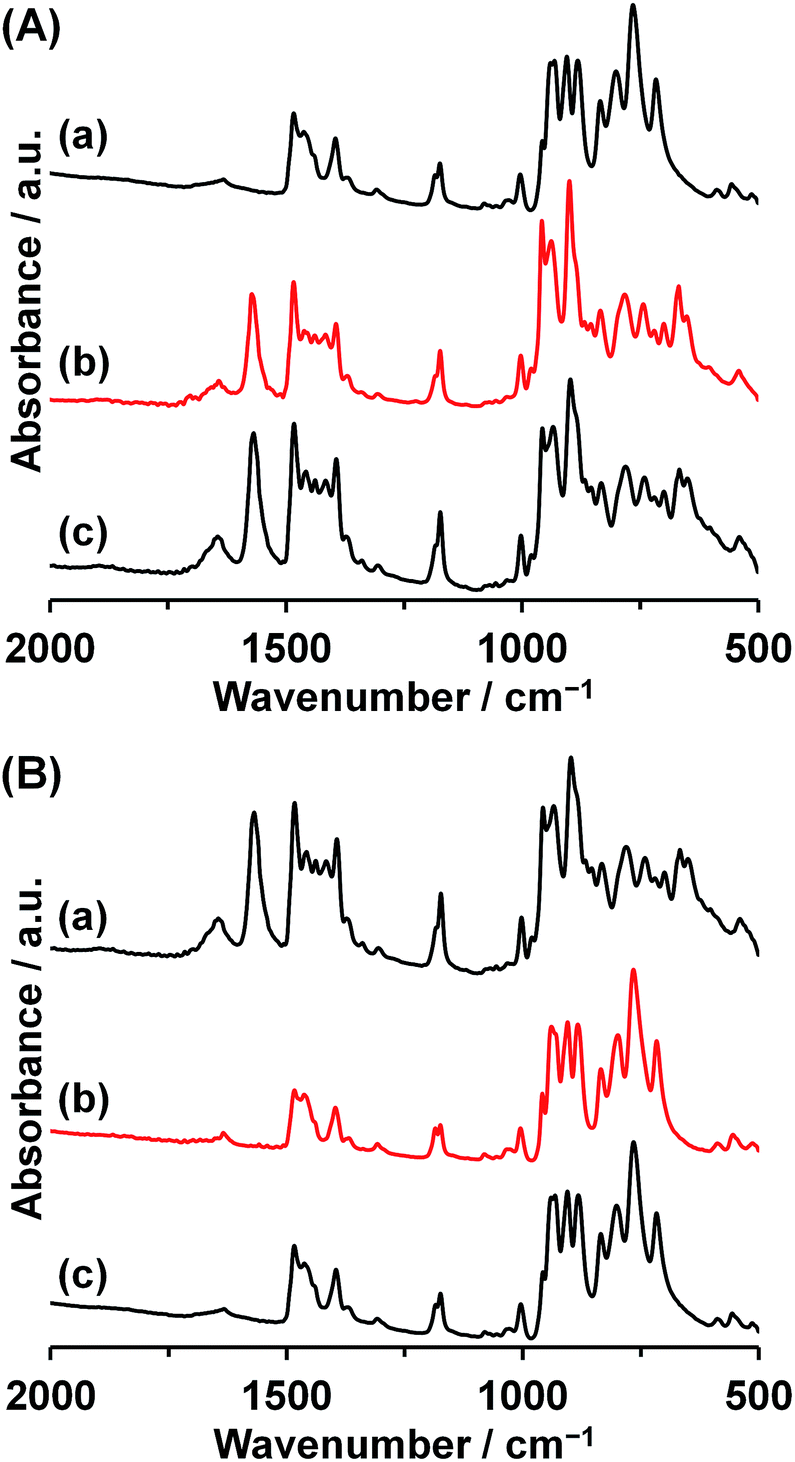 | ||
| Fig. 4 (A) IR spectra of (a) Mn2, (b) Mn2 treated with 1.0 equiv. of Mn(OAc)2, and (c) Mn3. (B) IR spectra of (a) Mn3, (b) Mn3 treated with 5.0 equiv. of {Et4N}[VO3], and (c) Mn2. | ||
We tried the synthesis of a dinuclear manganese complex with the same structure of Co2 by removing aquo ligands. The only difference between Mn2 and Co2 in formula is the presence or absence of aquo ligand on the dinuclear-metal-core, and the structural conversion between two types of dinuclear complexes may be possible by controlling the amount of water. By addition of water (ca. 20 equiv. of water respect to Mn2) to the synthetic solution of Mn2, the orange precipitates were obtained. The IR spectrum of the product was identical to that of Mn2. On the other hand, the dinuclear-cobalt complexes was not converted to a Mn2 type complex under a completely dehydrated condition.
