
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and properties of a trapezoid shaped macrocycle with different [n]yne units†
Maude Desroches and
Jean-François Morin *
*
Département de Chimie and Centre de Recherche sur les Matériaux Avancés (CERMA), Université Laval, 1045 Ave de la Médecine, Québec, Canada G1V 0A6. E-mail: jean-francois.morin@chm.ulaval.ca
First published on 17th March 2017
Abstract
A new alkyne-based macrocycle that combines monoyne, diyne and tetrayne is reported. Despite its unusual trapezoid shape, the bending of the tetrayne unit allows the macrocycle to be unstrained. The macrocycle is synthesized by a combination of selective deprotection of silylated alkyne, Castro–Stephen–Sonogashira coupling and copper-mediated oxidative homocoupling. The final intramolecular cyclization proceeded in surprisingly high yield (72%) using modified Eglinton conditions in moderate dilution.
Introduction
From the naturally occurring diamond and graphite to the man-made carbon nanotubes and fullerenes, carbon allotropes have been a subject of particular interest for the past 25 years.1 The astonishing structural diversity of carbon-based materials has continuously fuelled the imagination of chemists, allowing them to create new arrangements of sp3, sp2 and sp carbon atoms. Unlike sp2 carbon-based materials, molecules and materials containing sp-hybrized carbon atoms (alkyne) have been much less studied, although they have shown interesting properties for different applications such as nonlinear optics,2 sensors3 and molecular switch.4 More recently, alkyne-containing molecules and macrocycles (dehydrobenzo[18]annulenes) have been used as starting materials for the preparation of π-conjugated molecules5 and nanoarchitectures6 owing to their reactivity in the crystalline or xerogel state. It has been shown through numerous examples that small structural changes on alkyne-rich molecules may have dramatic effects on their electro-optical properties, making them an exciting subject of investigation.Our longstanding interest in carbon-rich materials motivated us to undertake the bottom-up synthesis of carbon nanotubes through topochemical polymerization of pre-organized phenylacetylene-based macrocycles in a xerogel phase.7,8 Most recently, we reported the synthesis of rhomboidal macrocycles serving as a monomeric unit of graphdiyne-like nanoribbons (Fig. 1, left).9 All of these examples underline the fundamental importance and the potential applicability of carbon-rich nanostructures for the development of new functional materials. In view of developing a new and unique macrocyclic architecture, we have been inspired by Tobe's report of a trapezoid-shaped tris(dehydrobenzo[12]annulene) (Fig. 1, right) to develop a novel macrocyclic structure in which each vertex is connected to a different sp-hybridized sub unit.10 More specifically, the flexibility and polyvalence of sp–sp2 connection has motivated us to prepare a new kind of macrocycle that bears three different types of [n]ynes in which monoyne, diyne and tetrayne are combined in a single trapezoid ring by a straightforward synthesis involving a key intramolecular cyclization step. Surprisingly, high yield was obtained with modified Eglinton conditions and has lead to an unprecedented and stable macrocycle. Despite the unusual shape and potential strain in the macrocycle structure, the flexibility of the tetrayne chain allows a relaxed planar structure, as evidenced by the X-ray diffraction study and quantum mechanical calculations. The remarkably high stability of this macrocycle in both solution and solid states suggests that even long [n]yne moieties can be used as building block for the preparation of carbon-rich materials, more specifically graph[n]ynes, with unique properties (Fig. 2).
