
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Corannulene–fullerene C70 noncovalent interactions and their effect on the behavior of charge transport and optical property†
Yan-Zhi Liua,
Kun Yuan *ab,
Zhao Yuanc,
Yuan-Cheng Zhua,
Sheng-Dun Zhao*b and
Ling-Ling Lva
*ab,
Zhao Yuanc,
Yuan-Cheng Zhua,
Sheng-Dun Zhao*b and
Ling-Ling Lva
aCollege of Chemical Engineering and Technology, Tianshui Normal University, Tianshui 741001, China. E-mail: tsnuyk@yeah.net
bInstitute for Chemical Physics, Department of Chemistry, School of Science, State Key Laboratory of Electrical Insulation and Power Equipment, School of Mechanical Engineering, Xi'an Jiaotong University, Xi'an 710049, China. E-mail: sdzhao@mail.xjtu.edu.cn
cDepartment of Chemical and Biomedical Engineering, Florida State University, Tallahassee, 32306, USA
First published on 26th May 2017
Abstract
Due to the special geometry structures of C70 (an ellipsoidal shape with the highest aspect ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.12) among fullerenes family) and bowl-shaped aromatic hydrocarbons, there are great opportunities for the theoretical computation to deeply explore the “ellipsoid-in-bowl” supra-molecule and their effect on the behavior of charge transport and optical properties. In this study, a new molecular system comprising the non-covalently functionalized complexes of fullerene C70 with corannulene is investigated via the dispersion-corrected density functional theory calculations. Based on the interaction modes, two and three different kinds of configurations have been located on the potential surfaces of the 1:1 and 2:1 corannulene@C70 complexes, respectively. A comprehensive study of binding energy, ionization energy, electron affinity, intermolecular weak interaction regions, the frontier molecular orbitals and gaps, and absorption spectra unravels the structure–property relationship of the complexes. By using the charge hopping rate based on Marcus theory, the charge transport properties of the complexes were discussed. The results shows that the electron transport was more efficient and fast than the hole when C70 interacts with two corannulene molecules by its polar positions. In additional, the modification of C70 on its equatorial position with two corannulene is better for acquiring relative high charge mobility than on polar position with one or two. The electronic transitions and UV-vis absorption spectra of the complexes are mainly determined by the constituent molecule of C70 but hardly dependent on corannulene moiety. Meanwhile, it is found that the more numbers of corannulene noncovalently bonded, the more red-shifted of electron absorption of C70 is.
:1.12) among fullerenes family) and bowl-shaped aromatic hydrocarbons, there are great opportunities for the theoretical computation to deeply explore the “ellipsoid-in-bowl” supra-molecule and their effect on the behavior of charge transport and optical properties. In this study, a new molecular system comprising the non-covalently functionalized complexes of fullerene C70 with corannulene is investigated via the dispersion-corrected density functional theory calculations. Based on the interaction modes, two and three different kinds of configurations have been located on the potential surfaces of the 1:1 and 2:1 corannulene@C70 complexes, respectively. A comprehensive study of binding energy, ionization energy, electron affinity, intermolecular weak interaction regions, the frontier molecular orbitals and gaps, and absorption spectra unravels the structure–property relationship of the complexes. By using the charge hopping rate based on Marcus theory, the charge transport properties of the complexes were discussed. The results shows that the electron transport was more efficient and fast than the hole when C70 interacts with two corannulene molecules by its polar positions. In additional, the modification of C70 on its equatorial position with two corannulene is better for acquiring relative high charge mobility than on polar position with one or two. The electronic transitions and UV-vis absorption spectra of the complexes are mainly determined by the constituent molecule of C70 but hardly dependent on corannulene moiety. Meanwhile, it is found that the more numbers of corannulene noncovalently bonded, the more red-shifted of electron absorption of C70 is.
1. Introduction
Noncovalent functionalization of sp2 carbon materials has received tremendous interest since it offers the possibility of attaching a functionality while maintaining the integrity of the sp2-hybridized carbon network, that is, without disturbing the electronic properties of the substrate.1 The non-covalently functionalized complexes of carbon nanomaterials are among the various derivatives which find a myriad of applications in organic electronics,2,3 light-harvesting4 and organic field-effect transistors.5 On the other hand, a supramolecular combination of carbon nano-materials, such as single-wall carbon nanotubes and fullerenes,6–8 nano-ring and fullerene,9–12 constitutes a unique class of molecular assemblies and devices. The structure of this complex is composed of curved π-systems with a concave–convex interface, and its assembly is driven essentially by van der Waals interactions.13,14Although sp2 carbon materials have extraordinary mechanical and electronic proprieties, one of the main drawbacks of these materials is their poor solubility in the common solvents or matrix used in the applications. In recent years, supramolecular approaches15–17 have been employed to overcome this problem even to improve their performances. Based on noncovalent interactions, pyrene derivatives are used as a dispersing agent for favoring nanotube dispersion in different solvents.18 Moreover, Joshi and Ramachandran19 have studied the noncovalent interactions in the complexes of indigo wrapped over carbon nanotubes (CNT) by means of the dispersion corrected density functional theory method. It was found that indigo forms stable noncovalent complexes with carbon nanotubes. These complexes showed distinct electronic properties compared to the component species. Unlike bare CNT and indigo, the complexes absorb in a broad range in the visible region, making them suitable for solar cell applications; the charge transport properties of a complex can be tuned by changing the orientation of the adsorbed molecule.
Particularly, it is important to prevent the aggregation of unfunctionalized fullerene in photovoltaic blends for replacing the commonly employed butyric acid methyl ester derivatives functionalized fullerene with economical and light harvesting advantages and performances. Recently, Cominetti et al.20 have demonstrated that the use of a pyrene derivative (1-pyrenebutyric acid butyl ester, PyBB) is effective in preventing the aggregation of fullerene in photovoltaic blends made of regioregular poly(3-hexylthiophene). In additional, as indicated by the photo-induced electron transfer in C60-pyrene films21 or by the energy transfer in pyrene appended C60 and C70 derivatives,22 the π–π interaction between pyrene and fullerene molecules can promote the dispersion of the acceptor in the polymer matrix at the molecular level and favors electronic processes. Very recently, Carati, et al.23 have explored the interactions between PyBB and unfunctionalized C60 or C70 blended with poly({4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl}{3-fluoro-2-[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl}) as electron donor. Both the spectroscopic and the electrical investigation indicated that PyBB addition has important consequences on the morphology of the blends as well as on their charge transport properties.