EXAFS studies
Mn2 and Mn3 were reversibly transformed by the reaction although each compound possessed quite different anion structures. To eliminate the potential possibility that the solution state structures of Mn2 and Mn3 were different from those of the crystal structure. The EXAFS analyses were performed. The FT magnitudes of the solid and solution samples of Mn2 and Mn3 are shown in Fig. 5. The coordination number, the interatomic distance, and the Debye–Waller factor were determined by the least-squares fitting of the theoretical EXAFS function to the experimental one in r-space. The EXAFS parameters except for these structure parameters were estimated by using the FEFF code for the crystal structure of Mn2 and Mn3. The fitting parameters of the EXAFS results are given in Table S3.† The solution sample of Mn2 showed the three peaks at 2.16, 3.2, and 3.45 Å assignable to the Mn–O, Mn⋯Mn, and Mn⋯V, respectively. The peaks of the Mn–O, Mn⋯Mn, and Mn⋯V were observed at 2.15, 3.20, and 3.50 Å, respectively, by the curve fitting of the EXAFS data for the solid sample of Mn2. The Mn–O bond lengths of Mn3 were 2.07 and 2.22 Å for the solid authentic sample, and 2.05 and 2.20 Å for the solution sample. The Mn⋯Mn distances of Mn3 were 3.1 and 3.4 Å for the solid sample, and 3.1 and 3.479 Å for the solution sample. The Mn⋯V distances of Mn3 was 3.5 Å in solution, which was slightly longer than that of solid sample. From the EXAFS analyses, the solution state structure of Mn2 and Mn3 were corresponds to the solid state structures of each compound. | ||
| Fig. 5 Mn-K-edge Fourier transform of (A) Mn2 (a) in propylene carbonate and (b) in BN pellets and (B) Mn3 (a) in propylene carbonate and (b) in BN pellets. Experimental and simulated data are shown as solid and dotted lines, respectively. | ||
Magnetic studies
The magnetic measurements of Co2, Co3, Mn2, and Mn3 were carried out using the polycrystalline sample (applied magnetic field 1000 Oe, 2–300 K). The temperature dependence of χMT and the field dependence of magnetization of the complexes are shown in Fig. 6a–d. | ||
| Fig. 6 Temperature dependence of χMT values of (a) Co2, (b) Co3, (c) Mn2, and (d) Mn3 in the temperature range of 2.0–300 K under 1000 Oe field. Inset represents the field dependence of magnetization data. Solid lines represent the fitting data. | ||
The χMT value of Co2 at 300 K was 6.85 cm3 K mol−1 (μeff = 7.40 μB). The value indicates that the magnetic interaction is caused by the independent spin with the presence of orbital angular momenta (6.75 cm3 K mol−1, μ = 7.35 μB; μ = [2 × {L(L + 1) + 4S(S + 1)}]1/2; L = 3, S = 3/2), and it deviates significantly from the spin-only value (3.75 cm3 K mol−1, μspin-only = 5.48 μB; μspin-only = [2 × 4S(S + 1)]1/2, S = 3/2). When decreasing the temperature, the χMT slightly decreases to show a minimum (5.70 cm3 K mol−1) near 35 K, but below 35 K, the χMT increases to show a maximum (6.36 cm3 K mol−1) close to 6.5 K, and then the χMT decreases to the minimum (5.37 cm3 K mol−1) at 2 K. For the curve-fittings of the magnetic data, temperature dependent susceptibility data and field dependent magnetization data were simultaneously analyzed, considering the distortion around each cobalt(II) ion, the local spin–orbit coupling, and the exchange interaction between cobalt(II) ions.65–68 The best fitting parameters were obtained as J = 8.05 cm−1, λ = −145 cm−1, κ = 0.93, Δ = −796 cm−1, θ = −1.01 cm−1, and TIP = 997 × 10−6 cm3 mol−1, where J is the interaction parameter (Hex = −JS1S2), λ, κ, Δ are the local parameters of spin–orbit coupling, orbital reduction factor, and the axial splitting, respectively, θ is the Weiss constant, and TIP is the temperature-independent paramagnetism for each cobalt(II) ion. The definition of the local parameters is adopted from Kahn's equations.69 The obtained local parameters are within the range of typical values for octahedral high-spin cobalt(II) ions. The obtained positive J value indicates the ferromagnetic interaction between cobalt(II) ions, and this is correlate well with the small Co–O–Co angle of <93°. According to the obtained parameters, the slight decrease in χMT from 300 K to 35 K is due to the local spin–orbit coupling, and the increase below 35 K is due to the ferromagnetic interaction.