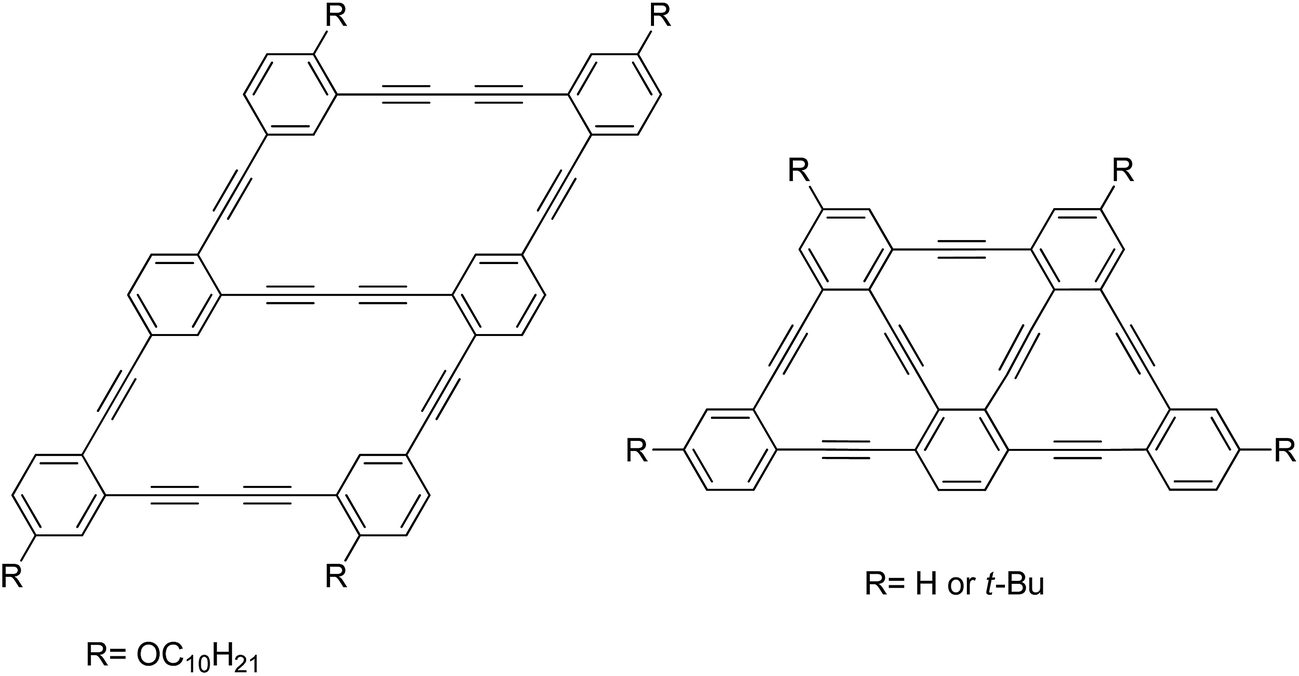 | ||
| Fig. 1 Monomeric unit of graphdiyne-like nanoribbons (left)9 and trapezoid-shaped tris(dehydrobenzo[12]annulene) (right).10 | ||
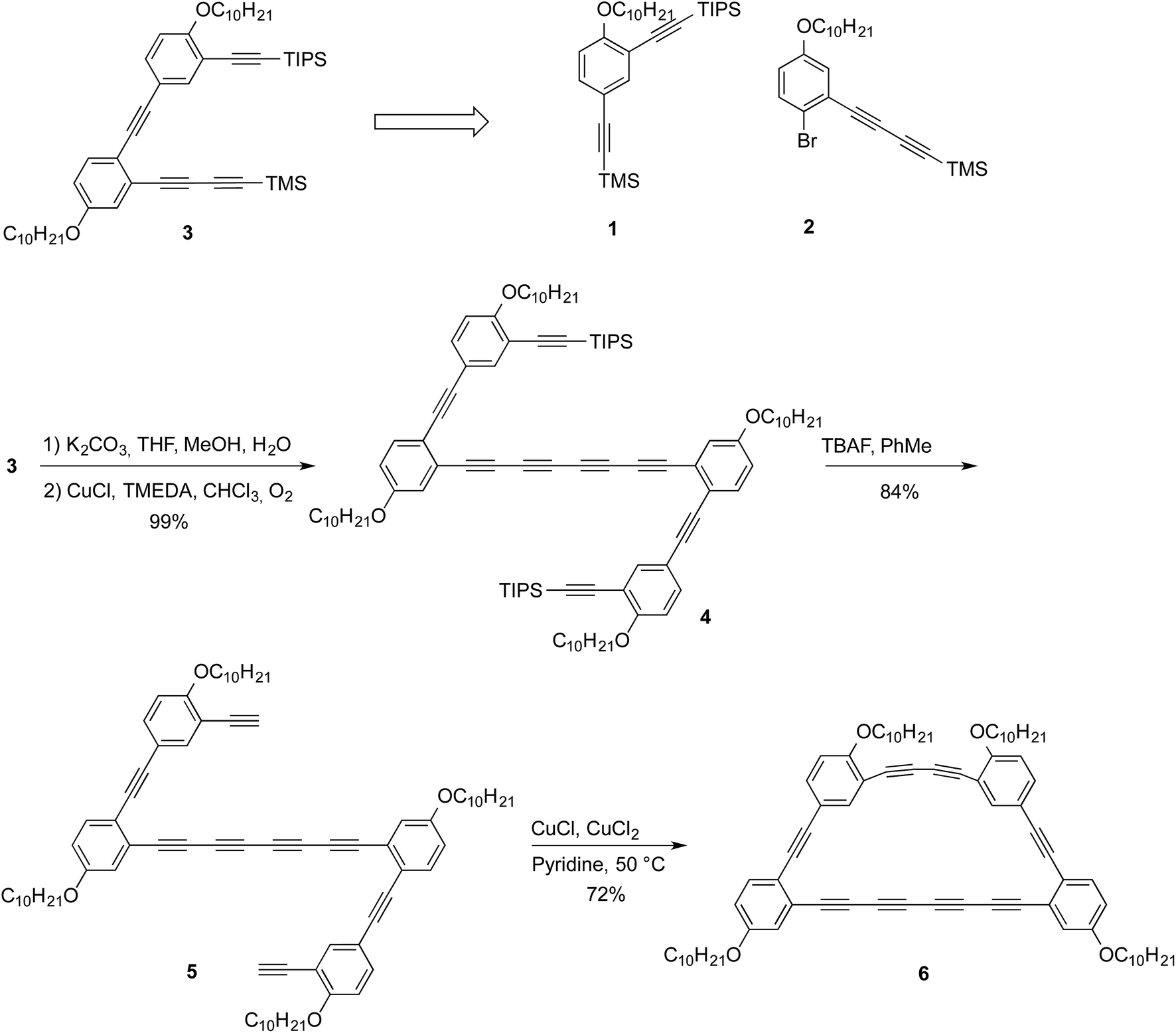 | ||
| Fig. 2 Synthetic scheme of macrocycle 6. | ||
Results
Synthesis
A retrosynthetic analysis allowed to identify compounds 1 and 2 as suitable precursors to the half-macrocycle.9 The alkyne moieties in both 1 and 2 are installed in high yield using Castro–Stephen–Sonogashira coupling from commercially available starting materials while the alkyl chains, which are intended to provide increased solubility, are installed by a simple Williamson etherification from the phenol (see ESI† for synthetic details). Compound 3 can then be assembled through simple Castro–Stephen–Sonogashira coupling of compounds 1 and 2.9 This compound was selectively deprotected with potassium carbonate in THF and methanol to afford the terminal diyne. Then, the crude product was homocoupled using Glaser–Hay conditions in chloroform in 99% yield (for 2 steps) to afford compound 4. The remaining TIPS groups were removed using TBAF in toluene with 84% yield to afford compound 5. The final macrocycle 6 was obtained using modified Eglinton conditions with a 72% yield. The yield of the final macrocyclization was surprisingly high considering that the reaction was performed using moderate dilution (10−3 M) conditions. The high yield of the intramolecular cyclization can be explained by the fact that alkynes from a same molecule are in a nearby space and that the steric congestion prevents other molecules to react and form oligomers. The success of this reaction can also be attributed to the catalyst employed for this macrocyclization. Indeed, work conducted by Haley and coworkers showed that the Cu-acetylide intermediate is in a pseudo-trans configuration versus the palladium oxidative homocoupling which involved a cis geometry of the intermediate.11X-ray diffraction