Supramolecular order determines to a large extent the performance of these carbon nanostructures within a device. It is known that corannulene belongs to a typical curved carbon π-systems, and fullerene C70 has an ellipsoidal shape with long (0.796 nm) and short axes (0.712 nm) and has the highest aspect ratio (1:1.12) among fullerenes family.24 Therefore, stable complexes could be formed by host–guest interactions between C70 and corannulene. Concurrently, corannulene is a typical bucky bowl carbon-rich organic molecule, which is the smallest nonplanar fullerene fragment (C20H10). Inherently, it has well geometric matching with fullerenes. Therefore, the interaction between fullerenes C70 and corannulene with an anisotropic shape is of great interest because, unlike for spheroidal C60, several geometrically distinct orientations are possible. By means of density functional theory (DFT) protocols, Casella and Saielli25 presented a theoretical investigation of the complexation thermodynamics of complexes of C70 and C60 fullerenes with bowl-shaped hexabenzocoronene derivatives. For C70 they have considered two different orientations with respect to the bowl surface: either with the long axis of C70 parallel to the bowl or perpendicular to it. In recent, a class of outstanding fullerene receptors was developed by Denis and his co-worker.26 They designed and studied new fullerene receptors which are constructed employing porphyrins, single or multi-corannulene pincers and metallic centers. Theoretical calculations indicated that if porphyrin and corannulene pincers are merged, the strongest hosts for fullerenes can be built.
Recently, Josa et al. have carried out several comprehensive studies for the stacking interactions between corannulene or sumanene (including their derivatives) with different sizes and fullerene, for both C60 and C70 by using dispersion-corrected DFT.27–29 No doubt, these works would be very essential for providing considerable improvement in the task of fullerene recognition, or helpful for finally finding the best buckybowl to improve the efficiency and/or selectivity of future buckycatchers for fullerene. Although the interaction behaviors between fullerenes and corannulene or sumanene have been deeply studied, their supramolecular spectral and electronic properties are seldom explored. Moreover, the filling of the interior space of the corannulene with nanoscale materials results in novel nano-hybrid with interesting properties and unique functions including electron and optical characters, which may be very different from the individual components. In this work, we theoretically investigate the structures and properties of 1:1 and 2:1 supramolecular complexes formed with corannulene and C70. Especially, the behaviors of charge transport and optical properties of the corannulene@C70 supramolecular systems at a molecular level by quantum chemical method are discussed. We hope that the present study would be helpful for the deep understanding to the experimental results of π–π noncovalent wrapped fullerene-based electric device and materials.
2. Computational methods
In the current work, the density functional theory of Grimme's DFT-D3 (ref. 30) was mainly employed for the study of corannulene@C70 systems. DFT-D3 method provides an empirical dispersion correction for DFT.30,31 The ability of this new density functional to predict and explain the supramolecular chemistry at van der Waals distances is very encouraging since density functional theory can be used conveniently for supramolecular systems.32,33 All the geometric configurations were fully optimized at the B3LYP-D3/6-31G(D) levels. No symmetry constraints were applied during optimizations. Harmonic frequency analyses were performed at the same level to confirm that these structures were local minima or transition state on the potential energy surfaces. The intermolecular interaction energies (ΔEcpint) with basis set superposition errors (BSSE) corrected were calculated by the counterpoise method.34 The ionization energy and the electron affinity of the complexes were calculated from the energies of the optimized geometries of corresponding charged and neutral systems. The transport of charge carriers at room temperature was studied using the hopping model based on Marcus theory.35,36 The transfer integral (t) and the internal reorganization energy (λ) of the carriers were computed to assess the rate constant of charge transfer (kCT).36 The foregoing parameters along with the diffusion coefficient (D) and the charge carrier mobility (μ) were determined using the equations given in ref. 36. The excited state calculations were done using the TD-DFT by the same method and basis set. Additionally, a visual study of intermolecular noncovalent interaction between host and guest was performed via calculating the reduced density gradient (RDG),37 coming from the electron density (ρ(r)) and its first derivative (RDG(r) = 1/(2(3π2)1/3)|∇ρ(r)|/ρ(r)4/3), and the second largest eigenvalue of Hessian matrix of electron density (λ2) functions by using Multiwfn program.38,39 All the other calculations were performed with the Gaussian 09 program.403. Results and discussion
Geometric configurations
Bowl-shaped aromatic hydrocarbons exemplify well Euler's rule concerning the insertion of five-membered rings in a hexagonal net to induce curvature.41 Corannulene, C20H10, with one central five-membered ring, is the parent structure to which other bowls are often compared.42 As having been mentioned, the encapsulation of fullerenes C70 by corannulene with an anisotropic shape is of great interest because, unlike for spheroidal C60, two geometrically distinct orientations are possible. All the geometric configurations of the 1:1 and 2:1 corannulene@C70 complexes obtained at B3LYP-D3/6-31G(D) level are shown in Fig. 1. In addition, the Cartesian coordinates of optimized complexes included in this work are available in the ESI.†. As expectedly, two and three different kinds of configurations can be located on the potential surfaces of the 1:1 and 2:1 corannulene@C70 complexes, respectively. In order to ensure that these complexes are global minimums, we made a 36° rotation of C70 around its long axis for the complexes be re-optimized (36° rotation was selected because the C70 belongs to D5h point group. It can be imagined that when C70 rotates by 72° around its long axis in the cave of corannulene, an equivalent complex would be obtained, but when it rotates by 36°, a conformational isomer complex may be obtained). However, it is found that the new initial structures all automatically returned to the pre-rotated configurations. Namely, the new obtained complex is nearly with the same geometry parameters and energy to the originally optimized structure, indicating that the obtained complexes are global minimums. For ease of discussion, these different configurations are denoted as nCor@C70-S, -L and -SL (n = 1 or 2), which correspond to C70-standing and -lying orientations in the cavities of corannulene, respectively.
