
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene oxide-modified electrospun polyvinyl alcohol nanofibrous scaffolds with potential as skin wound dressings
Qiang
Zhang
a,
Qiaoyue
Du
a,
Yanan
Zhao
a,
Feixiang
Chen
a,
Zijian
Wang
a,
Yaxing
Zhang
a,
Hong
Ni
b,
Hongbing
Deng
c,
Yinping
Li
*a and
Yun
Chen
 *ad
*ad
aDepartment of Biomedical Engineering, School of Basic Medical Sciences, Wuhan University, Wuhan 430071, China. E-mail: liyinping@whu.edu.cn; yunchen@whu.edu.cn
bSchool of Life Science, Hubei University, Wuhan 430062, China
cDepartment of School of Environmental Sciences, Resorece and Environmental Sciences, Wuhan 430065, China
dHubei Province Key Laboratory of Allergy and Immune Related Dieases, Wuhan University, Wuhan 430071, China
First published on 31st May 2017
Abstract
A series of ultrafine polyvinyl alcohol (PVA)/graphene oxide (GO) nanofibrous scaffolds (PGNSs) with different GO content (0, 0.25, 0.5, and 1.0%) was fabricated via an electrospinning process. The morphology, composition, mechanical behavior, surface wettability and protein adsorptivity of these scaffolds were investigated. The results showed that the diameter of the nanofibers in the PGNSs was 230 ± 68, 197 ± 46, 188 ± 38 or 164 ± 54 nm when the GO content in the scaffolds was 0, 0.25, 0.5 or 1.0%, respectively. The ATR-Fourier transform infrared spectra, Raman spectra, and X-ray diffraction results revealed the successful introduction of GO into the PVA matrix. Mechanical testing indicated that GO increased the tensile strength while reducing the ductility of the scaffolds. The biological properties of the scaffolds were evaluated by water contact angle measurements, protein adsorption assays, hemolysis ratio testing, cell co-culturing and in vivo skin repair in animal models. The results showed that the GO-modified scaffolds had favorable hydrophilicity and enhanced protein-enrichment ability. No visible hemolysis was observed for any of the scaffolds. Cellular behavior on the scaffolds revealed that the PGNSs were suitable for the growth and adhesion of L929 fibroblasts. Moreover, the PGNS containing a 0.25% GO content was the most suitable scaffold for cell viability and adhesion. In vivo wound repair assessments showed that the wound contraction rate when treated by the PGNS containing a 0.25% GO content exceeded 90% within 9 days, suggesting excellent potential of the PGNS for skin tissue engineering.
1 Introduction
The skin's utmost important function is to form a natural barrier to pathogens and to any damage between the internal and external environment in bodily defense.1 If the damaged skin is not treated in time, open skin wounds are prone to be invaded by microorganisms. Thus, a wound infected by microorganisms will cause much pain and disfigurement, and could even be life threatening.2 Therefore, adequate wound treatment for rapid healing is indispensable to prevent infection. To heal a skin defect, the transplantation of autologous, xenogeneic and allogeneic skin grafts and tissue engineered skin replacements has been widely used.3 However, the transplantation of autografts, xenografts and allografts is not economical and is also limited by several potential disadvantages, such as requiring an extra incision, the availability of a compatible donor, and a mismatch in size between the donor skin and the injured skin.4,5 To resolve these problems, commercialized tissue engineered skin-substitutes, such as Dermagraft, Integra, Alloderm, and Apligraft, have been developed.5 However, in order to develop more and better products to meet the demands of the market, these substitutes need to be further optimized.6 Therefore, ideal dressing materials for skin tissue repair are still lacking.Nanoscale biomaterials can mimic the structure of extracellular matrices (ECMs) and provide a platform for cell attachment, differentiation and proliferation. The amount of research on nano-biomaterials as skin tissue engineering scaffolds is increasing tremendously. Five methods, including phase separation, drawing, template assisted synthesis, self-assembly, and electrospinning, have been commonly used to fabricate nanofibrous scaffolds for skin tissue engineering.7 Of these techniques, electrospinning is one of the most simple and cost-effective methods to prepare a variety of polymeric nanofibers.8 As a result, the fabrication of ultrafine fibers from various natural and synthetic polymers by electrospinning has been extensively investigated.9
Polyvinyl alcohol (PVA) has excellent water absorption ability and biocompatibility,10 and can provide a suitable environment for cell growth because of its semi-permeability, flexibility and pH-stability.11 Based on these advantages, electrospun PVA nanofibrous scaffolds have been used for skin tissue engineering.12,13 However, owing to its insufficient functional groups and cell recognition sites, the bioactivity of PVA, including its protein adsorption ability, cell compatibility and skin repair efficiency, needs to be further improved.13,14 One of the most effective methods is to modify PVA using some special bioactive composites. Graphene oxide (GO) is a good candidate with bioactive performance due to its large surface area, abundant functional groups and protein adsorption properties.15 Several studies have shown that a small amount of GO (less than 5%) could significantly improve many properties of the matrix, such as mechanical strength,16 electronic conductivity,17 and protein affinity.18 For example, GO has been used to bind proteins and vitamins to improve cell proliferation.19 Moreover, the addition of GO endows the matrix with some new functions such as improving the drug loading capacity,20 directing cell behaviors,21 promoting wound healing,22 and enhancing bone tissue formation.18 Shin et al. have demonstrated that GO-incorporated nanofibrous films could promote the adhesion of human fibroblasts, and accelerate skin regeneration.15 In addition, because of its favorable antibacterial properties, GO can prevent infection in wounds.23
Therefore, in this work, we introduced GO as a modifier to improve the bioactivity of PVA-based nanofibrous scaffolds. It was expected that the bioactivity of PVA could be enhanced because of the addition of GO. In order to verify our hypothesis and further investigate the effects of the GO content on their biological properties, a series of PVA/GO nanofibrous scaffolds (PGNSs) with different GO content was fabricated via an electrospinning process, and then the physicochemical properties and the in vitro biocompatibility of the PGNSs were investigated. Finally, the in vivo wound repair efficiency of the PGNSs was evaluated in a defective rat wound model by macroscopical observation and histological analysis.
