
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First synthesis of rugosaflavonoid and its derivatives and their activity against breast cancer†
Ninad V. Puranik and
Pratibha Srivastava*
and
Pratibha Srivastava*
Bioprospecting Group, Agharkar Research Institute, G. G. Agarkar Road, Pune, 411004, Maharashtra, India. E-mail: ninadv_puranik@yahoo.co.in; psrivastava@aripune.org
First published on 29th June 2017
Abstract
Rugosaflavonoid, is a secondary metabolite isolated from the plant Rosa rugosa was synthesized in five simple steps from commercially available 3,5-dihydroxy benzoic acid involving domino aldol-Michael-oxidation reaction. This is the first report of the synthesis of rugosaflavonoid (6a). A series of its derivatives were also synthesized, characterized and evaluated for the cytotoxicity against the breast cancer MCF-7 and normal NIH3T3 cell lines. The synthetic derivatives of rugosaflavonoid showed comparable activity in both the cell lines and compounds 6d, 6e and 6f, which were found to be cytotoxic towards MCF-7 cell lines but nontoxic to NIH3T3 cell lines at 5 μM concentration. In an attempt to explore the mode of action of the best active compounds, docking on the ATP binding site of EGFR (1M17) was performed considering that EGFR over-expressed in most of the tumors. The docking score (Gscore) of 6f and standard quercetin was found to be −8.608 and −8.310 respectively.
1. Introduction
Cancer still continues to be one of the major threats to human society because it is widely spreading day by day and there is no complete cure for it. The three most commonly diagnosed types of cancer among women in 2010 were cancers of the breast, lung, and colorectum, accounting for 52% of cancer cases in this group. Breast cancer (BC) represents the most common cancer among women; there were 232![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 672 estimated new cases and 40000 estimated deaths in the United States in 2014.1–3 The toxicity allied with conventional cancer chemotherapy arises primarily from the lack of specificity for tumor cells. It leads to a low therapeutic index, which results in undesirable damage to healthy organs and consequently puts restrictions on the dose of the drug that can be administered. The majority of the currently available anticancer drugs are designed to have specific toxicity toward tumor cells.4,5 Several trials are being considered to handle this predicament and thus improve the effectiveness and tumor cell specificity of anticancer drugs. Among these approaches, many studies have focused on natural compounds that inhibit precisely the growth of cancer cells more selectively than normal cells. Thus, phytoconstituents have become the dignified category of anticancer drugs. Over 75% of non-biological anticancer drugs approved between 1981 and 2007 were either natural products or were developed based on them.6 Therefore, the search for new anticancer agents continues to draw attention to the research community. Nature is the biggest lab where millions of chemical reactions are taking place in milliseconds. Medicinal plants are one of the best equipment for the biosynthesis of various drug based molecules. Chromone is a valid scaffold7 in the field of medicinal chemistry, due to the wide range of its biological activities, and their structure–activity relationships have generated curiosity among medicinal chemists, and this has culminated into the breakthrough of the clinical anticancer agent flavopiridol, as well as several lead molecules in other disease areas.8 Rosa rugosa Thunb. belongs to the family of Rosaceae is a common ornamental flower distributed in the temperate regions of eastern Asia and widely cultivated in Yunnan Province.9 The petals and buds of R. rugosa are frequently used as food, incense, and Chinese medicinal materials for the cure of stomachache, diarrhea, and gynecological ailments.10 The literature survey has shown the presence of tannins, terpenoids,11–13 and flavonoids14,15 in this genus. Anti-inflammatory, cytotoxic and anti-HIV activities have observed with selected chemical ingredients isolated from R. rugosa.16 Hu et al.17 have recently isolated and characterized cytotoxic oxepinochromenone and flavonoids from R. rugosa. Rugosaflavonoid is a new flavonoid, isolated from Rosa rugosa which showed cytotoxicity against NB4, SHSY5Y, and MCF-7 cells. Till today the synthesis of rugosaflavonoid (Scheme 1) is not reported in the literature. Therefore, we have focused to synthesize recently isolated naturally occurring rugosaflavonoid (6a) and its derivatives by simple and convenient method. All the synthesized derivatives were evaluated for their cytotoxicity against the breast cancer cell lines MCF-7 and the normal cell lines NIH3T3.
672 estimated new cases and 40000 estimated deaths in the United States in 2014.1–3 The toxicity allied with conventional cancer chemotherapy arises primarily from the lack of specificity for tumor cells. It leads to a low therapeutic index, which results in undesirable damage to healthy organs and consequently puts restrictions on the dose of the drug that can be administered. The majority of the currently available anticancer drugs are designed to have specific toxicity toward tumor cells.4,5 Several trials are being considered to handle this predicament and thus improve the effectiveness and tumor cell specificity of anticancer drugs. Among these approaches, many studies have focused on natural compounds that inhibit precisely the growth of cancer cells more selectively than normal cells. Thus, phytoconstituents have become the dignified category of anticancer drugs. Over 75% of non-biological anticancer drugs approved between 1981 and 2007 were either natural products or were developed based on them.6 Therefore, the search for new anticancer agents continues to draw attention to the research community. Nature is the biggest lab where millions of chemical reactions are taking place in milliseconds. Medicinal plants are one of the best equipment for the biosynthesis of various drug based molecules. Chromone is a valid scaffold7 in the field of medicinal chemistry, due to the wide range of its biological activities, and their structure–activity relationships have generated curiosity among medicinal chemists, and this has culminated into the breakthrough of the clinical anticancer agent flavopiridol, as well as several lead molecules in other disease areas.8 Rosa rugosa Thunb. belongs to the family of Rosaceae is a common ornamental flower distributed in the temperate regions of eastern Asia and widely cultivated in Yunnan Province.9 The petals and buds of R. rugosa are frequently used as food, incense, and Chinese medicinal materials for the cure of stomachache, diarrhea, and gynecological ailments.10 The literature survey has shown the presence of tannins, terpenoids,11–13 and flavonoids14,15 in this genus. Anti-inflammatory, cytotoxic and anti-HIV activities have observed with selected chemical ingredients isolated from R. rugosa.16 Hu et al.17 have recently isolated and characterized cytotoxic oxepinochromenone and flavonoids from R. rugosa. Rugosaflavonoid is a new flavonoid, isolated from Rosa rugosa which showed cytotoxicity against NB4, SHSY5Y, and MCF-7 cells. Till today the synthesis of rugosaflavonoid (Scheme 1) is not reported in the literature. Therefore, we have focused to synthesize recently isolated naturally occurring rugosaflavonoid (6a) and its derivatives by simple and convenient method. All the synthesized derivatives were evaluated for their cytotoxicity against the breast cancer cell lines MCF-7 and the normal cell lines NIH3T3.