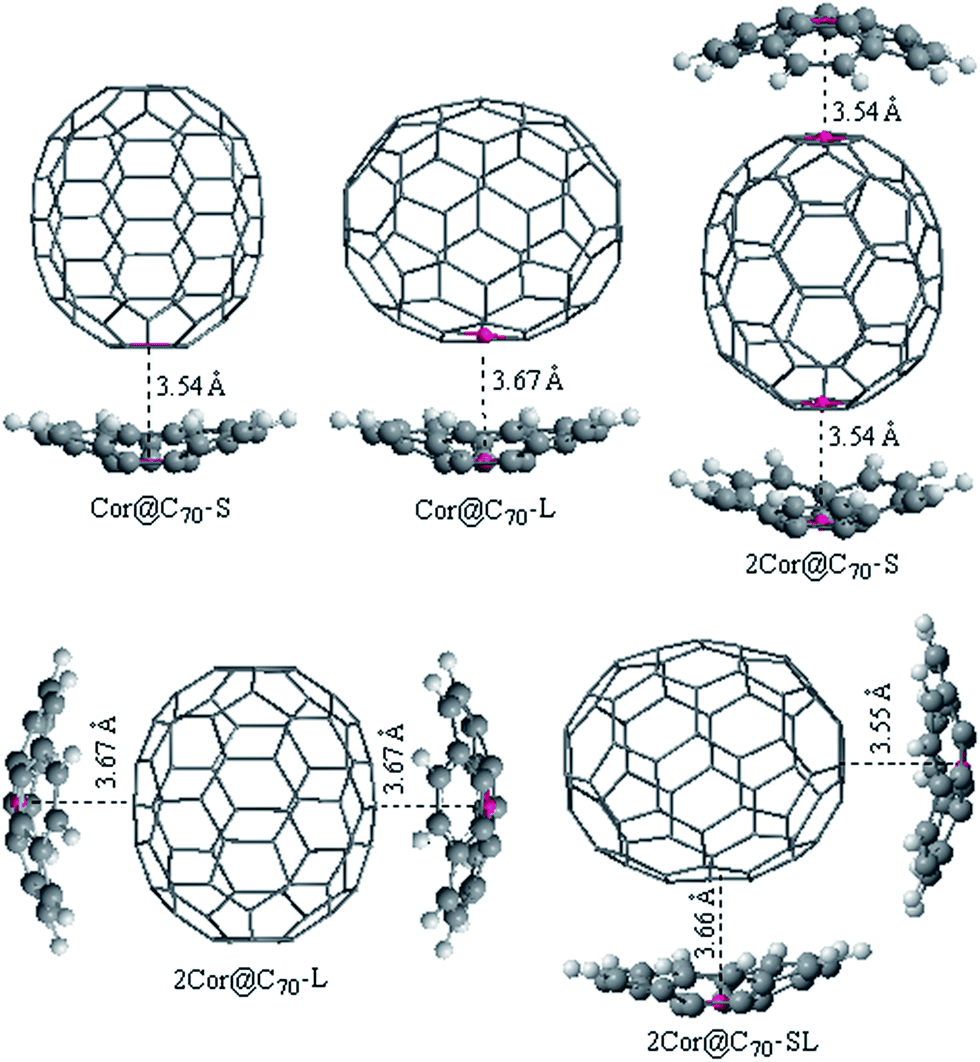 | ||
| Fig. 1 The optimized geometries of Cor@C70-S, Cor@C70-L, 2Cor@C70-S, 2Cor@C70-L and 2Cor@C70-SL. | ||
As shown in Fig. 1, the interfacial distances (di) between corannulene and C70 of the complexes, which is an important parameter for the charge transport calculations and will be discussed later, is defined as lengths between the centroids (red dots in Fig. 1) of the pentagon unit of corannulene and the nearest centroid of a hexagon or pentagon of C70. It is known that the interplanar π–π van der Waals interaction distance between graphite sheets is 3.4 Å. In fact, 3.4 Å is regard as a benchmark and the most nice π–π van der Waals interaction distance either in planar–planar or in convex–concave π–π systems.32,43,44 Generally, if the distance between host and guest is smaller than 3.4 Å, the repulsion would be increase. In the C70-standing configurations, di are found to be within the ranges of 3.54–3.55 Å; those in C70-lying configurations are within the ranges of 3.66–3.67 Å, which are very close to the equilibrium distances defined by Josa et al. using the self-consistent charge density functional tight-binding method together with an empirical correction for the dispersion (SCC-DFTB-D).27 Meanwhile, those interfacial distances are not significantly far away from the 3.4 Å, a van der Waals distance between graphite sheets, thereby accounting for strong π–π interactions and well mutual-fitting between corannulene and C70 either in case of C70-standing or C70-lying orientation.