2 Materials and methods
2.1 Materials
The L929 mouse fibroblast cell line used in this study was from the Shanghai Institutes for Biological Sciences (SIBS), Chinese Academy of Sciences (Shanghai, China). Alpha minimum essential medium (α-MEM) and fetal bovine serum (FBS) were obtained from Sigma Aldrich. Polyvinylalcohol (PVA, polymerization degree PD = 1750 ± 50) was purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Graphene oxide (GO, diameter of 0.5–5 μm and thickness of 0.6–1.0 nm, purity > 99%) was obtained from Suzhou Hengqiu Technology Co., Ltd (Suzhou, China). 3-(4,5-Dimethyl-2-thiazoly (2,5-diphenyl-2H-tetrazolium bromide)) (MTT) was obtained from Invitrogen Corporation (Gibco BRL, Grand Island, NY, USA) and 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) was obtained from Beijing Solarbio Science & Technology Co., Ltd. (Beijing, China). The other chemicals were of analytical grade and used without further treatment.2.2 Fabrication of electrospun GO-modified PVA nanofibrous scaffolds
The fabrication procedure of the PVA/GO composite solution is as follows: PVA solution was prepared by dissolving 2 g of PVA granules in a 20 mL acetic acid/distilled water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solvent mixture with constant stirring at 80 °C for 4 h. Similarly, 0.4 g GO powder was added to 20 mL of deionized water and stirred by magnetic stirring for 30 min, and then ultrasonically dispersed for 30 min. After the preparation of both solutions, 0, 0.25, 0.5, and 1.0 mL of the GO solution (20 mg mL−1) was dispersed into 20 mL of PVA solution with magnetic stirring. After 24 h, homogeneous composite solutions were obtained, which were used for electrospinning according to a previous method.24 Briefly, the PVA/GO composite solution was loaded into a 5 mL syringe with a needle with an inner diameter of 0.69 mm, and then was electrospun at a positive DC voltage of 18 kV and a flow rate of 1.2 mL h−1. A piece of aluminum foil used as a collector was placed under the capillary needle tip at a distance of 14 cm. The electrospun PVA/GO nanofibrous scaffolds (PGNSs) were cross-linked and stabilized in glutaraldehyde steam in a ventilation cabinet for 36 h. The obtained fibrous scaffolds were stored in a desiccator for further characterization. The PVA/GO nanofibrous scaffolds with different GO content were coded as PGNS-n, where n = 0, 0.25, 0.5 or 1.0, corresponding to the weight percent of GO in the scaffolds. For example, PGNS-0.25 indicates the PVA/GO nanofibrous scaffold with a 0.25% GO content and PGNS-0 indicates the neat PVA nanofibrous scaffold without GO.
:1, v/v) solvent mixture with constant stirring at 80 °C for 4 h. Similarly, 0.4 g GO powder was added to 20 mL of deionized water and stirred by magnetic stirring for 30 min, and then ultrasonically dispersed for 30 min. After the preparation of both solutions, 0, 0.25, 0.5, and 1.0 mL of the GO solution (20 mg mL−1) was dispersed into 20 mL of PVA solution with magnetic stirring. After 24 h, homogeneous composite solutions were obtained, which were used for electrospinning according to a previous method.24 Briefly, the PVA/GO composite solution was loaded into a 5 mL syringe with a needle with an inner diameter of 0.69 mm, and then was electrospun at a positive DC voltage of 18 kV and a flow rate of 1.2 mL h−1. A piece of aluminum foil used as a collector was placed under the capillary needle tip at a distance of 14 cm. The electrospun PVA/GO nanofibrous scaffolds (PGNSs) were cross-linked and stabilized in glutaraldehyde steam in a ventilation cabinet for 36 h. The obtained fibrous scaffolds were stored in a desiccator for further characterization. The PVA/GO nanofibrous scaffolds with different GO content were coded as PGNS-n, where n = 0, 0.25, 0.5 or 1.0, corresponding to the weight percent of GO in the scaffolds. For example, PGNS-0.25 indicates the PVA/GO nanofibrous scaffold with a 0.25% GO content and PGNS-0 indicates the neat PVA nanofibrous scaffold without GO.
2.3 Characterization
| Hemolysis ratio (%) = [(AS − AN)/(AP − AN)] × 100 | (1) |
2.3.5.1 Cell culture. L929 mouse fibroblasts were seeded in α-MEM supplemented with 10% FBS and 1% antibiotics in a 25 cm2 cell culture flask and incubated at 37 °C in a humidified atmosphere containing 5% CO2. The culture medium was replaced every three days. Cells were subcultured upon 80% confluency with 0.25% trypsin-0.1% EDTA.
2.3.5.2 Cell viability. The extracts from the PGNSs were prepared according to ISO 10993-12:2007. The PGNSs were immersed in cell culture medium (0.2 g of membrane per 1 mL of α-MEM) and incubated at 37 °C for 72 h. The extracts were collected and stored at 4 °C before use.
Cell viability was evaluated by an MTT assay at 24, 48 and 72 h after cell seeding. Briefly, L929 cells were re-suspended in culture medium and incubated in a 96-well tissue culture plate at a density of 1 × 103 cells per well (200 μL per well) at 37 °C in a humidified atmosphere of 5% CO2 for 24 h. Then the medium was replaced with the extracts. A culture medium with the same number of cells was used as a control. After incubating for 24, 48, or 72 h, the cells were treated with 20 μL of MTT per well and incubated for another 4 h at 37 °C in a humidified 5% CO2 atmosphere. Afterwards, MTT solutions were removed and 200 μL of dimethyl sulfoxide was added to dissolve the formazan crystals. The plate was incubated at room temperature on a shaker for 30 min to enhance the dissolution of the formazan crystals. A 100 μL aliquot from each well was transferred to another 96-well tissue culture plate. Then absorbance values were tested at a wavelength of 490 nm using a multi-well microplate reader (Tecan GENios, Tecan Austria GmbH, Salzburg, Austria). The cell viability was calculated using the following equation:
| Cell viability (%) = (Atest/Acontrol) × 100 | (2) |
2.3.5.3 Cell adhesion and morphology. To evaluate the potential application of PGNSs as cell culture scaffolds, L929 cells were used to observe their adhesion, proliferation and distribution. A 10 mm stainless ring and PGNSs were placed in a 24-well plate and immersed in 70% ethanol, washed twice with sterile PBS and irradiated with UV light for 2 h. L929 cells were then seeded onto the surfaces of the PGNSs at a density of 2 × 104 cells per well and cultured with α-MEM at 37 °C in a 5% CO2 humidified atmosphere. Three days later, the cells were fixed in 2.5% glutaraldehyde and then washed with PBS three times. Finally, the samples were stained with 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) and analyzed with a fluorescence microscope (IX73, OLYMPUS, Japan).