 | ||
| Scheme 1 Synthesis of rugosaflavonoid and its derivatives using 3,5-dihydroxybenzoic acid as starting material. | ||
2. Results and discussion
The synthesis of rugosaflavonoid (6a) and its derivatives was accomplished in 5 simple steps by selecting 3,5-dihydroxybenzoic acid (1) as a building block (Scheme 1). In the first step 3,5-dihydroxybenzoic acid was methylated using dimethyl sulfate in presence of the base,18 which produced methoxy ester (2) with 92% yield. The resultant ester was further acylated using acetyl chloride in carbon disulfide into methyl 2-acetyl-3,5-dimethoxybenzoate (3) with 52% yield.19 Demethylation of methyl 2-acetyl-3,5-dimethoxybenzoate (3) using BBr3 in dichloromethane was attempted as per the procedure reported,20 however, unfortunately only monomethylated product was obtained. Thus, AlCl3 was used21 instead of BBr3 to get the desired demethylated product in good yield. Attempt to cyclize intermediate methyl 2-acetyl-3,5-dihydroxybenzoate (4) to rugosaflavonoid (6a) by the literature method22 was unsuccessful, because the reaction resulted into the formation of methyl 7-hydroxy-2-(4-methoxyphenyl)-4-oxo-3,4-dihydro-2H-chromene-5-carboxylate (5) rather than rugosaflavonoid (6a), which was accentuated by spectral data. This reaction was attempted 3–4 times to get the required compound (6a), but it couldn't be obtained. Eventually, the final step was carried out using the intermediate 5 with I2 and DMSO (Scheme 1), which provided the desired product (6a) during 1 hour only. Rugosaflavonoid was obtained as a yellow solid. A molecular formula of C18H14O6 was confirmed by LC-MS m/z 326.30 [M+] and HRMS m/z 327.0853 [M + 1]+. The 1H NMR and 13C NMR data of 6a were obtained and compared (Table 2) with the reported data17 and were found in agreement with natural product rugosaflavonoid. It revealed that the compound 6a has 18 carbon and 14 protons. The 1H NMR of 6a showed proton signals for methoxycarbonyl group δ (3.81, s, 3H), a methoxy group (3.86, s, 3H), and other proton peaks at δ 6.79 (s, 1H, 3-H), 6.82 (d, 1H, 8-H, J = 1.6 Hz), 7.10 (d, 1H, 6-H, J = 2 Hz), 7.12 (d, 2H, 3′, 5′-H, J = 7.2 Hz), 8.04 (d, 2H, 2′, 6′-H, J = 7.2 Hz), and a phenolic hydroxylic proton at 11.14 (s, 1H, OH). The 13C NMR of compound 6 displayed carbon signals at δ 52.86 (C-11, OCH3), 55.97 (C-4′, OCH3), 104.25 (C-8), 105.58 (C-3), 113.55 (C-10), 113.94 (C-6), 114.81 (C-3′), 114.98 (C-5′), 123.49 (C-1′), 127.11 (C-6′), 128.59 (C-2′), 134.55 (C-5), 157.7 (C-9), 158.55 (C-4′) 162.39 (C-2), 162.54 (C-7), 169.19 (C-11), 175.68 (C-4). The IR exhibited the peak at 3446 (OH), and 1735 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1624 (CO). Several derivatives of the rugosaflavonoid using different aromatic aldehydes were also synthesized. Hu et al. had reported17 13.6 mg of rugosaflavonoid (6a) from 8 kg of plant material after several steps of purification. But in the current experiment, 250 mg rugosaflavonoid (6a) was obtained from 1 g of methyl 2-acetyl-3,5-dihydroxybenzoate (4) via the intermediate 5. The derivatives were also synthesized by replacing the ester group with the methyl group in rugosaflavonoid moiety. These derivatives were synthesized using orcinol (1′) as a starting material (Scheme 2), which was acylated followed by the previously stated procedure of cyclization to yield the compounds (6g–j). The rugosaflavonoid and its derivatives displayed comparative results in the MTT cytotoxicity assay. The details are presented in the Table 1. The synthetic rugosaflavonoid (6a) showed 50% cytotoxicity to MCF-7 cells at 5 μM concentration, but its cytotoxicity reduced after enhancing concentration up to 20 μM with 68% cell viability of MCF-7 cells. It was found to be non-toxic to NIH3T3 normal cell line with 87% cell viability at the lower concentration of 5 μM. However, the toxicity increased with the higher concentration. The derivative 6b showed dose dependent cytotoxicity towards MCF-7 and NIH3T3 cell lines. Compounds 6c and 6i showed marginal cytotoxicity towards MCF-7 and NIH3T3 cell lines at lower concentrations, whereas they displayed high cytotoxicity at 20 μM concentration. When the methoxy substituent at 4′ position of rugosaflavonoid was replaced with halogen, the compounds 6d, 6e and 6f expressed 50% cytotoxicity of MCF-7 cells at the lower concentration. The synthesized derivatives of rugosaflavonoid showed dose-dependent cytotoxicity on MCF-7 cell lines and most of them were non-toxic to NIH3T3 cells. The dimethoxy derivatives 6c and 6j showed inhibition of growth of MCF-7, but they were toxic to normal cells. The images of MCF-7 and NIH3T3 cells with 6f before the treatment and after the treatment are shown in the Fig. 1.