Interaction energies and thermodynamic properties
Binding energy is very valuable and necessary for measuring the stability and strength of intermolecular noncovalent interaction of Cor@C70 systems. In Table 1, with and without BSSE corrected binding energies (ΔECP and ΔE) are tabulated. For the Cor@C70-L complex, the ΔECP is about 17.11 kcal mol−1, which is close to the previous works of other groups computing at B97-D2/TZVP level.27–29 It is noted that the ΔECP of Cor@C70-L is slightly larger than that in Cor@C70-S by 1.53 kcal mol−1, indicating that the stability of Cor@C70-L is stronger than that of Cor@C70-S, namely C70 is preferred to adopt the lying orientation in the cavity of corannulene. In additional, it is noted that, for the same C70 orientation, the ΔECP in 2:1 complexes are close to twice as much as those of 1:1 complexes, suggesting an additive nature of binding energy against number of the involved molecules. Obviously, the stabilization of the complexes is derived from the concave–convex π–π interactions.45
| Complexes | ΔE | BSSE | ΔECP | ΔG | ΔH | ΔS |
|---|---|---|---|---|---|---|
| Cor@C70-S | −21.09 | 5.51 | −15.58 | −22.94 | −86.11 | −50.64 |
| Cor@C70-L | −23.05 | 5.94 | −17.11 | −34.02 | −92.22 | −46.65 |
| 2Cor@C70-S | −42.11 | 11.00 | −31.11 | −5.32 | −124.18 | −95.28 |
| 2Cor@C70-L | −45.97 | 11.79 | −34.18 | −14.59 | −135.48 | −96.91 |
| 2Cor@C70-SL | −45.39 | 11.69 | −33.70 | −18.89 | −131.75 | −90.47 |
The thermodynamic information of the encapsulations of C70 by the corannulene at 298.15 K and 1 atm obtained by using (DFT) calculations at the B3LYP-D3/6-31G(D) level of theory are also given in Table 1. It can be seen that the relative order of the ΔG and ΔH of the 1:1 complexes are all well consistent with those of the ΔECP. The binding process of C70 by corannulene in the vacuum are exergonic and spontaneous, with ΔG values −22.94 and −34.02 kJ mol−1 for Cor@C70-S and Cor@C70-L, respectively. For two 1:1 complexes, ΔG of Cor@C70-L is larger (more negative) than that of Cor@C70-S, manifesting the spontaneous trend of formation of C70-lying configuration is stronger than that of C70-standing configuration. Moreover, it is worthy to note that the ΔG of 2:1 complexes are obviously smaller than those of two 1:1 complexes, meaning that the thermodynamic spontaneous trend of C70 binding the second corannulene molecule is weaker than that of C70 binding the first one. Especially, the ΔG of 2Cor@C70-S is only −5.32 kJ mol−1, suggesting that it is not easy to binding two corannulene molecules at the regions of two polar positions of C70. Because of the number of the free molecules decreasing by a half or two-thirds after the formations of the 1:1 or 2:1 complexes, the entropies of the present five complexes decrease by 47–97 J mol−1 K−1. According to the ΔH values, all the Cor@C70 binding reactions are found to be exothermic. All these thermodynamic information indicate that the binding of C70 by corannulene in the vacuum is enthalpy-driven and entropy-opposed, which is utterly same to the carbon nanoring@fulleren systems.9,10
Weak interaction regions and frontier orbital features
Intermolecular weak interactions can be detected and visualized in real space based on the electron density ρ and its derivatives,37 viz. the reduced density gradient (RDG), coming from the electron density (ρ(r)) and its first derivative (RDG(r) = 1/(2(3π2)1/3)|∇ρ(r)|/ρ(r)4/3), and the second largest eigenvalue (λ2) of Hessian matrix of electron density functions. Fig. 2 shows the visualized weak interaction regions of the Cor@C70-S and Cor@C70-L complexes and their corresponding scatter graphs. The dish-shaped region marked with green accompanied with light-brown around C70 can be identified as weak interaction region (Fig. 2, left) between the corannulene and C70. In these regions, it mirrors concave–convex π–π van der Waals interaction. The spike marked with green circle (Fig. 2, right) is correlating with π–π van der Waals interaction of the host and guest, and the electron density around this spike is low to zero.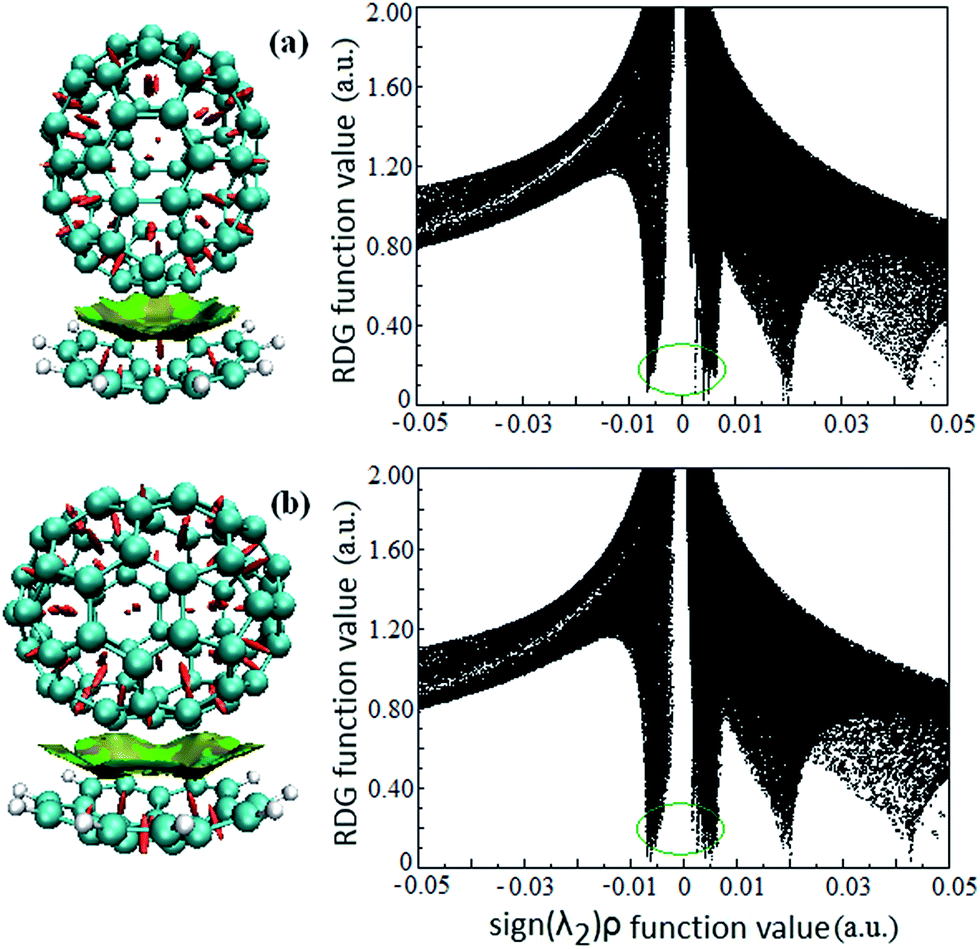 | ||
| Fig. 2 The visualized weak interaction regions (left) and the scatter graph (right) of the (a) Cor@C70-S and (b) Cor@C70-L complexes. | ||
It is noted that the edge of the weak interaction regions between corannulene and C70 present wavelike-shapes (Fig. 2, left), this is derived from the characteristic of corannulene molecular structure with five-membered rings in a hexagonal net. Additionally, qualitatively seen from Fig. 2 (a and b, left), the π–π van der Waals interaction area in Cor@C70-L is larger than that in Cor@C70-S, which is well consistent with the relative order of the binding energies.