The morphology of the cells cultured on the PGNSs was observed by SEM using 20 kV as the accelerating voltage. Briefly, the scaffolds with cultured cells were fixed in 2.5% glutaraldehyde overnight. Then, the scaffolds were washed with deionized water and dehydrated with graded concentrations (50, 70, 80, 90, and 100% v/v) of ethanol. Finally, the scaffolds were vacuum-dried and coated with gold for cell morphology observations.
| Wound contraction rate (%) = (Ao − Aa)/Ao × 100 | (3) |
3 Results and discussion
3.1 Fabrication of the PGNSs
According to the general literature, water was used as the solvent for PVA for electrospinning.9,14 In the pre-experiment, we found that a series of uniform electrospun PGNSs could be obtained when the concentration of the PVA solution was in a range of 8–15%. The final optimized concentration of the PVA solution for electrospinning was 10% in this work. However, we found that the tip of the needle was occasionally blocked by the electrospinning solution when water was used as the solvent. In order to avoid the blocking phenomenon, we tried to introduce 50% acetic acid into the water which significantly enhanced the spinnability of the PVA solution. Due to the nature of PVA, the electrospun PVA fibers easily dissolved in water. To avoid the dissolution of the PGNSs into water, glutaraldehyde steam was used to cross-link and stabilize them. As a result, a series of water-insoluble PVA nanofibrous scaffolds with or without GO were fabricated by an electrospinning process.3.2 Morphology and structure of the PGNSs
SEM images of the PGNSs are shown in Fig. 1. The PGNSs were formed by randomly oriented fibers. These fibers interweaved with each other and showed a smooth surface with no aggregation or nodes. The diameters of PGNS-0, PGNS-0.25, PGNS-0.5, and PGNS-1.0 were 230 ± 68, 197 ± 46, 188 ± 38, and 164 ± 54 nm, respectively. It was clear that the PGNSs were of nanoscale diameter. After GO was introduced into the scaffolds, the diameter of the nanofibers decreased gradually in the range from 197 ± 46 nm to 164 ± 54 nm with increased GO content. The decrease in fiber diameter is probably attributed to the following reasons: (1) the change in the properties of the electrospinning solution, such as surface tension and solution viscosity26 and (2) GO improved the electronic and ionic conductivity of the electrospinning solution so that the nanofibers were well elongated, split off, and separated into thinner nanofibers under the same processing conditions.21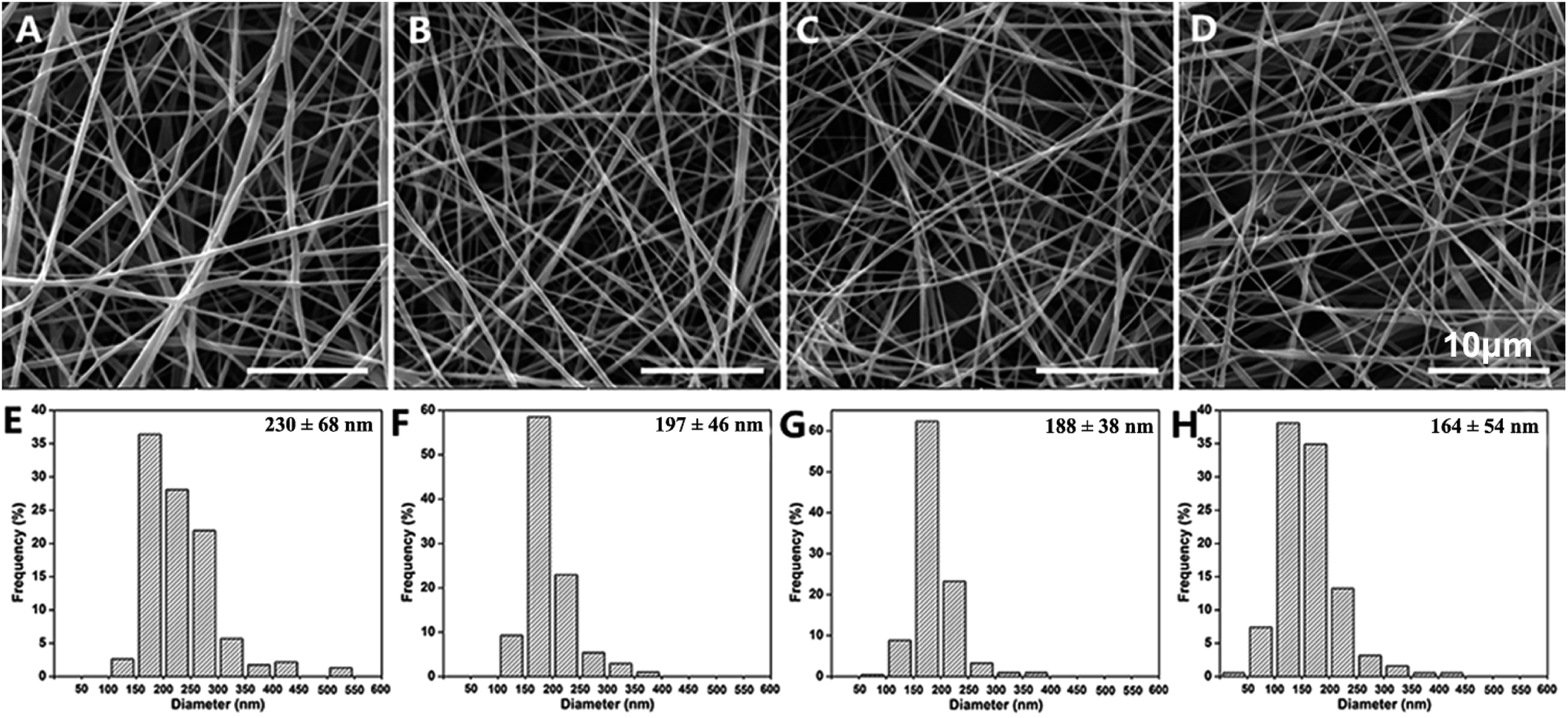 | ||
| Fig. 1 SEM images and the diameter distribution of the electrospun fibers in PGNS-0 (A and E); PGNS-0.25 (B and F); PGNS-0.5 (C and G); and PGNS-1.0 (D and H). | ||
3.3 Surface structure of the PGNSs
First, ATR-FTIR was used to confirm the existence of GO in the PVA matrix. As shown in Fig. 2A, the characteristic peaks at 3401, 1727, 1624, 1396, and 1065 cm−1 of GO are assigned to the stretching vibrations of hydroxyl groups, the stretching vibrations of carboxylic acid, skeletal vibrations of unoxidized graphitic domains, O–H deformations of the C–OH groups, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, respectively. However, in the ATR-FTIR spectra of PGNS-0.25, PGNS-0.5 and PGNS-1.0, the characteristic band of GO (1624 cm−1) was not observed. Thus, more sensitive Raman spectra were further used to confirm the incorporation of GO and these Raman spectra are shown in Fig. 2B. The Raman spectra of the PGNSs showed the characteristic D band and G band of GO at 1396 and 1624 cm−1, respectively, suggesting the presence of GO in PGNS-0.5 and PGNS-1.0.27 However, the characteristic D and G bands of GO were not observed in PGNS-0.25, which was due to insufficient GO loading. Afterwards, the crystalline characteristics of the PGNSs were investigated by XRD as shown in Fig. 2C. The characteristic XRD diffraction peaks of neat GO and PGNS-0 appear at 2θ = 10.4° and 19.46°, respectively. After being incorporated into the PVA matrix, the characteristic peak of GO disappeared, while PGNS-0.25, PGNS-0.5 and PGNS-1.0 only show the PVA diffraction peak at about 2θ = 19.5°. The results revealed that GO has a limited effect on the XRD pattern, resulting in an invisible change in the crystalline characteristics.