The χMT value of Co3 at 300 K was 10.14 cm3 K mol−1 (μeff = 9.01 μB). This value was close to the calculated value (10.13 cm3 K mol−1, μ = 9.00 μB; L = 3, S = 3/2) estimated from the existence of the orbital and spin angular momenta independently. When decreasing the temperature, the χMT decreases monotonously, and starting below 20 K, the χMT drops to show a minimum (2.33 cm3 K mol−1) at 2 K. The magnetic susceptibility data and the magnetization data were simultaneously analyzed, considering the distortion around cobalt(II) ions, the spin–orbit coupling, and the exchange interactions on the isosceles triangle model (Hex = −JSA1SA2 − J′[SA1SB + SA2SB]).70 The best fitting parameters were obtained as J = −1.59 cm−1, J′ = 13.6 cm−1, λA = λB = −159 cm−1, κA = κB = 0.93, ΔA = ΔB = 118 cm−1, θ = 0.00 cm−1, and TIPA,B = 325 × 10−6 cm3 mol−1. The obtained local parameters are typical values for octahedral high-spin cobalt(II) ions. The difference of the sign of the interaction in which the ferromagnetic interaction between Co(2) and Co(3) ions and the antiferromagnetic interaction between Co(1) and Co(2) ions and Co(1) and Co(3) ions reflect the difference of Co–O–Co angles: the ferromagnetic interaction was observed with the smaller angles of Co(2)–O–Co(3) in the range of 87° to 93°, while the antiferromagnetic interaction was observed with the larger angles of Co–O–Co bridging in the range of 97° to 101°.71–73
The χMT value of Mn2 at 300 K was 8.88 cm3 K mol−1 (μeff = 8.43 μB), and close to the expected value of 8.75 cm3 K mol−1 (μspin-only = 8.37 μB) for two noninteracting high-spin Mn(II) ions. When decreasing the temperature, the χMT increases to the maximum (12.35 cm3 K mol−1, 9.94 μB) at 2 K. The value is close to the spin-only value when two high-spin Mn(II) ions are ferromagnetically coupled (15.01 cm3 K mol−1, 10.95 μB). The data were analyzed by a conventional dinuclear model (Hex = −JS1S2), and the best fitting parameters were obtained as J = 0.76 cm−1, g = 2.00, θ = 0.00 cm−1, and TIP = 0 × 10−6 cm3 mol−1. Using these parameters, the magnetization data was also fitted. The week ferromagnetic interaction coincides with the small Mn–O–Mn bridging angles (<97°).
The χMT value of Mn3 at 300 K was 12.78 cm3 K mol−1 (μeff = 10.11 μB), and close to the theoretical spin-only value of 13.13 cm3 K mol−1 (μspin-only = 10.25 μB) for three high-spin Mn(II) ions. When decreasing the temperature, the χMT monotonously decreases to the minimum (4.58 cm3 K mol−1, 6.05 μB) at 2 K. This value is close to the spin-only value for S = 5/2 state (4.38 cm3 K mol−1, 5.92 μB). The data were analyzed by the isosceles triangle model (Hex = −JSA1SA2 − J′[SA1SB + SA2SB]), and the best fitting parameters were obtained as J = −2.20 cm−1, J′ = 0.07 cm−1, g = 2.00, θ = 0.00 cm−1, and TIP = 0 × 10−6 cm3 mol−1. Using these parameters, the magnetization data was also fitted. The obtained positive J value indicates the ferromagnetic interaction between Mn(2) and Mn(3) ions, which correspond to the small Mn(2)–O–Mn(3) angles (88–94°).71,74 The obtained negative J′ value indicates the antiferromagnetic interactions between Mn(1) and Mn(2) ions and between Mn(1) and Mn(3) ions, which correspond to the large Mn–O–Mn bridging angles (99–102°).71,74 Therefore, in the ground state, Mn(2) and Mn(3) are ferromagnetically coupled and Mn(1) and Mn(2), and Mn(1) and Mn(3) are antiferromagnetically coupled, affording S = 5/2 state. This is consistent with the χMT value at 2 K.