O), 1624 (CO). Several derivatives of the rugosaflavonoid using different aromatic aldehydes were also synthesized. Hu et al. had reported17 13.6 mg of rugosaflavonoid (6a) from 8 kg of plant material after several steps of purification. But in the current experiment, 250 mg rugosaflavonoid (6a) was obtained from 1 g of methyl 2-acetyl-3,5-dihydroxybenzoate (4) via the intermediate 5. The derivatives were also synthesized by replacing the ester group with the methyl group in rugosaflavonoid moiety. These derivatives were synthesized using orcinol (1′) as a starting material (Scheme 2), which was acylated followed by the previously stated procedure of cyclization to yield the compounds (6g–j). The rugosaflavonoid and its derivatives displayed comparative results in the MTT cytotoxicity assay. The details are presented in the Table 1. The synthetic rugosaflavonoid (6a) showed 50% cytotoxicity to MCF-7 cells at 5 μM concentration, but its cytotoxicity reduced after enhancing concentration up to 20 μM with 68% cell viability of MCF-7 cells. It was found to be non-toxic to NIH3T3 normal cell line with 87% cell viability at the lower concentration of 5 μM. However, the toxicity increased with the higher concentration. The derivative 6b showed dose dependent cytotoxicity towards MCF-7 and NIH3T3 cell lines. Compounds 6c and 6i showed marginal cytotoxicity towards MCF-7 and NIH3T3 cell lines at lower concentrations, whereas they displayed high cytotoxicity at 20 μM concentration. When the methoxy substituent at 4′ position of rugosaflavonoid was replaced with halogen, the compounds 6d, 6e and 6f expressed 50% cytotoxicity of MCF-7 cells at the lower concentration. The synthesized derivatives of rugosaflavonoid showed dose-dependent cytotoxicity on MCF-7 cell lines and most of them were non-toxic to NIH3T3 cells. The dimethoxy derivatives 6c and 6j showed inhibition of growth of MCF-7, but they were toxic to normal cells. The images of MCF-7 and NIH3T3 cells with 6f before the treatment and after the treatment are shown in the Fig. 1.
 | ||
| Scheme 2 Synthesis of rugosaflavonoid derivatives using orcinol as starting material. | ||
| S. no. | Conc. used | Residue involved in binding with 1M17 | Docking score | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MCF7 (% cell viability) | NIH3T3 (% cell viability) | |||||||||
| 5 μM | 10 μM | 15 μM | 20 μM | 5 μM | 10 μM | 15 μM | 20 μM | |||
| a All the samples run in triplicate and average of three results are presented here. | ||||||||||
| 6a | 43 | 58 | 60 | 68 | 87 | 74 | 71 | 54 | Met 769, Leu 768, Asp 831, Gly 772, Leu 694, Glu 738 | −5.040 |
| 6b | 58 | 47 | 41 | 40 | 90 | 67 | 66 | 62 | Met 769, Leu 768, Leu, 694, Gly 772, Asp 831, Thr 830 | −6.159 |
| 6c | 70 | 52 | 49 | 32 | 73 | 83 | 84 | 82 | Met 769, Leu 768, Gln 767, Asp 831, Thr 830, Leu 694 | −6.661 |
| 6d | 64 | 62 | 45 | 36 | 99 | 97 | 93 | 91 | Met 769, Leu 768, Asp 831, Glu 831 | −6.549 |
| 6e | 50 | 49 | 41 | 31 | 84 | 79 | 33 | 30 | Met 769, Leu 768, Asp 831, Glu 738 | −6.483 |
| 6f | 52 | 45 | 39 | 31 | 96 | 85 | 70 | 62 | Met 769, Leu 768, Leu 694, Asp 831, Lys 721, Glu 738 | −8.310 |
| 6g | 56 | 50 | 48 | 44 | 83 | 71 | 64 | 51 | Met 769, Leu 768, Gln 767, Asp 831, Thr 830, Leu 694 | −4.557 |
| 6h | 65 | 46 | 43 | 40 | 89 | 74 | 60 | 43 | Met 769, Leu 768, Leu, 694, Gly 772, Asp 831, Thr 830 | −4.743 |
| 6i | 71 | 53 | 48 | 41 | 92 | 88 | 82 | 85 | Met 769, Leu 768, Asp 831, Glu 831 | −4.743 |