O stretching vibrations, respectively. However, in the ATR-FTIR spectra of PGNS-0.25, PGNS-0.5 and PGNS-1.0, the characteristic band of GO (1624 cm−1) was not observed. Thus, more sensitive Raman spectra were further used to confirm the incorporation of GO and these Raman spectra are shown in Fig. 2B. The Raman spectra of the PGNSs showed the characteristic D band and G band of GO at 1396 and 1624 cm−1, respectively, suggesting the presence of GO in PGNS-0.5 and PGNS-1.0.27 However, the characteristic D and G bands of GO were not observed in PGNS-0.25, which was due to insufficient GO loading. Afterwards, the crystalline characteristics of the PGNSs were investigated by XRD as shown in Fig. 2C. The characteristic XRD diffraction peaks of neat GO and PGNS-0 appear at 2θ = 10.4° and 19.46°, respectively. After being incorporated into the PVA matrix, the characteristic peak of GO disappeared, while PGNS-0.25, PGNS-0.5 and PGNS-1.0 only show the PVA diffraction peak at about 2θ = 19.5°. The results revealed that GO has a limited effect on the XRD pattern, resulting in an invisible change in the crystalline characteristics.
 | ||
| Fig. 2 ATR-FTIR spectra (A), Raman spectra (B) and XRD patterns (C) of the GO powder and electrospun nanofibers. | ||
3.4 Mechanical properties
A successful tissue engineering scaffold must have sufficient mechanical integrity to withstand manual manipulation.28 The tensile strength and elongation at break of native human skin were 5–30 MPa and 35–115%, respectively.29 If a scaffold could match the native skin's mechanical properties, it would be beneficial for the repair of skin wounds. The tensile strain–stress curves, and the tensile strength and elongation at break of the PGNSs, are shown in Fig. 3. The tensile strength and elongation at break were 2.0 MPa and 69.22% for PGNS-0, 2.74 MPa and 58.15% for PGNS-0.25, 4.12 MPa and 52.93% for PGNS-0.5, and 5.17 MPa and 36.74% for PGNS-1.0, respectively. Notably, the addition of GO increased the tensile strength of the scaffolds, but reduced the elongation at break. These results might be explained as follows. Firstly, GO made the scaffolds more effective in absorbing energy against an applied load. Secondly, due to the hydrogen interactions between the hydroxyl groups of PVA and the oxygen-containing functional groups of GO, more energy would be needed to disrupt the fibers, resulting in increased tensile strength. However, due to the 2D topological planar structure of GO, parts of GO under stress might tend to be perpendicular to the fibers. Therefore, these parts of GO cannot transfer the force. In addition, the interaction and large aspect ratio between the PVA matrix and GO restrict the movement of the polymer chains,30 decreasing the elongation at break.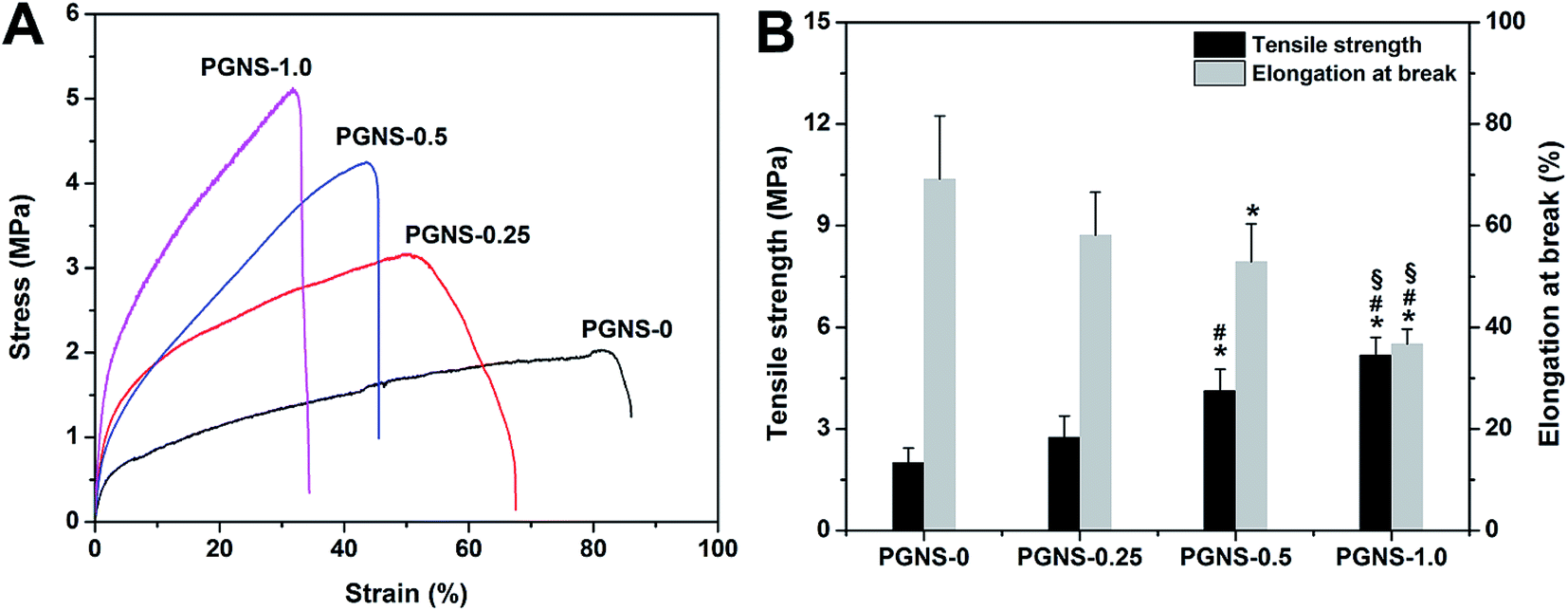 | ||
| Fig. 3 Typical stress–strain curves (A) and tensile strength and elongation at break (B) of the PGNSs. All values are the mean ± standard deviation (n = 3). *P < 0.05 (compared to PGNS-0), #P < 0.05 (compared to PGNS-0.25) and §P < 0.05 (compared to PGNS-0.5). | ||
In this study, all elongation at break values for the PGNSs were within the range of human skin's elongation at break, while the values of tensile strength of PGNS-0.5 and PGNS-1.0 were close to that of human skin. This indicated that the mechanical properties of PGNS-0.5 and PGNS-1.0 matched those of native skin, showing their good potential as skin wound dressings. However, for skin tissue engineering applications, soft skin grafts with low tensile strength should be more suitable to treat damaged skin. Barnes's study showed that a scaffold with low tensile strength was more advantageous for cell culture because of its similarity to soft tissues such as collagen and elastin.31 In addition, the grafts were seldom under high tensile stress when immobilized at the wound site.32 Therefore, PGNS-0.25, with a lower tensile strength than human skin, may be satisfactory as a wound dressing and may be a good potential candidate for a skin tissue engineering substitute.