Conclusions
The interconversion reactions between a dinuclear and a trinuclear Mn(II) or Co(II) complex are achieved by the polyoxovanadate ligand which demonstrates a versatility of the flexible inorganic ring to be able to bind each metal cores by arranging in a different conformation and a coordination mode. Each of the dinuclear complex (Et4N)6[M2(H2O)n(V10O30)] (M = Co, n = 2 for Co2; M = Mn, n = 0 for Mn2) was converted to the corresponding trinuclear complex (Et4N)5[M3(H2O)(OAc)(V10O30)] by adding 1 equiv. of M(OAc)2. The reverse reaction was also achieved by the reaction with [VO3]− as a metal elimination reagent. Both of the forward and reverse reactions proceed quantitatively. In trinuclear complexes, thirteen coordination sites on the edge-shared M3O13 triad in Co3 and Mn3 complexes are occupied from ten oxido-groups from the 10-membered vanadium ring and the rest of the three sites are coordinated by additional water and acetate ligands. The decameric ring in the transformation of cobalt complexes was maintained, while the sandwich structure by two 5-membered vanadium-rings in Mn2 has transformed to Mn3 in the same 10-membered vanadium-ring of Co3. Cyclic polyoxovanadates can be regarded as flexible ligands to accept a different type of multinuclear-metal-cores by rearranging and changing the conformation flexibly.Acknowledgements
This work was supported in part by JST presto Grant Number JPMJPR16S5, Japan, JSPS KAKENHI Grant Number 15K05445, Kanazawa University SAKIGAKE Project, and Sasakawa Scientific Research Grant from the Japan Science Society. We thank Prof. N. Mizuno (the University of Tokyo) and their co-workers for their help with the magnetic measurement.Notes and references
- P. J. Steel, Acc. Chem. Res., 2005, 38, 243–250 CrossRef CAS PubMed.
- A. T. Wagner and P. W. Roesky, Eur. J. Inorg. Chem., 2016, 2016, 782–791 CrossRef CAS.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J.-R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- T. L. Olson, E. Espiritu, S. Edwardraja, C. R. Simmons, J. C. Williams, G. Ghirlanda and J. P. Allen, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 539–547 CrossRef CAS PubMed.
- Y. Yamada, Y. Maeda, T. Konno, K. Fujisawa and K. Okamoto, Bull. Chem. Soc. Jpn., 2000, 73, 1831–1837 CrossRef CAS.
- M.-N. Collomb Dunand-Sauthier, A. Deronzier, A. Piron, X. Pradon and S. Ménage, J. Am. Chem. Soc., 1998, 120, 5373–5380 CrossRef.
- M. Lama, O. Mamula, G. S. Kottas, L. De Cola, H. Stoeckli-Evans and S. Shova, Inorg. Chem., 2008, 47, 8000–8015 CrossRef CAS PubMed.
- N. Yoshinari, Y. Hirai, T. Kawamoto, A. Igashira-Kamiyama, K. Tsuge and T. Konno, Chem. Lett., 2009, 38, 1056–1057 CrossRef CAS.
- W. Cullen, C. A. Hunter and M. D. Ward, Inorg. Chem., 2015, 54, 2626–2637 CrossRef CAS PubMed.
- M. J. Burke, G. S. Nichol and P. J. Lusby, J. Am. Chem. Soc., 2016, 138, 9308–9315 CrossRef CAS PubMed.
- S. Takemoto, Y. Yamazaki, T. Yamano, D. Mashima and H. Matsuzaka, J. Am. Chem. Soc., 2012, 134, 17027–17035 CrossRef CAS PubMed.
- K. Isele, P. Franz, C. Ambrus, G. Bernardinelli, S. Decurtins and A. F. Williams, Inorg. Chem., 2005, 44, 3896–3906 CrossRef CAS PubMed.
- C. Ganesamoorthy, M. S. Balakrishna, P. P. George and J. T. Mague, Inorg. Chem., 2007, 46, 848–858 CrossRef CAS PubMed.