| 6j | 54 | 49 | 49 | 47 | 55 | 55 | 62 | 65 | Met 769, Leu 768, Asp 831, Glu 738 | −4.965 |
| Std I quercetin | 90 | 75 | 67 | 50 | 89 | 76 | 72 | 68 | Met 769, Lys 721, Glu 738, Asp 831 | −8.608 |
| Paramater | Rugosaflavonoid synthesized | Rugosaflavonoid isolated |
|---|---|---|
| Mp | 226–228 °C | Not reported |
| HRMS | m/z 327.0863 [M + 1]+ (calcd for C18H15O6, 327.0863) | HRESIMS m/z 349.0682 [M + Na]+ (calcd for C18H14NaO6, 349.0688) |
| IR (cm−1) | 3446, 1735, 1624, 1600, 1543, 1436, 1435, 1253, 1180, 1029, 894 | 3416, 1702, 1657, 1610, 1565, 1456, 1432, 1287, 1182, 1028, 893 |
| 1H NMR | (Solvent DMSd6) δ 3.81, (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 6.79 (s, 1H, 3-H), 6.82 (d, 1H, 8-H, J = 1.6 Hz), 7.10 (d, 1H, 6-H, J = 2 Hz), 7.12 (d, 2H, 3′, 5′-H, J = 7.2 Hz), 8.04 (d, 2H, 2′, 6′-H, J = 7.2 Hz), 11.14 (s, 1H, OH) | (Solvent pyridine-d5, 500 MHz) δ 3.80 (s, 3H, OCH3), 3.95 (s, 3H, OCH3), 6.68 (s, 1H, 3-H), 6.74 (d, 1H, 8-H, J = 1.8 Hz), 6.89 (d, 1H, 6-H, J = 1.8 Hz), 7.00 (d, 2H, 3′,5′-H, J = 8.8 Hz), 7.76 (d, 2H, 2′,6′-H, J = 8.8 Hz) |
| 13C NMR | 52.86 (OCH3), 55.97 (C-4′, OCH3) 104.25 (C-8), 105.58 (C-3), 113.55 (C-10), 113.94 (C-6), 114.81 (C-3′), 114.98 (C-5′), 123.49 (C-1′), 127.11 (C-6′), 128.59 (C-2′), 134.55 (C-5), 157.7 (C-9), 158.55 (C-4′), 162.39 (C-2), 162.54 (C-7), 169.19 (C-11), 175.68 (C-4) | 52.4 (OCH3), 55.6 (C-4′, OCH3), 103.8 (C-8), 105.2(C-3), 113.1 (C-6), 115.6 (C-3′), 115.6 (C-5′), 122.9 (C-1′), 131.0 (C-2′), 131.0 (C-6′), 136.8 (C-5), 158.8 (C-9), 163.2 (C-2), 165.0 (C-7), 168.3 (C-11), 181.5 (C-4) |
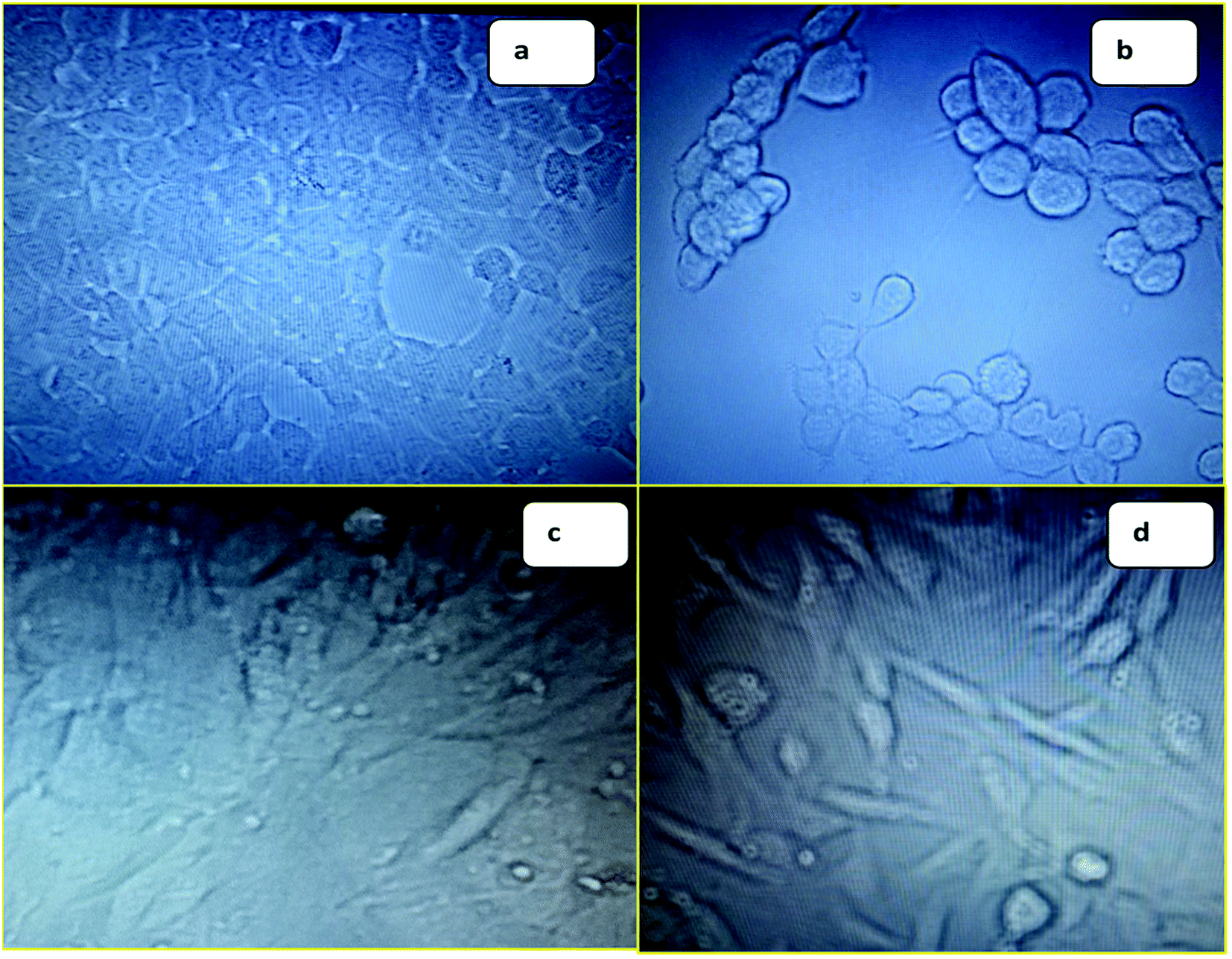 | ||
| Fig. 1 (a) Image of MCF-7 before treatment with 6f; (b) image of MCF-7 after treatment with 6f; (c) image of NIH3T3 before treatment with 6f; (d) image of NIH3T3 after treatment with 6f. | ||
The tyrosine kinase epidermal growth factor receptor (EGFR) is a transmembrane receptor central to numerous cellular process comprising cell migration, adhesion, apoptosis and cell proliferation. The EGFR is over-expressed in almost 90% of tumors.23,24 Protein–ligand interaction of 1M17 with EGFR-specific inhibitor25 and anticancer agent, erlotinib, demonstrated computationally that Met 769 formed hydrogen bond with tyrosine kinase inhibitor, whereas Leu 820, Leu 768, Gly 772, Met 769, and Leu 694 indicated hydrophobic interaction with tyrosine kinase inhibitor, erlotinib. Therefore, interaction studies of rugosaflavonoid compounds were carried out with EGFR (1M17) and compared with the molecular docking of quercetin with 1M17. Interestingly, almost all the synthesized compounds showed non bonded interactions (Fig. 2) with the same residues such as Leu 768, Gly 772, Met 769 and Asp 831 as observed in the crystal structure of 1M17 with erlotinib. The protein–ligand interaction profile of 6f revealed that Lys 721, Glu 738, Met 769 and Asp 831, amino acids involved in the hydrogen bond and π–π interactions in addition to hydrophobic interaction. Molecular docking score of quercetin and 6f with 1M17 were found to be −8.310 and −8.608 respectively. This result is in agreement with the data published by Singh and Bast.26 Overall, docking analysis of standard quercetin and rugosaflavonoid derivatives with 1M17 indicated that these derivatives had equal binding affinity which was also well noticed from experimental cytotoxicity results (Table 1).