It is known that molecular orbital are not physical observables. However, the properties of the frontier orbital are often closely related to the photo-electron behaviors and spectrum properties. Fig. 3 presents the compositions and energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of C70 and the five different complexes. As Fig. 3 showing, the energies of the HOMOs and LUMOs of the five complexes are close to those of the free C70. Consequently, the compositions of HOMOs and LUMOs of the complexes completely derive from the HOMO and LUMO of C70, respectively. The frontier molecular orbital of the complexes are completely localized on the C70 but independent of the moiety of corannulene, indicating the lack of charge transfer between the corannulene and C70 during the formations of the complexes,46 which is different from indigo@carbon-nanotubes systems.19 Significantly, what above suggests that the lowest-energetic electron transition (HOMO → LUMO) of the complexes would take place in the intra-molecule of C70 rather than between the corannulene and C70 molecule, that is no photoinduced charge transfer phenomenon occurring during photoexcitation.
 | ||
| Fig. 3 The frontier molecular orbital and the corresponding energy level diagram of the C70 and five complexes. | ||
The HOMO–LUMO energy gaps of the complexes are close to that of C70 but much smaller than that of corannulene (listed in Table 2). Meaningfully, compared to that of free C70, the gapHOMO–LUMO of Cor@C70-S and 2Cor@C70-S are slightly increased by 0.01–0.02 eV, while those of other three complexes are slightly decreased by 0.02–0.04 eV. Therefore, the introducing an additional molecule of corannulene onto the C70 at different position does not change the energy gap significantly predicting that the electronic transition properties of the C70 would be less affected upon further addition of corannulene.
| Systems | IPv | IPa | EAv | EAa | GapHOMO–LUMO |
|---|---|---|---|---|---|
| Corannulene | 7.49 | 7.38 | −0.04 | −0.17 | 4.39 |
| C70 | 7.10 | 7.02 | −2.07 | −2.14 | 2.69 |
| Cor@C70-S | 6.92 | 6.83 | −2.02 | −2.11 | 2.70 |
| Cor@C70-L | 6.87 | 6.76 | −2.03 | −2.12 | 2.66 |
| 2Cor@C70-S | 6.86 | 6.69 | −2.05 | −2.06 | 2.71 |
| 2Cor@C70-L | 6.73 | 6.62 | −2.00 | −2.10 | 2.65 |
| 2Cor@C70-SL | 6.76 | 6.65 | −1.98 | −2.08 | 2.67 |
Electronic properties and behavior of charge transport
Electronic behaviors and properties of the complexes can be obtained by investigating the molecular ionization potential and electron affinity which can be calculated by removing or adding an electron from the monomers or supramolecular complexes (the Cartesian coordinates of those optimized anion and cation complexes are available in the ESI†). Table 2 lists the computed values of vertical ionization energy (IPv), adiabatic ionization energy (IPa), vertical electron affinity (EAv) and adiabatic electron affinity (EAa) of corannulene, C70 and their complexes. It is found that either the vertical ionization energy or adiabatic ionization energy of free corannulene is larger than those of free C70, indicating that either the electron-denoting or electron-accepting of C70 is stronger than corannulene in their isolated state. Thereby, gapHOMO–LUMO of corannulene is distinctly larger than that of C70. As having been noted, the frontier molecular orbital of the five complexes are completely derived from those of C70 but independent of corannulene. Either ionization energies or electron affinities of the complexes should be mainly determined by moiety of C70. In fact, as listed in Table 2, both the ionization energies and electron affinities of the complexes are all close to those of C70, while they are all smaller than those of free C70.In additional, the ionization energies of C70-lying configurations are smaller than those of C70-standing ones, while the electron affinities of C70-lying configurations are similar to those of C70-standing ones. Therefore, comparing to by polar position, noncovalent functionalization of C70 with corannulene by its equatorial position is more favorable to increase the capability of electron-denoting but less affects that of electron-accepting. Furthermore, ionization energies of the three 2:1 complexes are smaller than those of the two 1:1 complexes; meanwhile, electron affinities of them are slightly smaller than those of two 1:1 complexes, meaning that the capability of electron-denoting of C70 would be enhanced but capability electron-accepting would be weakened with the number of wrapped corannulene increasing from one to two. By contrast, the relatively low value of ionization energy and electron affinity for the complexes with respect to introducing the noncovalent binding of corannulene would favor to the creation of holes but against to injection of electrons for fullerene C70.
Charge mobility calculations were performed to obtain more insight into the charge transport properties of these supramolecular systems. The carrier mobility depends on various parameters including the transfer integral, reorganization energy, rate constant for charge carrier transport and the distance between the molecules. The calculated values of these parameters are listed in Table 3, where + and − signs represent the hole and the electron, respectively. The high value of the transfer integral for the hole transport can be correlated with the high values of the rate constant and the diffusion coefficient which in turn increases the hole mobility.19 Because the reorganization energies of electrons (λ+ = 0.174 eV) and holes (λ− = 0.177 eV) are nearly the same in 2Cor@C70-SL, the transfer rate and carrier mobility depend mainly on the transfer integral. Most interestingly, t− in all complexes except for 2Cor@C70-L shows a smaller value than t+, higher k− was obtained for 2Cor@C70-L. Therefore, the electron transport was more dominant than the hole transport in Cor@C70-L complex. That is, the electron transport was more efficient and fast than the hole when C70 interacts with two corannulene molecules by its polar positions. The distance between the molecules affects the diffusion coefficient and hence the carrier mobility.36 For the 2Cor@C70-SL, separated by a longer distance of 3.66 Å, the hole and electron mobilities are 2.63 × 10−3 and 1.85 × 10−4 cm2 V−1 s−1 higher, respectively.