3.5 Surface wettability and protein adsorptivity
The attachment and growth of cells need scaffolds with a relatively hydrophilic surface.33 In general, high hydrophilicity was favorable for cell adhesion. Moreover, scaffolds with improved protein adsorption properties will be more conducive to cell adhesion, growth, and wound healing.34 As shown in Fig. 4, GO reduces the hydrophilicity of scaffolds, while increasing protein adsorption. Moreover, the higher the GO content, the lower the hydrophilicity, but this also causes an invariable protein adsorption level. This was mainly because the covalent interactions between hydrophilic OH, C–O–C and COOH groups in GO and the OH group in PVA chains were enhanced through the cross-linking of glutaraldehyde while the number of free hydrophilic groups decreased. Thus, GO weakened the interaction between oxygen-containing groups and water molecules, resulting in decreased hydrophilicity. Besides, the addition of GO increased the specific surface area of the PGNSs and improved their affinity to proteins, leading to enhanced protein enrichment. However, due to steric hindrance, the amount of protein adsorbed was not significantly changed when the GO content increased. These results were consistent with Lampin's report.35 Finally, it should be noted that all of the PGNSs were still hydrophilic so they may be suitable for cell adhesion.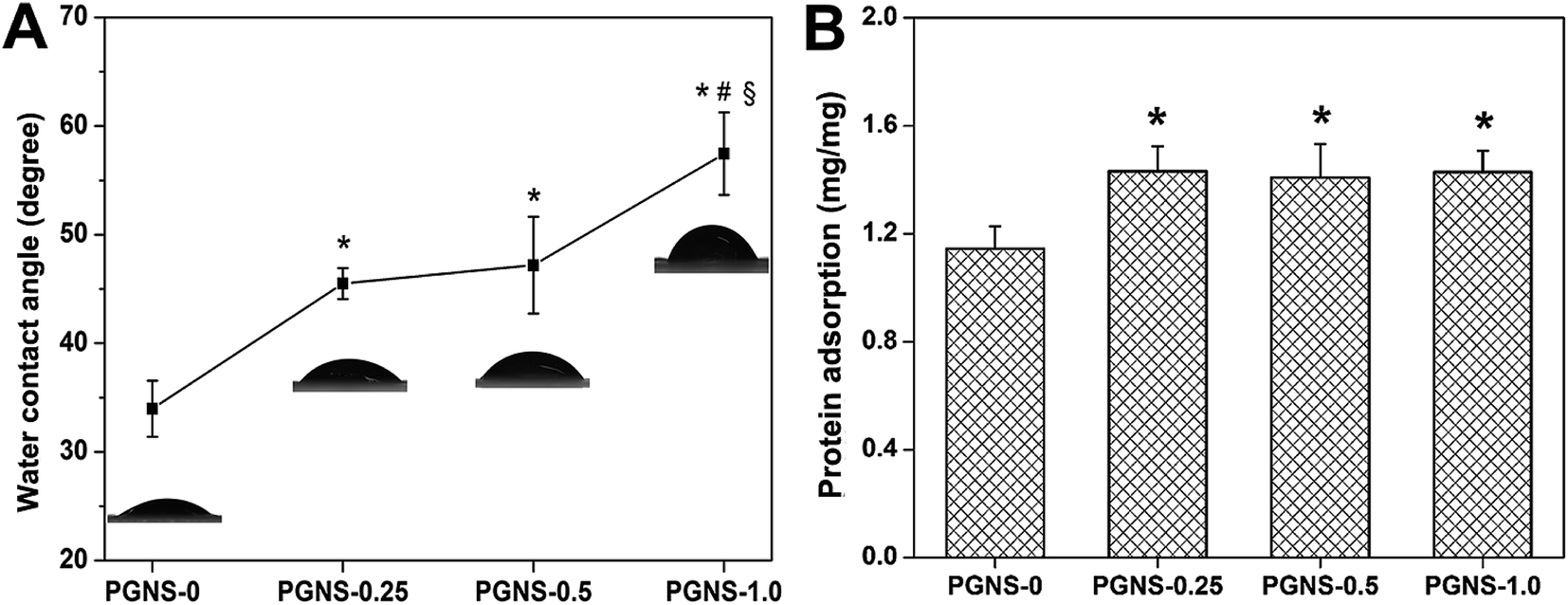 | ||
| Fig. 4 Water contact angle (A) and protein adsorption (B) of the PGNSs. *P < 0.05 (compared to PGNS-0), #P < 0.05 (compared to PGNS-0.25) and §P < 0.05 (compared to PGNS-0.5). | ||
3.6 Evaluation of the hemolysis ratio
The hemolysis ratio is a major concern for evaluating the hemocompatibility of biomaterials, as scaffolds will come into direct contact with blood in vivo.18 A scaffold which could cause obvious hemolysis is not suitable for tissue engineering. The results of the hemolysis assays of water (positive control), PBS (negative control), and the PGNSs are shown in Fig. 5. Obvious hemolysis was observed in water, while no visible hemolytic phenomenon was found in PBS and in the PGNS groups. The relative hemolysis percentage for all of the scaffolds was less than 2%, suggesting that no hemolysis occurs.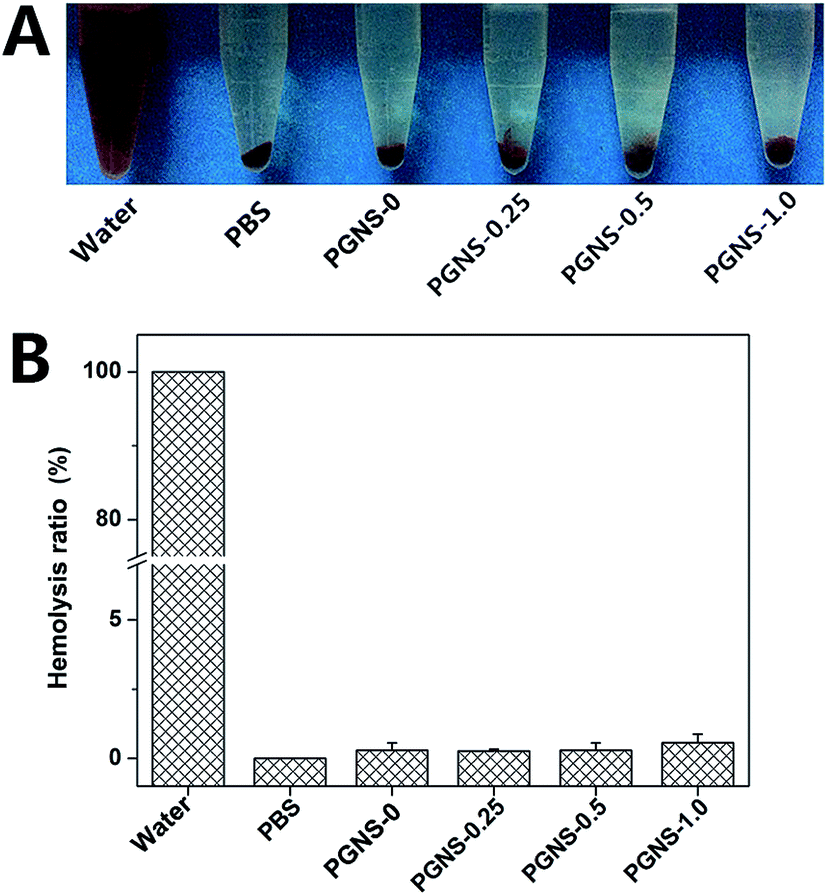 | ||
| Fig. 5 (A) Photo of a solution of rabbit red blood cells treated with water, PBS, and the nanofibrous scaffolds, followed by centrifugation. (B) The hemolysis ratio of rabbit red blood cells after being incubated with water, PBS, PGNS-0, PGNS-0.25, PGNS-0.5 or PGNS-1.0. | ||
3.7 Evaluation of cytocompatibility
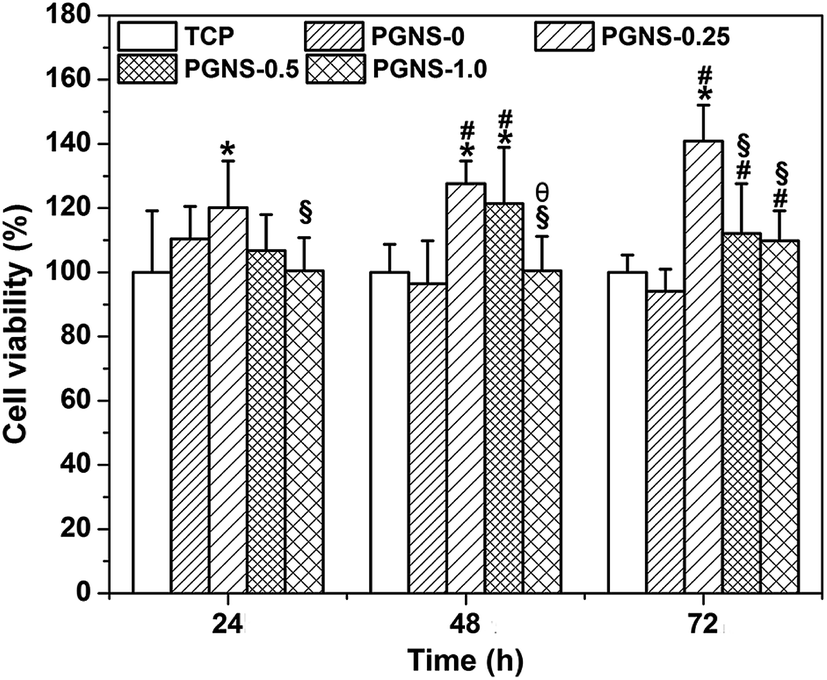 | ||
| Fig. 6 Cell viability of L929 cells cultured in extracts from the PGNSs for 24, 48, and 72 h. *P < 0.05 (compared to TCP), #P < 0.05 (compared to PGNS-0), §P < 0.05 (compared to PGNS-0.25) and θP < 0.05 (compared to PGNS-0.5). | ||
 | ||