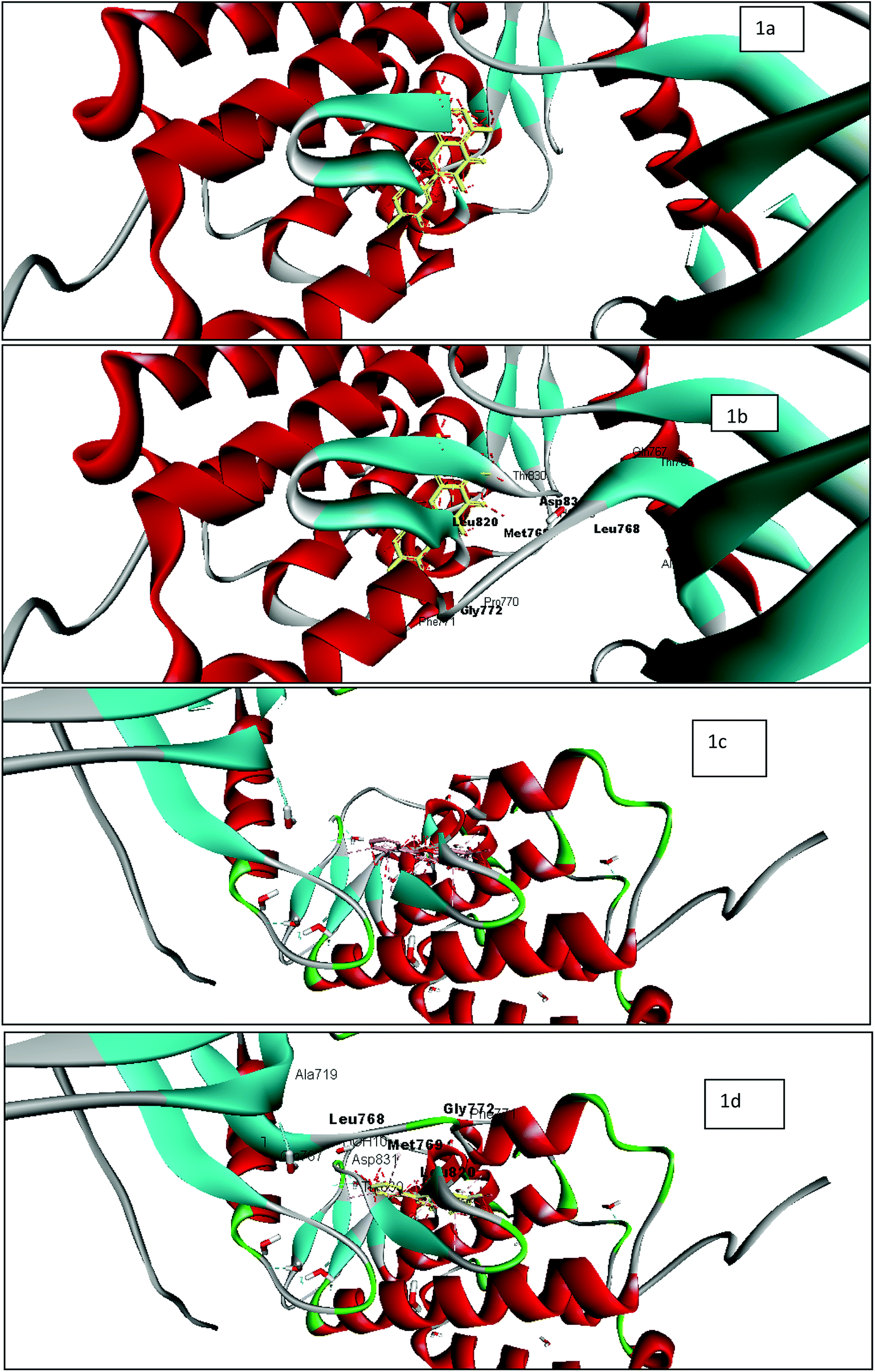 | ||
| Fig. 2 Docking studies of rugosaflavonoid derivatives with EGFR (1M17) using discovery studio client version 4.0 (a) image of 1M17 with quercetin without active site pocket; (b) image of 1M17 with quercetin with active site pocket (c) image of 1M17 with 6f without active site pocket; (d) image of 1M17 with 6f with active site pocket. | ||
3. Experimental section
3.1 Materials and method
All the chemicals used during the reactions were procured from Spectrochem, India. 1H NMR and 13C NMR spectra were recorded at the room temperature on Varian 400 MHz spectrometer and 100 MHz respectively. Chemical shift values were reported with reference to TMS as an internal standard. The samples were prepared by dissolving the synthesized compounds in DMSO-d6, chemical shifts were expressed in δ (ppm) and coupling constants (J) in hertz. The splitting pattern abbreviations are as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, unresolved multiplet; dd, doublet of doublet. The column chromatography was performed on Merck silica gel 60 (230–400 mesh). Analytical thin layer chromatography was carried out on the precoated Merck silica gel 60F254 and iodine was used for development of compounds. IR was recorded on FTIR IR Affinity −1 Shimadzu spectrophotometer. CHNS analysis was recorded on Elementar Vario El-III. Dulbecco's Modified Eagle medium (DMEM), Fetal Bovine Serum (FBS) and phosphate buffer saline were procured from Invitrogen. Trypsin and antibiotic solutions were procured from Sigma-Aldrich.3.2 General procedure
:30) to obtain methyl 2-acetyl-3,5-dimethoxybenzoate in 52% yield.:20) to acquire clean methyl 2-acetyl-3,5-dihydroxybenzoate with 68% yield.:50) and methyl 7-hydroxy-2-(4-methoxyphenyl)-4-oxo-3,4-dihydro-4H-chromene-5-carboxylate was achieved in 45% yield.:60) and obtained methyl 7-hydroxy-2-(4-methoxyphenyl)-4-oxo-4H-chromene-5-carboxylate in 60% yield.
Methyl 7-hydroxy-2-(4-methoxyphenyl)-4-oxo-4H-chromene-5-carboxylate (6a). Mp: 226–228 °C; IR (KBr, cm−1), 3446 (OH), 1735 (C
O), 1624 (CO); 1H NMR (400 MHz, DMSO-d6) δ 3.81 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 6.79 (s, 1H, 3-H), 6.82 (d, 1H, 8-H, J = 1.6 Hz), 7.10 (d, 1H, 6-H, J = 2 Hz), 7.12 (d, 2H, 3′, 5′-H, J = 7.2 Hz), 8.04 (d, 2H, 2′, 6′-H, J = 7.2 Hz), 11.14 (s, 1H, OH); 13C NMR, (100 MHz, DMSO-d6) δ 52.86 (OCH3) 55.97 (C-4′, OCH3), 104.25 (C-8), 105.58 (C-3), 113.55 (C-10), 113.94 (C-6), 114.81 (C-3′), 114.98 (C-5′), 123.49 (C-1′), 127.11 (C-6′), 128.59 (C-2′), 134.55 (C-5), 157.7 (C-9), 158.55 (C-4′), 162.39 (C-2), 162.54 (C-7), 169.19 (C-11), 175.68 (C-4); LCMS (ESI) m/z calculated for C18H14O6: 326.3 and found 327.0; HRMS m/z 327.0863 [M + 1]+ elemental analysis calculated for C18H14O6: C, 66.25, H, 4.32; found: C, 66.31, H, 4.28.