| Systems | Distance/Å | t+ | t− | λ+ | λ− | kCT+ | kCT− | D+ | D− | μ+ | μ− |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cor@C70-S | 3.54 | 3.0 × 10−4 | 8.0 × 10−5 | 0.142 | 0.16 | 1.01 × 109 | 5.67 × 107 | 6.3 × 10−7 | 3.6 × 10−8 | 2.43 × 10−5 | 1.38 × 10−6 |
| Cor@C70-L | 3.67 | 3.0 × 10−3 | 2.0 × 10−3 | 0.171 | 0.163 | 3.08 × 1010 | 3.41 × 1010 | 2.1 × 10−5 | 2.3 × 10−5 | 8.03 × 10−4 | 8.89 × 10−4 |
| 2Cor@C70-S | 3.54 | 4.0 × 10−4 | 4.0 × 10−4 | 0.212 | 0.095 | 7.42 × 108 | 3.46 × 109 | 4.6 × 10−7 | 2.2 × 10−6 | 1.80 × 10−5 | 8.39 × 10−5 |
| 2Cor@C70-L | 3.67 | 4.9 × 10−3 | 7.1 × 10−3 | 0.166 | 0.174 | 1.97 × 1011 | 3.73 × 1011 | 1.3 × 10−4 | 2.5 × 10−4 | 5.14 × 10−3 | 9.74 × 10−3 |
| 2Cor@C70-SL | 3.55 | 3.7 × 10−3 | 1.0 × 10−3 | 0.174 | 0.177 | 1.01 × 1011 | 7.13 × 109 | 6.4 × 10−5 | 4.5 × 10−6 | 2.48 × 10−3 | 1.74 × 10−4 |
| 3.66 | 6.8 × 10−5 | 4.8 × 10−6 | 2.63 × 10−3 | 1.85 × 10−4 |
For 1:1 complexes, both the hole and electron mobilities of the Cor@C70-L are one or two order higher than those of the Cor@C70-S. For 2:1 complexes, those of the 2Cor@C70-L are nearly two orders higher than those of 2Cor@C70-S, suggesting that the noncovalent binding fullerene C70 with corannulene can obviously alter its transport properties. For 2Cor@C70-SL, its charge mobilities are only slightly smaller than those of 2Cor@C70-L but significantly larger than those of 2Cor@C70-S. Therefore, the above results indicate that the modification of C70 on its equatorial position (C5 symmetry axis of corannulene parallel to the long axis of C70) with two corannulene is better for acquiring relative high charge mobility than on polar position with one or two.
Electronic transitions and UV-vis absorption spectra
The UV-vis absorption spectra and the main corresponding transition composition of C70 and corannulene molecules and their complexes are determined using the time-dependent density functional theory (TD-DFT) method and depicted in Fig. 4 (free moiety and 1:1 complexes) and Fig. 5 (2:1 complexes). The characteristic peaks of the free corannulene are substantially different from that of the fullerene C70. It is clear that the absorption peaks of C70 appear in the longer range of 300–600 nm (UV-vis region), in which the highlighted peaks have been labeled as 327, 365, 452 and 527 nm, correlating with the contributions of the π → π* transitions with different energy levels (S0 → Sn, Fig. 4(a)), respectively, but those of free corannulene appear in the shorter range of 150–300 nm (UV region) which mirror to the sigma (σ) and π electron transitions. Therefore, the electronic transitions of corannulene are more difficult than C70. This is well coincide to the fact of significant difference of gapHOMO–LUMO between C70 and corannulene (Table 2). In additional, corannulene is a kind of chemically inert noncovalent functional reagent of C70. The electronic transitions and UV-vis absorption spectra of the nCor@C70-S, -L and -SL (n = 1 or 2) would be mainly determined by the constituent molecule of C70 but hardly dependent on corannulene.
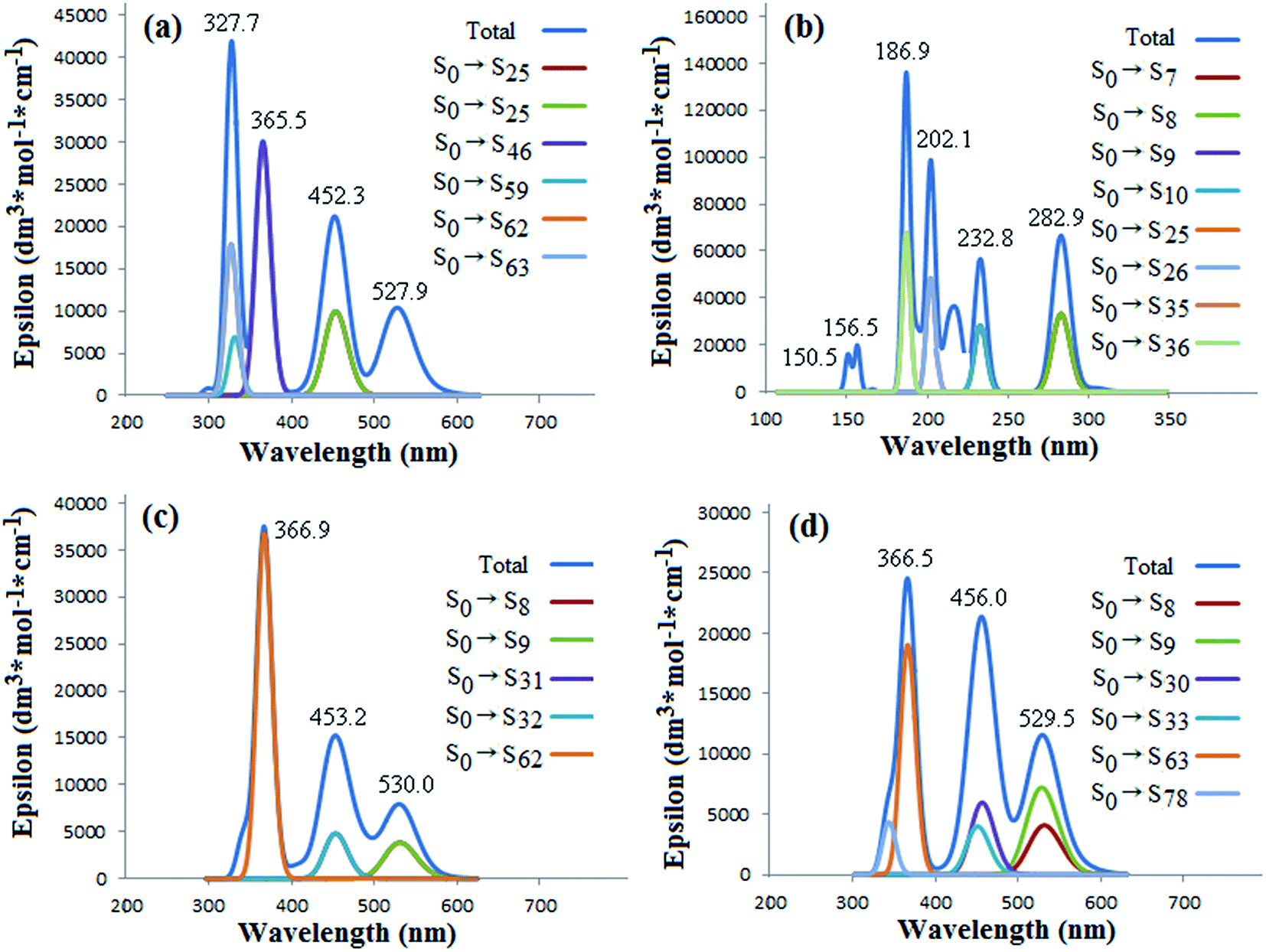 | ||
| Fig. 4 Simulated UV-vis absorption spectra and the main corresponding transition composition (a) C70, (b) Cor, (c) Cor@C70-S and (d) Cor@C70-L. | ||
 | ||
| Fig. 5 Simulated UV-vis absorption spectra and the main corresponding transition composition (a) 2Cor@C70-S, (b) 2Cor@C70-L and (c) 2Cor@C70-SL. | ||
As Fig. 4(c) and (d) showing, in the absorption spectra of the Cor@C70-S and Cor@C70-L complexes, the characteristic peaks of corannulene do not appear, and the three prominent peaks of them appear at the positions similar to those of free C70 only with no more than 4 nm red-shifts. The largest contributions for the peak at 366 nm of Cor@C70-S and Cor@C70-L derived from transitions of S0 → S62 and S0 → S63, respectively (Fig. 4(c) and (d)). Although the maximum absorption wave lengths of Cor@C70-S and Cor@C70-L are nearly the same, the corresponding absorption strengths and transition composition are of some difference, which maybe brings by the difference of the electron transition probabilities in different configurations.