Crystals suitable for an X-ray diffraction analysis were obtained from a slow evaporation of compound 6 in chloroform. The macrocycle crystallized in a P-1 triclinic space group. Surprisingly, no geometrical strain is present in the structure since the internal angle for this trapezoid-shaped macrocycle range from 118.74° to 122.20°, close to the ideal 120° value for sp2 hybridized carbons. The ring shows bond length alternation as expected for alkyne.12 The terminus C–C bonds are longer than the internal ones (1.429/1.431 Å vs. 1.366/1.367 Å) with the shortest central bond, 1.358 Å. The average C–C triple bond and single bond in macrocycle 6 is 1.201 Å and 1.390 Å, respectively, which is shorter than for a linear tetrayne (1.208 Å for triple bond and 1.365 for single bond).13 While this is most likely due to the fact that the tetrayne needs to accommodate a macrocyclic structure, the small difference is consistent with minimal strain. A close look at the tetrayne angle reveals that it is partly straight with an angle of 178.3°, which is almost on the average for a C–C–C triple bond (177.8°).13 However, the tetrayne is not exactly linear, even in the macrocycle, as a slight bow was observed. This is also in good agreement with the calculated structure (see ESI†). One end of the tetrayne shows a more pronounced bending (angle of 171.9°). All the structural features are nicely replicated in a DFT model of macrocyclic 6, which was optimized in the gas phase at the B3LYP/6-31G** level of theory (see ESI†).14–17Interestingly, the macrocycles pack in dimers with the tetrayne moieties slipped from each other, hypothetically because of weak dispersion interactions. Single point DFT calculations at the B97D/6-31G* using the crystal structure's geometry as an input revealed that dimer formation was indeed endothalpic by 11.7 kcal mol−1 in the gas phase, which is consistent with significant stabilization. The close packing distance of ∼3.75 Å prompted us to investigate the topochemical dimerization of the tetrayne units to provide a polydiacetylene. After one week of irradiation at 254 nm, no reaction occurred even though the color of the crystal changes from orange to deep red. In fact, the crystals were solubilized after irradiation, but no sign of the formation of a new compound was detected by NMR and the spectrum only showed the presence of the starting material. The most plausible explanation for this change of color over time is the reaction of a very small portion of the tetraynes that yield dimers in traces amount. To push this reaction further, we irradiated the powder of macrocycle 6 for 24 hours using a UV lamp at 300 nm. Unfortunately, the mass spectrometry analysis after irradiation only showed the starting material. Our hypothesis for the lack of reactivity toward topochemical reaction is that the rigidity of the macrocycle might prevent the rehybridization of the carbons required for the reaction to occur. Nonetheless, the lack of reactivity indicates that tetrayne moieties can be embedded within a graph[n]yne structure to provide stable materials.
Optical properties