Methyl 7-hydroxy-2-(4-methylphenyl)-4-oxo-4H-chromene-5-carboxylate (6b). Mp: 240–242 °C; IR (KBr, cm−1), 3516 (OH), 1737 (C
O), 1627 (CO); 1H NMR (400 MHz, DMSO-d6) δ 2.39 (s, 3H, CH3), 3.81 (s, 3H, OCH3), 6.83 (s, 1H, 3-H), 6.84 (s, 1H, 8-H), 7.10 (s, 1H, 6-H), 7.38 (d, 2H, J = 8.0 Hz, 3′, 5′-H), 7.95 (d, 2H, J = 8.0 Hz, 2′, 6′-H), 11.18 (s, 1H, OH); 13C NMR, (100 MHz, DMSO-d6) δ 21.51 (CH3), 52.88 (OCH3), 104.27 (C-8), 106.46 (C-3), 113.43 (C-10), 113.68 (C-6), 126.68 (C-2′, 6′), 128.55 (C-1′), 130.14 (C-3′, 5′), 130.25 (C-4′), 134.58 (C-5), 142.42 (C-9), 157.78 (C-2), 162.49 (C-7) 169.14 (C-11), 175.76 (C-4); LCMS (ESI) m/z calculated for C18H14O5: 310.3 and found 311.0. Elemental analysis calculated for C18H14O5: C, 69.66, H, 4.54; found: C, 69.61, H, 4.49.
Methyl 2-(3,4-dimethoxyphenyl)-7-hydroxy-4-oxo-4H-chromene-5-carboxylate (6c). Mp: 233–236 °C; IR (KBr, cm−1), 3444 (OH), 1737 (C
O), 1627 (CO); 1H NMR (400 MHz, DMSO-d6) δ 3.78 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 6.79 (s, 1H, 3-H), 6.84 (s, 1H, 8-H), 7.13 (m, 2H, 5′-H, 6-H), 7.52 (s, 1H, 2′-H), 7.63 (d, 1H, 6′-H, J = 8 Hz), 11.08 (s, 1H, OH); 13C NMR, (100 MHz, DMSO-d6) δ 52.87 (OCH3), 56.17 (OCH3), 56.32 (OCH3), 104.34 (C-8), 105.89 (C-3), 109.83 (C-6′), 112.14 (C-5′), 113.39 (C-10), 113.53 (C-6), 120.29 (C-1′), 123.59 (C-2′), 134.51 (C-5), 149.47 (C-3′), 152.34 (C-4′), 157.74 (C-9), 162.36 (C-2), 162.48 (C-7), 169.19 (C-11), 175.74 (C-4); LCMS (ESI) m/z calculated for C19H16O7: 356.32 and found 357.0; elemental analysis calculated for C19H16O7: C, 64.04, H, 4.52; found: C, 64.16, H, 4.59.
Methyl 2-(4-chlorophenyl)-7-hydroxy-4-oxo-4H-chromene-5-carboxylate (6d). Mp: 262–268 °C; IR (KBr, cm−1), 3645 (OH), 1714 (C
O), 1697 (CO); 1H NMR (400 MHz, DMSO-d6) δ 3.78 (s, 3H, OCH3), 6.80 (s, 1H, 3-H), 6.89 (s, 1H, 8-H), 7.08 (s, 1H, 6-H), 7.60 (d, 2H, 2′, 6′-H, J = 8.4 Hz), 8.07 (d, 2H, 3′, 5′-H, J = 8.8 Hz), 11.19 (s, 1H, OH); 13C NMR, (100 MHz, DMSO-d6) δ 52.91 (OCH3), 104.32 (C-8), 107.48 (C-3), 113.36 (C-10), 113.86 (C-6), 128.60 (C-2′, 6′), 129.62 (C-3′, 5′), 130.28 (C-1′), 134.60 (C-5), 136.99 (C-9), 157.79 (C-4′), 161.24 (C-2), 162.63 (C-7), 169.06 (C-11), 175.75 (C-4); LCMS (ESI) m/z calculated for C17H11ClO5: 330.71 and found 331.0; elemental analysis calculated for C17H11ClO5: C, 61.73, H, 3.34; found: C, 61.67, H, 3.28.
Methyl 2-(4-bromophenyl)-7-hydroxy-4-oxo-4H-chromene-5-carboxylate (6e). Mp: 275–277 °C; IR (KBr, cm−1), 3564 (OH), 1737 (C
O), 1627 (CO); 1H NMR (400 MHz, DMSO-d6) δ 3.78 (s, 3H, OCH3), 6.81 (s, 1H, 8-H), 6.90 (s, 1H, 6-H), 7.07 (s, 1H, 3-H), 7.75 (d, 2H, 2′, 6′-H, J = 8 Hz), 8.0 (d, 2H, 3′, 5′-H, J = 8 Hz), 11.18 (s, 1H, OH); 13C NMR, (100 MHz, DMSO-d6) δ 53.03 (OCH3) 104.38 (C-8), 107.41 (C-3), 113.35 (C-10), 113.93 (C-6), 126.01 (C-2′, C-6′), 128.78 (C-3′, 5′), 130.59 (C-1′), 132.62 (C-5), 134.62 (C-9), 157.84 (C-4′), 161.52 (C-2), 162.70 (C-7), 169.19 (C-11), 175.90 (C-4); LCMS (ESI) m/z calculated for C17H11BrO5: 375.17 and found 376.9, 378.9. Elemental analysis calculated for C17H11BrO5: C, 54.42, H, 2.95; found: C, 54.48, H, 2.95.