For UV-vis absorption spectra of the three 2:1 complexes (Fig. 5), three prominent peaks are also presented, respectively. The highlighted peaks have been labeled as 368, 467, and 532 nm for 2Cor@C70-S, also correlating with the contributions of the π → π* transitions with different energy levels (S0 → Sn, Fig. 5(a)). It is noted that wave lengths of the three maximum peaks are all longer than those of 1:1 complex of Cor@C70-S; meanwhile, those of 2Cor@C70-L are longer than those of Cor@C70-S. So does for those of 2Cor@C70-SL. What above results suggest the more numbers of corannulene noncovalent-bond, the more red-shifted of electron absorption of C70. Additionally, it is found that the largest contributions for the maximum peaks at 368.3 nm of 2Cor@C70-S and 459.6 nm of 2Cor@C70-L derived from transitions of S0 → S74 (Fig. 5(a)) and S0 → S33 (Fig. 5(b)), respectively. Interestingly, in the total absorption curve, 2Cor@C70-SL shows a very similar features as 2Cor@C70-S in 300–400 nm region and as 2Cor@C70-L in 400–600 nm region, suggesting that corannulene located on the polar position affects electronic transitions of 2Cor@C70-SL in UV region and that located on the equatorial position affects electronic transitions in visible region.
4. Conclusion
Because of the anisotropic feature of the fullerene C70, the supramolecular complexes formed with bowl-shaped curved aromatic corannulene (C20H10) and C70 are diversity and interesting. In this paper, the noncovalent interactions in the 1:1 and 2:1 complexes formed with corannulene and fullerene C70 are studied by means of the dispersion corrected density functional theory method (DFT-B3LYP-D3). It was found that C70 is preferred to adopt the lying orientation in the cavity of corannulene due to a larger concave–convex π–π interactions area than the standing orientation. The binding energies between corannulene and C70 in 2:1 complexes are close to twice as much as those of 1:1 complexes, suggesting an additive nature of binding energy against number of the involved molecules. The thermodynamic information indicates that the noncovalent wrapping of C70 by corannulene in the vacuum is enthalpy-driven and entropy-opposed.
The frontier molecular orbital of the complexes are completely localized on the C70 but independent of the moiety of corannulene, indicating the lack of charge transfer between the corannulene and C70 during the formations of the complexes. The ionization energies and the electron affinities shows that, comparing to by polar position, noncovalent functionalization of C70 with corannulene by its equatorial position is more favorable to increase the capability of electron-denoting but less affects that of electron-accepting. Moreover, the capability of electron-denoting of C70 would be enhanced but capability electron-accepting would be weakened with the number of wrapped corannulene increasing from one to two. The investigations on the charge transport properties of the complexes indicated the electron transport was more efficient and fast than the hole when C70 interacts with two corannulene molecules by its polar positions. In additional, the modification of C70 on its equatorial position with two corannulene is better for acquiring relative high charge mobility than on polar position with one or two. We hope that the present study would be helpful for the deep understanding to the effects of corannulene–fullerene noncovalent interactions on their behavior of charge transport and optical properties.