- M.-N. Collomb Dunand-Sauthier, A. Deronzier and A. Piron, J. Electroanal. Chem., 1999, 463, 119–122 CrossRef CAS.
- M.-N. Collomb and A. Deronzier, Eur. J. Inorg. Chem., 2009, 2025–2046 CrossRef CAS.
- G. Aromí, H. Stoeckli-Evans, S. J. Teat, J. Cano and J. Ribas, J. Mater. Chem., 2006, 16, 2635–2644 RSC.
- C. E. Sumner, Inorg. Chem., 1988, 27, 1320–1327 CrossRef CAS.
- K. Kamata, Bull. Chem. Soc. Jpn., 2015, 88, 1017–1028 CrossRef CAS.
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag, New York, 1983 Search PubMed.
- M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219–238 CrossRef CAS PubMed.
- J. M. Clemente-Juan and E. Coronado, Coord. Chem. Rev., 1999, 193–195, 361–394 CrossRef CAS.
- J. M. Clemente-Juan, E. Coronado and A. Gaita-Ariño, Chem. Soc. Rev., 2012, 41, 7464–7478 RSC.
- S.-T. Zheng and G.-Y. Yang, Chem. Soc. Rev., 2012, 41, 7623–7646 RSC.
- T. Yamase, Chem. Rev., 1998, 98, 307–326 CrossRef CAS PubMed.
- C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407–455 CrossRef CAS.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed.
- R. Neumann, in Progress in Inorganic Chemistry, John Wiley & Sons, Inc., 1998, vol. 47, pp. 317–370 Search PubMed.
- U. Kortz, A. Müller, J. van Slageren, J. Schnack, N. S. Dalal and M. Dressel, Coord. Chem. Rev., 2009, 253, 2315–2327 CrossRef CAS.
- K. Suzuki, M. Shinoe and N. Mizuno, Inorg. Chem., 2012, 51, 11574–11581 CrossRef CAS PubMed.
- Y. Kikukawa, K. Suzuki, K. Yamaguchi and N. Mizuno, Inorg. Chem., 2013, 52, 8644–8652 CrossRef CAS PubMed.
- Y. Kuriyama, Y. Kikukawa, K. Suzuki, K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2016, 22, 3962–3966 CrossRef CAS PubMed.
- J. Livage, Coord. Chem. Rev., 1998, 178–180, 999–1018 CrossRef CAS.
- Y. Hayashi, Coord. Chem. Rev., 2011, 255, 2270–2280 CrossRef CAS.
- K. Y. Monakhov, W. Bensch and P. Kögerler, Chem. Soc. Rev., 2015, 44, 8443–8483 RSC.
- T. Kurata, A. Uehara, Y. Hayashi and K. Isobe, Inorg. Chem., 2005, 44, 2524–2530 CrossRef CAS PubMed.
- S. Inami, M. Nishio, Y. Hayashi, K. Isobe, H. Kameda and T. Shimoda, Eur. J. Inorg. Chem., 2009, 5253–5258 CrossRef CAS.
- M. Nishio, S. Inami, M. Katayama, K. Ozutsumi and Y. Hayashi, Inorg. Chem., 2012, 51, 784–793 CrossRef CAS PubMed.
- M. Nishio, S. Inami and Y. Hayashi, Eur. J. Inorg. Chem., 2013, 1876–1881 CrossRef CAS.
- T. Maruyama, Y. Kikukawa, K. Kawamoto and Y. Hayashi, Eur. J. Inorg. Chem., 2017, 596–599 CrossRef CAS.
- A. Seliverstov, J. Forster, M. Heiland, J. Unfried and C. Streb, Chem. Commun., 2014, 50, 7840–7843 RSC.
- M. Heiland, A. Seliverstov, B. Schwarz, M. H. Anjass and C. Streb, Chem.–Eur. J., 2017, 23, 2201–2205 CrossRef CAS PubMed.
- T. Kurata, Y. Hayashi and K. Isobe, Chem. Lett., 2010, 39, 708–709 CrossRef CAS.
- L. F. Chibotaru, L. Ungur, C. Aronica, H. Elmoll, G. Pilet and D. Luneau, J. Am. Chem. Soc., 2008, 130, 12445–12455 CrossRef CAS PubMed.