Methyl 2-(2-fluorophenyl)-7-hydroxy-4-oxo-4H-chromene-5-carboxylate (6f). Mp: 222–225 °C; IR (KBr, cm−1), 3645 (OH), 1732 (C
O), 1697 (CO); 1H NMR (400 MHz, DMSO-d6) δ 3.81 (s, 3H, OCH3), 6.65 (s, 1H, 3-H), 6.86 (s, 1H, 8-H), 7.06 (s, 1H, 6-H), 7.41–7.5 (m, 2H, 5′-H, 6′-H), 7.63–7.70 (m, 1H, 4′-H), 8.02–8.06 (m, 1H, 3′-H), 11.22 (s, 1H, OH); 13C NMR, (100 MHz, DMSO-d6) δ 52.81 (OCH3), 104.15 (C-8), 110.07 (C-6). 110.71 (C-10), 116.12 (C-5′), 116.30 (C-3), 125.21 (C-3′), 128.93 (C-1′), 128.96 (C-6′), 131.36 (C-9), 136.50 (C-5), 159.11 (C-2′), 161.07 (C-4′), 163.43 (C-2), 164.45 (C-7), 169.39 (C-11), 185.83 (C-4); LCMS (ESI) m/z calculated for C17H11FO5: 314.26 and found 315.0. Elemental analysis calculated for C17H11FO5: C, 64.96, H, 3.52; found: C, 64.91, H, 3.56.
7-Hydroxy-2-(4-methoxyphenyl)-5-methyl-4-oxo-4H-chromene (6g). Mp: 245–249 °C; IR (KBr, cm−1), 3566 (OH), 1704 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 2.68 (s, 3H, CH3), 3.83 (s, 3H, OCH3), 6.63 (s, 1H, 3-H), 6.67 (s, 1H, 8-H), 6.81 (s, 1H, 6-H), 7.08 (d, 2H, 2′, 6′-H, J = 8.4 Hz), 7.97 (d, 2H, 3′, 5′-H, J = 8.8 Hz), 10.59 (s, 1H, OH); 13C NMR, (100 MHz, DMSO-d6) δ 22.92 (CH3), 55.96 (C-4′, OCH3), 101.38 (C-8), 106.82 (C-3), 114.95 (C-3′, 5′), 117.19 (C-6), 123.73 (C-1′), 128.27 (C-2′, 6′), 128.56 (C-10), 141.90 (C-5), 159.32 (C-9), 160.71 (C-4′), 161.57 (C-2), 162.26 (C-7) 178.91 (C-4); LCMS (ESI) m/z calculated for C17H14O4: 282.29 and found 283.0. Elemental analysis calculated for C17H14O4: C, 72.32, H, 4.99; found: C, 72.36, H, 4.93.
7-Hydroxy-2-(4-methylphenyl)-5-methyl-4-oxo-4H-chromene (6h). Mp: 252–255 °C; IR (KBr, cm−1), 3565 (OH), 1710 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 2.38 (s, 3H, CH3), 2.69 (s, 3H, CH3), 6.65 (s, 1H, 3-H), 6.72 (s, 1H, 8-H), 6.82 (s, 1H, 6-H), 7.36 (d, 2H, 3′, 5′-H, J = 8.4 Hz), 7.92 (d, 2H, 2′, 6′-H, J = 7.6 Hz), 10.62 (s, 1H, OH); 13C NMR, (100 MHz, DMSO-d6) δ 21.54 (CH3), 22.92 (CH3), 101.40 (C-8), 107.65 (C-3), 115.16 (C-1′), 117.27 (C-6), 126.41 (C-3′, C-5′), 128.76 (C-10), 130.12 (C-2′, C-6′), 141.96 (C-4′, C-5), 159.37 (C-9), 160.78 (C-2), 161.67 (C-7), 178.94 (C-4); LCMS (ESI) m/z calculated for C17H14O3: 266.29 and found 267.0. Elemental analysis calculated for C17H14O3: C, 76.67, H, 5.29; found: C, 76.62, H, 5.26.
2-(2-Fluorophenyl)-7-hydroxy-5-methyl-4-oxo-4H-chromene (6i). Mp: 252–255 °C; IR (KBr, cm−1), 3564 (OH), 1714 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 2.69 (s, 3H, CH3), 6.55 (s, 1H, 3-H), 6.67 (s, 1H, 6-H), 6.78 (s, 1H, 8-H), 7.41–7.48 (m, 2H, 5′, 6′-H), 7.62–7.63 (m, 1H, 4′-H), 7.98–8.0 (m, 1H, 3′-H), 10.71 (s, 1H, OH). 13C NMR, (100 MHz, DMSO-d6) δ 22.90 (CH3), 101.34 (C-8), 112.91 (C-6′), 113.01 (C-5′), 114.96 (C-4′), 117.24 (C-6), 117.47 (C-3), 125.71 (C-1′), 129.78 (C-3′), 133.73 (C-2′), 133.82 (C-10), 142.13 (C-5), 156.58 (C-9), 159.54 (C-2), 161.92 (C-7), 178.58 (C-4); LCMS (ESI) m/z calculated for C16H11FO3: 270.25 and found 271.0. Elemental analysis calculated for C16H11FO3: C, 71.10, H, 4.09; found: C, 71.16, H, 4.17.
2-(3,4-Dimethoxylphenyl)-7-hydroxy-5-methyl-4-oxo-4H-chromene (6j). Mp: 231–234 °C; IR (KBr, cm−1), 3564 (OH), 1704 (C
O); 1HMR (400 MHz, DMSO-d6) δ 2.69 (s, 3H, CH3), 3.86 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 6.76 (s, 1H, 3-H), 6.79 (s, 1H, 8-H), 6.91 (d, 1H, 6-H), 7.13 (d, 1H, 6′-H, J = 7.2 Hz), 7.17 (d, 1H, 2′-H, 7.2 Hz), 7.77 (d, 1H, 5′-H, J = 7.2 Hz), 10.60 (s, 1H, OH). 13C NMR, (100 MHz, DMSO-d6) δ 22.90 (CH3) 58.90 (OCH3), 59.30 (OCH3), 101.33 (C-8), 104.20 (C-3), 110.30 (C-6), 113.35 (C-1′), 114.27 (C-10), 116.57 (C-6′), 120.50 (C-2′), 121.86 (C-5′), 141.86 (C-5), 149.83 (C-3′), 153.23 (C-4′), 159.30 (C-9), 160.80 (C-2), 162.55 (C-7), 178.90 (C-4); LCMS (ESI) m/z calculated for C18H16O5: 312.31 and found 313.0. Elemental analysis calculated for C18H16O5: C, 69.21, H, 5.15; found: C, 69.17, H, 5.11.