UV-visible spectra of compounds 5 and 6 were obtained in chloroform and are shown in Fig. 3. Vibronic structures are present for both compounds. This can be attributed to a rigid structure as expected for these phenyl acetylene structures. The λmax value for compound 5 (436 nm) is almost the same as compound 6 (λmax 421 nm). Upon closure of the macrocycle (from compound 5 to 6) no significant shift in the absorption spectra is observed. The optical bandgap of compound 5 (2.67 eV) measured at the onset of the absorption band (463 nm) is almost the same as compound 6 (2.70 eV, onset = 458 nm). Once again, the calculated value of 2.84 eV for compound 6 is in good agreement with the experimental data. Indeed, the macrocycle formation does not extend the π-conjugation because of the meta configuration of the phenyl groups.9 Therefore, the UV-visible spectra of compounds 5 and 6 can be viewed as spectra of an isolated tetrayne chromophore combined with an ortho-substitution of a phenylacetylene moiety (Fig. 4).18,19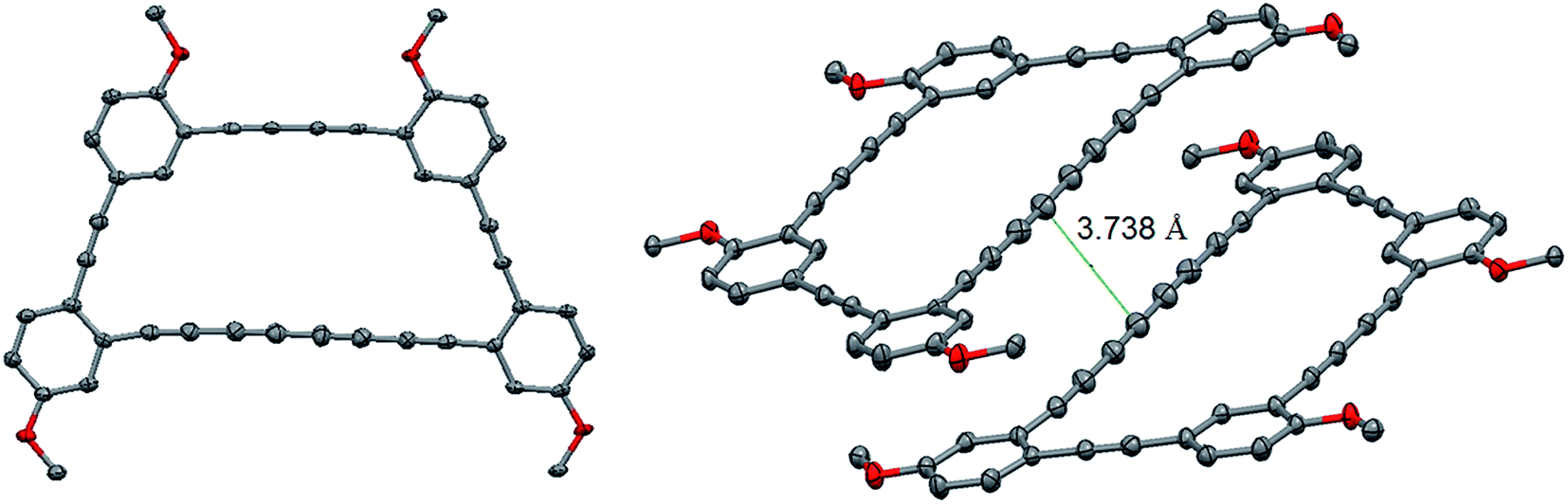 | ||
| Fig. 3 Crystal structure of macrocycle 6. Selected bond lengths and angles. Alkyl chains and H atoms are omitted for clarity. | ||
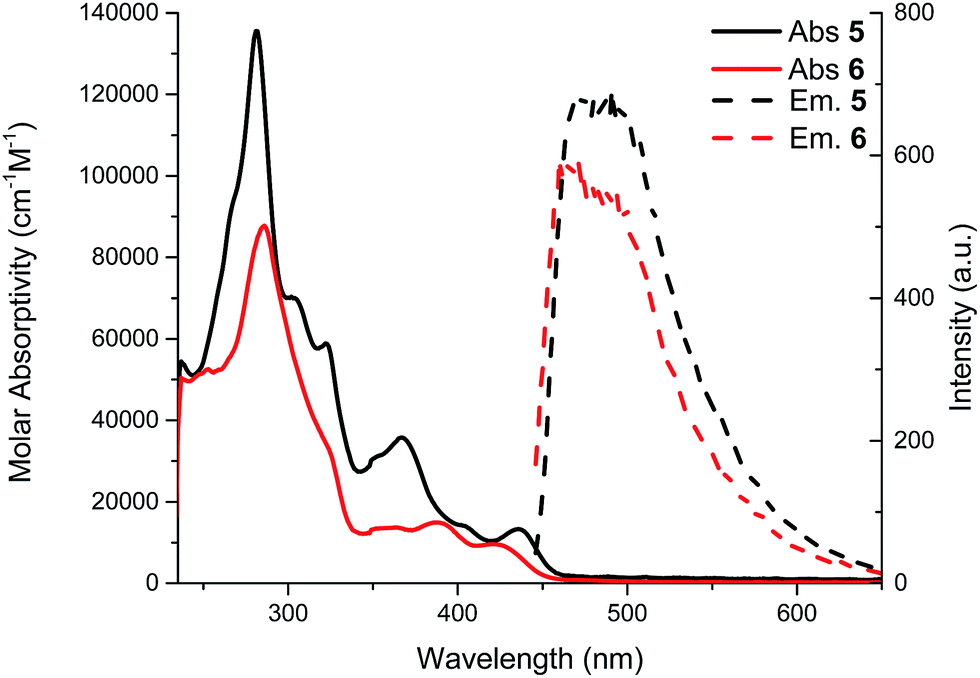 | ||
| Fig. 4 Absorption (solid lines) and emission (dashed lines) spectra of compounds 5 and 6 in CHCl3. | ||
A qualitative analysis of the electron density from the calculated Kohn–Sham orbitals is also informative, as we find that the LUMO is mostly localized on the electron withdrawing tetrayne subunit.20 Also, contributions to the HOMO arise from the triply bonded carbon atoms throughout the macrocyclic architecture. Results from a TD-DFT analysis can be matched nicely with the experimental UV-vis spectrum (see ESI†) and also suggest that the HOMO–LUMO transition is reflected in the absorption band centered at 431 nm (Fig. 5).