- Y.-Y. Zhu, C. Cui, Y.-Q. Zhang, J.-H. Jia, X. Guo, C. Gao, K. Qian, S.-D. Jiang, B.-W. Wang, Z.-M. Wang and S. Gao, Chem. Sci., 2013, 4, 1802–1806 RSC.
- S. Ohkoshi and K. Hashimoto, J. Photochem. Photobiol., C, 2001, 2, 71–88 CrossRef CAS.
- O. Sato, Acc. Chem. Res., 2003, 36, 692–700 CrossRef CAS PubMed.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- C. Liu, B. C. Colon, M. Ziesack, P. A. Silver and D. G. Nocera, Science, 2016, 352, 1210–1213 CrossRef CAS PubMed.
- H. Sakiyama, J. Comput. Chem., Jpn., 2007, 6, 123–134 CrossRef CAS.
- H. Sakiyama, J. Comput. Chem. Jpn. Int. Ed., 2015, 1, 9–13 Search PubMed.
- H. Sakiyama, J. Comput. Chem. Jpn. Int. Ed., 2016, 2, 1–4 Search PubMed.
- H. Nakano, T. Ozeki and A. Yagasaki, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, m464–m465 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- CrystalClear, 1.3.6, Rigaku and Rigaku/MSC, The Woodlands, Texas, USA Search PubMed.
- CrystalStructure, 4.1, Rigaku and Rigaku/MSC, The Woodlands, Texas, USA Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Applications in Coordination, Organometallic, and Bioinorganic Chemistry, John Wiley & Sons Ltd., 2009 Search PubMed.
- J. E. Tackett, Appl. Spectrosc., 1989, 43, 483–489 CrossRef CAS.
- S. G. Telfer, R. Kuroda, J. Lefebvre and D. B. Leznoff, Inorg. Chem., 2006, 45, 4592–4601 CrossRef CAS PubMed.
- R. D. Köhn, M. Haufe, G. Kociok-Köhn and A. C. Filippou, Inorg. Chem., 1997, 36, 6064–6069 CrossRef.
- E. Y. Tsui, J. S. Kanady, M. W. Day and T. Agapie, Chem. Commun., 2011, 47, 4189–4191 RSC.
- T. Kurata, Y. Hayashi and K. Isobe, Chem. Lett., 2009, 38, 218–219 CrossRef CAS.
- H. Sakiyama, R. Ito, H. Kumagai, K. Inoue, M. Sakamoto, Y. Nishida and M. Yamasaki, Eur. J. Inorg. Chem., 2001, 2027–2032 CrossRef CAS.
- H. Sakiyama, R. Ito, H. Kumagai, K. Inoue, M. Sakamoto, Y. Nishida and M. Yamasaki, Eur. J. Inorg. Chem., 2001, 2705 CrossRef CAS.
- M. J. Hossain, M. Yamasaki, M. Mikuriya, A. Kuribayashi and H. Sakiyama, Inorg. Chem., 2002, 41, 4058–4062 CrossRef CAS PubMed.
- H. Sakiyama, Inorg. Chim. Acta, 2006, 359, 2097–2100 CrossRef CAS.
- O. Kahn, Molecular magnetism, VCH, New York, 1993 Search PubMed.
- H. Sakiyama, H. Adams, D. E. Fenton, L. R. Cummings, P. E. McHugh and H. Okawa, Open J. Inorg. Chem., 2011, 1, 33–38 CrossRef CAS.
- Z.-Y. Du, L. Zhang, B.-Y. Wang, S. J. Liu, B. Huang, C.-M. Liu and W. X. Zhang, CrystEngComm, 2017, 19, 1052–1057 RSC.
- S. M. Humphrey and P. T. Wood, J. Am. Chem. Soc., 2004, 126, 13236–13237 CrossRef CAS PubMed.
- J. Luo, Y. Zhao, H. Xu, T. L. Kinnibrugh, D. Yang, T. V. Timofeeva, L. L. Daemen, J. Zhang, W. Bao, J. D. Thompson and R. P. Currier, Inorg. Chem., 2007, 46, 9021–9023 CrossRef CAS PubMed.
- E. Ruiz, J. Cano, S. Slvarez and P. Alemany, J. Am. Chem. Soc., 1998, 120, 11122–11129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1546869. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra05941h |
| This journal is © The Royal Society of Chemistry 2017 |