3.3 Biology
3.4 Molecular docking studies
The molecular modelling studies were carried out on Windows 7 64-bit operating system using Maestro 11.2 software. The GLIDE docking application of Maestro 11.2 software was used to calculate GScore, which is based on an empirical scoring function and deploy a combination of several parameters. GScore (docking score) was calculated in kcal mol−1 and it included ligand–protein interaction energies, hydrophobic interactions, hydrogen bonds, internal energy, π–π stacking interactions, root mean square deviation (RMSD) and desolvation. The X-ray crystallographic structure of erlotinib cocrystallized with EGFR was obtained from the protein data bank (PDB ID: 1M17). The ATP binding site of EGFR was prepared for docking studies in which erlotinib was removed from the active site, hydrogen atoms were added to the structure with their standard geometry. Active sites were observed from the sequence analysis and software run and it was used in predicting interactions at the active site between the selected compounds 6f and quercetin with EGFR. The 2D structures of the docked compounds were generated, transformed to 3D, protonated, and energy was minimized by using Ligprep application. Grid generation and ligand docking was performed to obtain ligand interaction diagram with 1M17 EGFR and docking score. Receptor–ligand interaction images were obtained from discovery studio client version.4. Conclusion
In summary, we successfully completed a simple and convenient 5 steps synthesis for a naturally occurring rugosaflavonoid (6a) with better yield and evaluated its cytotoxicity against breast cancer cell lines. Molecular docking score of its derivatives with EGFR (1M17) showed that it interacted with active site pocket of 1M17. Some of the synthetic derivatives showed better dose dependent activity against MCF7 than the natural molecule rugosaflavonoid and were found to be non toxic to NIH3T3 cell lines at lower concentration. Though flavonoids are known from the years for their bioactive potential but there is still scope in modifications of natural analogs, which may provide us the lead molecules.Conflict of interest
There is no conflict of interest among the authors.Acknowledgements
Authors are really thankful to Hu et al. for the isolation and characterization of rugosaflavonoid because of his team work we are able to develop synthetic route for naturally occurring compound (6a). We acknowledge the unconditional support of Mr Vinod Devaraji to understand Maestro 11.2 software to perform the molecular docking studies. Authors are also thankful to Agharkar Research Institute, Pune, India, for providing financial support and infrastructure.Notes and references
- R. Siegel, J. Ma, Z. Zou and A. Jemal, Ca-Cancer J. Clin., 2014, 64, 9–29 CrossRef PubMed.
- C. Andreetta, A. M. Minisini, M. Miscoria and F. Puglisi, Crit. Rev. Oncol. Hematol., 2010, 76, 99–111 CrossRef PubMed.
- A. Jemal, R. Siegel, J. Xu and E. Ward, Ca-Cancer J. Clin., 2010, 60, 277–300 CrossRef PubMed.
- N. G. Anderson, T. Ahmad, K. Chan, R. Dobson and N. G. Bundred, Int. J. Cancer Res., 2001, 94, 774–782 CrossRef CAS PubMed.
- A. Arora and E. M. Scholar, J. Pharmacol. Exp. Ther., 2005, 315, 971–979 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed.
- A. Gaspar, M. J. Matos, J. Garrido, A. Uriarte and F. Borges, Chem. Rev., 2014, 114, 4960–4992 CrossRef CAS PubMed.
- A. K. Verma and R. Pratap, Nat. Prod. Rep., 2010, 27, 1571–1593 RSC.
- S. M. Gault and P. M. Synge, The Dictionary of Roses in Colour, Ebury Press, London, 1971 Search PubMed.
- L. Putian and Y. Jiang, Flora of China, Chinese Science Press, Beijing, 1977 Search PubMed.
- S. Ochir, B. J. Park, M. Nishizawa, T. Kanazawa, M. Funaki and T. J. Yamagishi, J. Nat. Med., 2010, 64, 383–387 CrossRef CAS PubMed.
- Y. Hashidoko, Phytochemistry, 1996, 43, 535–549 CrossRef CAS.
- Y. Hashidoko, S. Tahara and J. Mizutani, Phytochemistry, 1993, 32, 387–390 CrossRef CAS.
- H. J. An, I. T. Kim, H. J. Park, H. M. Kim, J. H. Choi and K. T. Lee, Int. Immunopharmacol., 2011, 11, 504–510 CrossRef CAS PubMed.
- Z. P. Xiao, H. K. Wu, T. Wu, H. Shi, B. Hang and H. A. Aisa, Chem. Nat. Compd., 2006, 42, 736–737 CrossRef CAS.
- X. Gao, L. Yang, L. Shu, Y. Shen, Y. Zhang and Q. Hu, Heterocycles, 2012, 85, 1925–1931 CrossRef CAS.
- Q. F. Hu, B. Zhou, J. M. Huang, Z. Y. Jiang, X. Z. Huang, L. Y. Yang, X. M. Gao, G. Y. Yang and C. T. Che, J. Nat. Prod., 2013, 76, 1866–1871 CrossRef CAS PubMed.
- J. McNulty and D. Mcleod, Tetrahedron Lett., 2013, 54, 6303–6306 CrossRef CAS.
- J. Maresh, J. Zhang, Y. L. Tzeng, N. A. Goodman and D. G. Lynn, Bioorg. Med. Chem. Lett., 2007, 17, 3281–3286 CrossRef CAS PubMed.
- J. Johann, PCT Int. Appl., 2010, 2010022953 Search PubMed.
- G. Bringmann, T. F. Noll, T. Gulder, M. Dreyer, M. Grune and D. Moskau, J. Org. Chem., 2007, 72, 3247–3252 CrossRef CAS PubMed.
- M. M. Naik, S. G. Tilve and V. P. Kamat, Tetrahedron Lett., 2014, 55, 3340–3343 CrossRef CAS.
- J. M. Useros and J. G. Foncillas, Oral Oncol., 2015, 51, 423–430 CrossRef PubMed.
- J. Wang, J. M. Yu, S. W. Jing, Y. Guo, Y. J. Wu, N. Li, W. P. Jiao, L. Wang and Y. J. Zhang, Asian Pac. J. Cancer Prev., 2014, 15, 5889–5893 CrossRef PubMed.
- J. Stamos, M. X. Sliwkowski and C. Eigenbrot, J. Biol. Chem., 2002, 277, 46265–46272 CrossRef CAS PubMed.
- P. Singh and F. Bast, Med. Chem. Res., 2014, 23, 5074–5085 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04971d |
| This journal is © The Royal Society of Chemistry 2017 |