| Fig. 7 Fluorescence microscopy images of cells stained with DAPI (A–E), cell density (F), and SEM images (G–K) of L929 cells cultured on TCP and on the surfaces of the PGNSs for 72 h. (A and G) TCP; (B and H) PGNS-0; (C and I) PGNS-0.25; (D and J) PGNS-0.5; (E and K) PGNS-1.0; (F) the statistical cell density on the PGNSs. *P < 0.05 (compared to TCP) and #P < 0.05 (compared to PGNS-0.25). | ||
It has been reported that GO could enhance the attachment and proliferation of mammalian cells,40 and low GO loading favored cell spreading.26 Similar results were obtained in this study. Good hydrophilicity is beneficial for cell attachment. As demonstrated in previous results, all of the PGNSs showed hydrophilicity, and PGNS-0.25 with a low GO content showed the highest hydrophilicity. Therefore, the scaffold with a GO content of 0.25% would be more beneficial for the spread and growth of L929 cells.
3.8 Repair of skin defects with electrospun nanofibrous scaffolds in vivo
The wound repair efficiency of the PGNSs was evaluated in an in vivo full thickness skin wound model. Fig. 8A shows the repair process of skin defects. The process of wound healing and wound contraction rates at different time points from both the PGNSs and control group are shown in Fig. 8B and C. During the 12 day in vivo experiment, the wound size of all of the groups shrank with time. Three days after surgery, the wound area in the PGNS-0.25 group was significantly smaller than that in other groups, and this trend was sustained for 9 days. The wound area was similar between PGNS-0.5 and the control group, and the wound area in the PGNS-1.0 group was obviously larger than that in the other groups. During the early process of wound healing, a small amount of scar formation was observed in the PGNS-0.25 group at day 3. Moreover, the wound contraction rate in the PGNS-0.25 group reached more than 70% after 6 days and up to 90% within 9 days. These results indicated that a low GO content promoted the healing of wounds.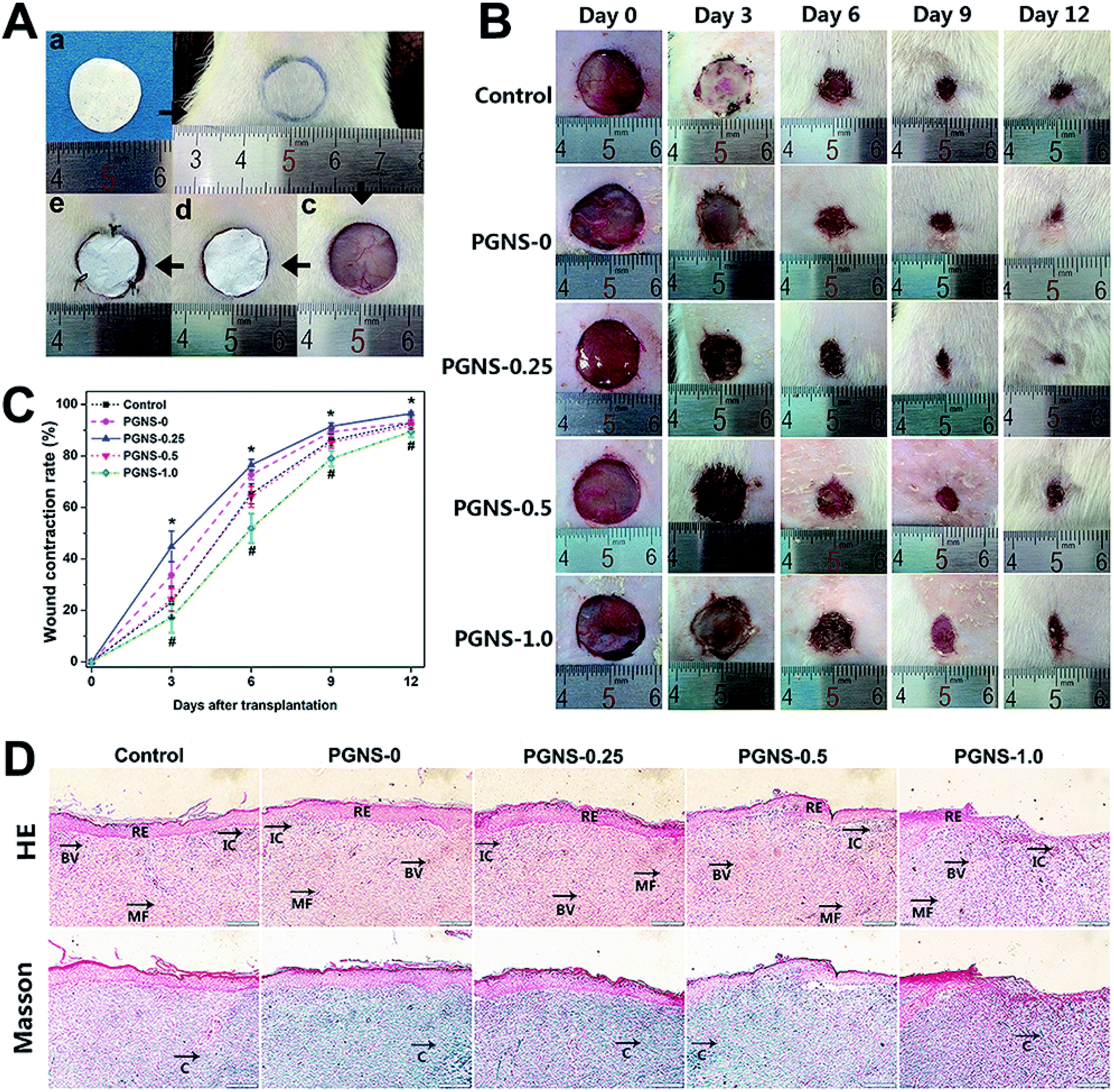 | ||
| Fig. 8 (A) The photographs of the surgery process. An electrospun scaffold (a), the circle pattern drawn on the dorsal skin of a rat (b), the blank wound defect without any treatment (c), the defect covered with an electrospun scaffold (d), and the wound defect covered with an electrospun scaffold and silicon sheet (e). (B) Repair of skin defects with electrospun scaffolds in SD rats for 12 days. (C) Wound contraction rate in each group (N = 5 in each group) at day 0, 3, 6, 9, and 12. *P < 0.05 (PGNS-0.25 compared to control group) and #P < 0.05 (PGNS-1.0 compared to control group). HE and Masson stained sections of the wound sites treated with electrospun scaffolds at day 12 (D). RE: re-epithelialization, IC: inflammatory cells, BV: blood vessel, MF: myofibroblasts and C: collagen. | ||