Acknowledgements
This project was funded by the National Natural Science Foundation of China (No. 21663024, 21362029 and 21663025), the China Postdoctoral Science Foundation (No. 2016M602809), and the Youth Science Research Funds of Tianshui Normal University (TSA1507).References
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- C. Romero-Nieto, R. Garcıa, M. A. Herranz, C. Ehli, M. Ruppert, A. Hirsch, D. M. Guldi and N. Martin, J. Am. Chem. Soc., 2012, 134, 9183–9192 CrossRef CAS PubMed.
- A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342–3405 RSC.
- J. Lohrman, C. Zhang, W. Zhang and S. Ren, Chem. Commun., 2012, 48, 8377–8379 RSC.
- Z. Dai, L. Yan, S. M. Alam, J. Feng, P. R. D. Mariathomas, Y. Chen, C. M. Li, Q. Zhang, L.-J. Li, K. H. Lim and M. B. Chan-Park, J. Phys. Chem. C, 2010, 114, 21035–21041 CAS.
- B. W. Smith, M. Monthioux and D. E. Luzzi, Nature, 1998, 396, 323–324 CrossRef CAS.
- M. Monthioux, Carbon, 2002, 40, 1809–1823 CrossRef CAS.
- A. de Juan and E. M. Perez, Nanoscale, 2013, 5, 7141–7148 RSC.
- K. Yuan, Y. J. Guo and X. Zhao, J. Phys. Chem. C, 2015, 119, 5168–5179 CAS.
- K. Yuan, Y. J. Guo and X. Zhao, Phys. Chem. Chem. Phys., 2014, 16, 27053–27064 RSC.
- K. Yuan, J. S. Dang, Y. J. Guo and X. Zhao, J. Comput. Chem., 2015, 36, 518–528 CrossRef CAS PubMed.
- K. Yuan, Y. J. Guo, T. Yang, J. S. Dang, P. Zhao, Q. Z. Li and X. Zhao, J. Phys. Org. Chem., 2014, 27, 772–782 CrossRef CAS.
- F. G. Klarner and T. Schrader, Acc. Chem. Res., 2013, 46, 967–978 CrossRef PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- Y. Matsuo, K. Morita and E. Nakamura, Chem.–Asian J., 2008, 3, 1350–1357 CrossRef CAS PubMed.
- G. J. Bahun and A. Adronov, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1016–1028 CrossRef CAS.
- C. Walgama, N. Means, N. F. Materer and S. Krishnan, Phys. Chem. Chem. Phys., 2015, 17, 4025–4028 RSC.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50, 1371–1374 CrossRef CAS PubMed.
- A. Joshi and C. N. Ramachandran, Phys. Chem. Chem. Phys., 2016, 18, 14040–14045 RSC.
- A. Cominetti, A. Pellegrino, L. Longo, A. Tacca, R. Po, C. Carbonera, M. Salvalaggio, M. Baldrighi and S. V. Meille, Mater. Chem. Phys., 2015, 159, 46–55 CrossRef CAS.
- M. I. Sluch, I. D. W. Samuel and M. C. Petty, Chem. Phys. Lett., 1997, 280, 315–320 CrossRef CAS.
- T. Gareis, O. Kothe and J. Daub, Eur. J. Org. Chem., 1998, 8, 1549–1557 CrossRef.
- C. Carati, N. Gasparini, S. Righi, F. Tinti, V. Fattori, A. Savoini, A. Cominetti, R. Po, L. Bonoldi and N. Camaioni, J. Phys. Chem. C, 2016, 120, 6909–6919 CAS.
- A. V. Nikolaev, T. J. S. Dennis, K. Prassides and A. K. Soper, Chem. Phys. Lett., 1994, 223, 143–148 CrossRef CAS.
- G. Casella and G. Saielli, New J. Chem., 2011, 35, 1453–1459 RSC.
- P. A. Denis and M. Yanney, RSC Adv., 2016, 6, 50978–50984 RSC.
- D. Josa, I. Gonzalez-Veloso, J. Rodriguez-Otero and E. M. Cabaleiro-Lago, Phys. Chem. Chem. Phys., 2015, 17, 6233–6241 RSC.
- D. Josa, L. A. dos Santos, I. Gonzalez-Veloso, J. Rodriguez-Otero, E. M. Cabaleiro-Lagoc and T. de Castro Ramalho, RSC Adv., 2014, 4, 29826–29833 RSC.
- D. Josa, J. Rodriguez-Otero, E. M. Cabaleiro-Lago, L. A. Santos and T. C. Ramalho, J. Phys. Chem. A, 2014, 118, 9521–9528 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- Y. Zhao and D. G. Truhlar, J. Am. Chem. Soc., 2007, 129, 8440–8442 CrossRef CAS PubMed.
- T. M. Simeon, M. A. Ratner and G. C. Schatz, J. Phys. Chem. A, 2013, 117, 7918–7927 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- H. Liu, S. Kang and J. Y. Lee, J. Phys. Chem. B, 2011, 115, 5113–5120 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- L. Zoppi, J. S. Siegel and K. K. Baldridge, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 1–12 CrossRef CAS.
- K. K. Baldridge and J. S. Siegel, Theor. Chem. Acc., 1997, 97, 67–71 CrossRef CAS.
- T. Iwamoto, Y. Watanabe, T. Sadahiro, T. Haino and S. Yamago, Angew. Chem., Int. Ed., 2011, 50, 8342–8344 CrossRef CAS PubMed.
- T. Iwamoto, Y. Watanabe, H. Takaya, T. Haino, N. Yasuda and S. Yamago, Chem.–Eur. J., 2013, 19, 14061–14068 CrossRef CAS PubMed.
- T. Kawase and H. Kurata, Chem. Rev., 2006, 106, 5250–5273 CrossRef CAS PubMed.
- I. Garcia Cuesta, T. B. Pedersen, H. Koch and A. Sanchez de Meras, ChemPhysChem, 2006, 7, 2503–2507 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03923a |
| This journal is © The Royal Society of Chemistry 2017 |