Twelve days after surgery, the regenerated skins were then subjected to histological analysis. The images of HE-stained and Masson-stained sections are shown in Fig. 8D. The multiple-layer epithelial cells formed a thick slice of epidermis layer in all of the groups, and the epidermis layer covered the wound site. Large amounts of fibroblasts and dense collagen deposition were observed in the wound area, revealing active healing processes. However, histological specimens in contact with the PGNS-0.5 and PGNS-1.0 groups showed the accumulation of inflammatory cells. This biological response may be caused by an overdose of GO, so an optimum content of GO should be used to prevent adverse effects during wound healing.38 In contrast to scaffolds with a higher GO content, the PGNS-0.25 group had dense new blood vessels and only a few inflammatory cells. These results suggested that the in vivo wound repair efficiency of the PGNS-0.25 group was the highest.
Electrospun nanofibers have proven to be promising for skin tissue engineering application due to ECM-like structures. The structures show great surface to volume ratios, providing abundant space for cell adhesion and growth. A small amount of GO has been used to modify the nanofibers. These modified nanofibers showed a soft surface and suitable hydrophilicity. Furthermore, these nanofibers provided a surface with improved bioactivity and cell affinity for skin wound regeneration. However, for the wound repair efficiency of the PGNSs, there are some explanations to be pointed out. Firstly, scaffolds with a soft surface and good hydrophilicity showed excellent cell affinity.16 Secondly, the scaffolds containing GO have better protein adsorption abilities, favoring the proliferation of various cells and enhancing collagen deposition and neovascularization.22,31 Thirdly, scaffolds containing a small amount of GO could decrease the expression of inflammation factor IL-1.38 Fourthly, a low GO content could significantly increase the deposition of collagen and upregulate expression levels of various proteins.16,21 Finally, a higher GO content could obviously increase the expression levels of the apoptosis-inducing factor, limiting cell proliferation and inhibiting cell viability.21,27 As a result, a GO content of 0.25% was selected as the optimal loading in this work. Therefore, PGNS-0.25 might be a suitable candidate for skin tissue engineering.
4 Conclusion
Four kinds of PVA/GO nanofibrous scaffold (PGNS) with different GO content were successfully fabricated by an electrospinning process in this work. The resulting nanofibrous scaffolds showed smooth surfaces and interweaved structures. Invisible hemolysis and suitable hydrophilicity were observed in the PGNSs. The PGNSs showed enhanced tensile strength, reduced ductility and improved bioactivity in terms of protein adsorption ability and cell viability. The animal experiments showed that the PGNS with a 0.25% GO content had the highest skin repair efficiency. These results suggested that PGNS-0.25 was optimal for facilitating skin regeneration and showed potential application as a skin wound dressing.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. NSFC 81471789) and the National High Technology Research and Development Program of China (863 program, No. 2015AA020313).References
- G. R. Jin, M. P. Prabhakaran, D. Kai, S. K. Annamalai, K. D. Arunachalam and S. Ramakrishna, Biomaterials, 2013, 34, 724–734 CrossRef CAS PubMed
.
- M. Sabitha and R. Sheeja, Polym. Eng. Sci., 2014, 55, 541–548 CrossRef
- S. Fu, X. Meng, J. Fan, L. Yang, Q. Wen, S. Ye, S. Lin, B. Wang, L. Chen, J. Wu, Y. Chen, J. Fan and Z. Li, J. Biomed. Mater. Res., Part B, 2014, 102, 533–542 CrossRef PubMed
- F. Groeber, M. Holeiter, M. Hampel, S. Hinderer and K. Schenke-Layland, Adv. Drug Delivery Rev., 2011, 63, 352–366 CrossRef CAS PubMed
- S. Gautam, C. F. Chou, A. K. Dinda, P. D. Potdar and N. C. Mishra, Mater. Sci. Eng., C, 2014, 34, 402–409 CrossRef CAS PubMed
- R. F. Pereira, C. C. Barrias, P. L. Granja and P. J. Bartolo, Nanomedicine, 2013, 8, 603–621 CrossRef CAS PubMed
- S. G. Kumbar, S. P. Nukavarapu, R. James, L. S. Nair and C. T. Laurencin, Biomaterials, 2008, 29, 4100–4107 CrossRef CAS PubMed
- N. Mahmoudi and A. Simchi, Mater. Sci. Eng., C, 2017, 70, 121–131 CrossRef CAS PubMed
- F. H. Zulkiflia, F. S. Hussaina, M. S. Rasad and M. Y. Mohd, Carbohydr. Polym., 2014, 114, 238–245 CrossRef PubMed
- A. Sionkowska, Prog. Polym. Sci., 2011, 36, 1254–1276 CrossRef CAS
- D. Killeen, M. Frydrych and B. Chen, Mater. Sci. Eng., C, 2012, 32, 749–757 CrossRef CAS
- K. Kataria, A. Gupta, G. Rath, R. B. Mathur and S. R. Dhakate, Int. J. Pharm., 2014, 469, 102–110 CrossRef CAS PubMed
- Y. O. Kang, I. S. Yoon, S. Y. Lee, D. D. Kim, S. J. Lee, W. H. Park and S. M. Hudson, J. Biomed. Mater. Res., Part B, 2010, 92, 568–576 Search PubMed
- S. Y. Lee, D. H. Jang, Y. O. Kang, O. B. Kim, L. Jeong, H. K. Kang, S. J. Lee, C. H. Lee, W. H. Park and B. M. Min, Appl. Surf. Sci., 2012, 258, 6914–6922 CrossRef CAS
- S. R. Shin, Y. C. Li, H. L. Jang, P. Khoshakhlagh, M. Akbari, A. Nasajpour, Y. S. Zhang, A. Tamayol and A. Khademhosseini, Adv. Drug Delivery Rev., 2016, 105, 255–274 CrossRef CAS PubMed
- J. Ruan, X. S. Wang, Z. Yu, Z. Wang, Q. Xie, D. D. Zhang, Y. Z. Huang, H. F. Zhou, X. P. Bi, C. W. Xiao, P. Gu and X. Q. Fan, Adv. Funct. Mater., 2016, 26, 1085–1097 CrossRef CAS
- M. J. Zhi, C. C. Xiang, J. T. Li, M. Li and N. Q. Wu, Nanoscale, 2013, 5, 72 RSC
- W. L. Shao, J. X. He, F. Sang, Q. Wang, L. Chen, S. Z. Cui and B. Ding, Mater. Sci. Eng., C, 2016, 62, 823–834 CrossRef CAS PubMed
- E. J. Lee, J. H. Lee, Y. C. Shin, D. G. Hwang, J. S. Kim, O. S. Jin, L. Jin, S. W. Hong and D. W. Han, Biomater. Res., 2014, 18, 18–24 CrossRef PubMed
- X. Yang, X. Zhang, Z. Liu, Y. Ma, Y. Huang and Y. Chen, J. Phys. Chem. C, 2008, 112, 17554–17558 CrossRef CAS
- J. Q. Song, H. C. Gao, G. L. Zhu, X. D. Cao, X. T. Shi and Y. J. Wang, Carbon, 2015, 95, 1039–1050 CrossRef CAS
- R. Deepachitra, V. Ramnath and T. P. Sastry, RSC Adv., 2014, 4, 62717 RSC
- G. M. Wang, F. Qian, C. W. Saltikov, Y. Q. Jiao and Y. Li, Nano Res., 2011, 4, 563–570 CrossRef CAS
- Q. Zhang, S. Lv, J. F. Lu, S. T. Jiang and L. Lin, Int. J. Biol. Macromol., 2015, 76, 94–101 CrossRef CAS PubMed
- Y. T. Zhao, M. He, L. Zhao, S. Q. Wang, Y. P. Li, L. Gan, M. M. Li, L. Xu, P. R. Chang, D. P. Anderson and Y. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 2781–2795 CrossRef CAS PubMed
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS PubMed
- C. J. Shuai, P. Feng, C. D. Gao, X. Shuai, T. Xiao and S. P. Peng, RSC Adv., 2015, 5, 25416 RSC
- J. A. Matthews, G. E. Wnek, D. G. Simpson and G. L. Bowlin, Biomacromolecules, 2002, 3, 232–238 CrossRef CAS PubMed
- C. Edwards and R. Marks, Clin. Dermatol., 1995, 13, 375–380 CrossRef CAS PubMed
- X. Zhao, Q. Zhang, D. Chen and P. Lu, Macromolecules, 2010, 43, 2357–2363 CrossRef CAS
- C. P. Barnes, S. A. Sell, E. D. Boland, D. G. Simpson and G. L. Bowlin, Adv. Drug Delivery Rev., 2007, 59, 1413–1433 CrossRef CAS PubMed
- G. Jin, M. P. Prabhakaran and S. Ramakrishna, Acta Biomater., 2011, 7, 3113–3122 CrossRef CAS PubMed
- M. A. Bhuttoa, T. Wu, B. B. Sun, H. EI-Hamshary, S. S. Al-Deyab and X. M. Mo, Colloids Surf., B, 2016, 144, 108–117 CrossRef PubMed
- H. Shen, M. Liu, H. X. He, L. M. Zhang, J. Huang, Y. Chong, J. W. Dai and Z. J. Zhang, ACS Appl. Mater. Interfaces, 2012, 4, 6317–6323 CrossRef CAS PubMed
- M. Lampin, R. Warocquier-Clérout, L. M. Degrange and M. F. Sigot-Luizard, J. Biomed. Mater. Res., Part B, 1997, 36, 99–108 CrossRef CAS
- K. Liao, Y. Lin, C. W. Macosko and C. L. Haynes, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CrossRef CAS PubMed
- Y. L. Chang, S. T. Yang, J. H. Liu, E. Dong, Y. W. Wang, A. N. Cao, Y. F. Liu and H. F. Wang, Toxicol. Lett., 2011, 200, 201–210 CrossRef CAS PubMed
- E. Nishida, H. Miyaji, H. Takita, I. Kanayama, M. Tsuji, T. Akasaka, T. Sugaya, R. Sakagami and M. Kawanami, Jpn. J. Appl. Phys., 2014, 53, 06JD04 CrossRef
- M. C. Matesanz, M. Vila, M. J. Feito, J. Linares, G. Gonçalves, M. Vallet-Regí, P. A. Marques and M. T. Portolés, Biomaterials, 2013, 34, 1562 CrossRef CAS PubMed
- O. N. Ruiz, K. A. S. Fernando, B. J. Wang, N. A. Brown, P. G. Luo, N. D. McNamara, M. Vangsness, Y. P. Sun and C. E. Bunker, ACS Nano, 2011, 5, 8100–8107 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2017 |