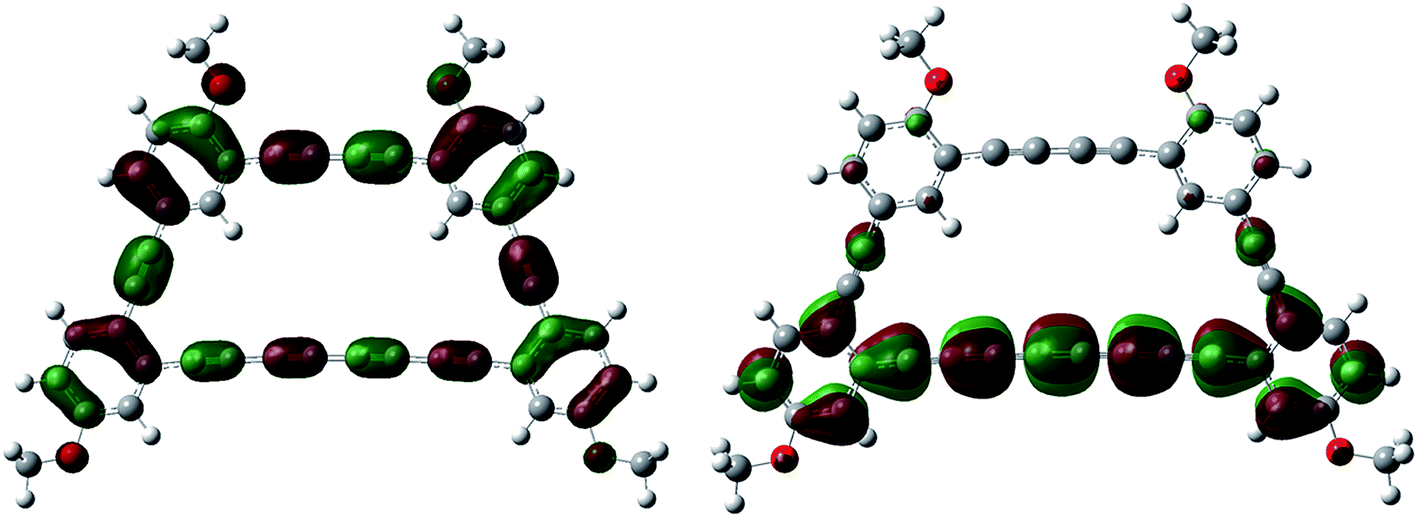 | ||
| Fig. 5 HOMO (left) and LUMO (right) orbitals of macrocycle 6. | ||
The molar absorptivity is not expected to increase upon closure of the macrocycle since it is driven by the longest conjugation pathway, in this case the tetrayne, which is unchanged from compound 5 to 6.21 The strongest absorption bands arising from the transitions to the tetrayne-localized LUMO are well correlated by TD-DFT studies, which are also in good agreement with the experimental UV-vis data. Finally, the Stokes shift is rather small (8 nm for compound 5 and 5 nm for compound 6) again indicating rigid π-conjugated structures. The NMR spectra are also consistent with the non-perturbed π-conjugation length. In fact, there is only a small shift (less than 0.1 ppm) of aromatic protons from compound 5 to macrocycle 6 to lower field, meaning a very weak contribution from a ring current.
Conclusion
In summary, we have synthesized an unprecedented macrocycle with monoynes, diyne and a tetrayne in high yield. The final intramolecular cyclization step involves a pseudo-trans geometry of the copper-mediated oxidative homocoupling that proved beneficial for the success of the reaction. The absorption spectra demonstrate that the macrocyclization does not extend the π-delocalization because of the meta substitution of the phenyl groups. The tetrayne of macrocycle 6 is shorter than the average length of linear tetrayne because of its cyclic structure. Upcoming work will focus on addition of orienting groups to change the packing motif of the macrocycle and to enable the topochemical polymerization.Experimental section
Materials and methods
Chemical reagents were purchased from Sigma-Aldrich Co. Canada and were used as received. THF and toluene used for organic synthesis were purified using a Solvent Purifier System (Vacuum Atmosphere Co., Hawthorne, USA). NMR spectra were recorded using a Varian Inova AS400 spectrometer (Varian, Palo Alto, USA) at 400 MHz and Agilent DD2 500 MHz. Signals are reported as m (multiplet), s (singlet), d (doublet), t (triplet), dd (doublet of doublet), and coupling constants are reported in hertz (Hz). The chemical shifts are reported in ppm (δ) relative to residual solvent peak. High-resolution mass spectra (HRMS) were recorded using an Agilent 6210 time-of-flight (TOF) LCMS apparatus equipped with an APPI ion source (Agilent Technologies, Toronto, Canada). UV-visible absorption spectra were recorded using a Varian diode-array spectrophotometer (Cary 500 model). Fluorescence spectroscopy was performed using a fluorescence spectrophotometer (Cary Eclipse model).Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (19 mL) were added 3 drops of water and K2CO3 (0.353 g, 2.55 mmol). The solution was stirred until the TLC showed a complete reaction. The solution was diluted in CHCl3, extracted three times with water and dried over sodium sulfate. The solvent was removed in vacuo. To a solution of CuCl (0.006 g, 0.064 mmol), TMEDA (0.03 mL, 0.192 mmol) in CHCl3 (22 mL) degassed with air for 1 h was added the unprotected alkyne. The reaction was stirred overnight. The solution was diluted in CHCl3, extracted three times with saturated aqueous NH4Cl, washed with water and brine, and dried over sodium sulfate. The crude product was recrystallized from EtOH/benzene 4:1 to afford compound 4 as a yellow solid (0.456 g, 99%). 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 2.1 Hz, 2H), 7.44 (dd, J = 8.6, 2.2 Hz, 2H), 7.39 (d, J = 8.7 Hz, 2H), 7.02 (d, J = 2.6 Hz, 2H), 6.91 (dd, J = 8.7, 2.6 Hz, 2H), 6.82 (d, J = 8.7 Hz, 2H), 3.99 (t, J = 6.2 Hz, 4H), 3.95 (t, J = 6.5 Hz, 4H), 1.84–1.73 (m, 8H), 1.48 (dd, J = 17.2, 8.2 Hz, 8H), 1.27 (d, J = 5.9 Hz, 48H), 1.14 (d, J = 1.5 Hz, 42H), 0.88 (q, J = 3.4 Hz, 12H). 13C NMR (101 MHz, CDCl3) δ 160.47, 158.48, 136.93, 133.34, 133.10, 124.06, 120.54, 118.21, 117.65, 114.90, 113.50, 111.77, 102.32, 95.41, 92.58, 86.45, 77.83, 76.81, 68.80, 68.50, 68.43, 64.24, 32.06, 29.78, 29.72, 29.71, 29.68, 29.52, 29.51, 29.48, 29.22, 26.36, 26.11, 22.85, 18.86, 14.28, 11.50. HRMS (APPI+): m/z calcd for C98H139O4Si2: 1436.0206; found: 1436.0258 [M + H]+.:3) to afford compound 5 as a light yellow solid (0.132 g, 84%). 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 2.1 Hz, 2H), 7.48 (dd, J = 8.6, 2.2 Hz, 2H), 7.39 (d, J = 8.7 Hz, 2H), 7.02 (d, J = 2.6 Hz, 2H), 6.91 (dd, J = 8.7, 2.6 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 4.03 (t, J = 6.6 Hz, 4H), 3.95 (t, J = 6.5 Hz, 4H), 3.27 (s, 2H), 1.87–1.73 (m, 8H), 1.45 (h, J = 8.3, 7.9 Hz, 8H), 1.39–1.20 (m, 48H), 0.91–0.85 (m, 12H). 13C NMR (126 MHz, CDCl3) δ 160.35, 158.56, 137.41, 133.74, 133.05, 124.17, 120.59, 118.11, 117.71, 115.18, 112.19, 112.08, 92.41, 86.71, 81.67, 79.33, 77.92, 69.07, 68.61, 68.54, 64.34, 32.06, 32.05, 29.71, 29.70, 29.50, 29.48, 29.21, 29.13, 26.10, 26.03, 22.84, 14.28. HRMS (APPI+): m/z calcd for C80H98O4: 1122.7465; found: 1122.7478 [M + H]+. UV/vis (CHCl3) λmax (ε) 436 (13323) nm. Fluorescence (CHCl3) λmax 463 nm.:1 to afford compound 6 as a yellow solid (0.040 g, 72%). 1H NMR (500 MHz, CDCl3) δ 7.73 (d, J = 2.2 Hz, 2H), 7.41 (dd, J = 8.6, 2.1 Hz, 2H), 7.38 (d, J = 8.6 Hz, 2H), 7.07 (d, J = 2.6 Hz, 2H), 6.90 (dd, J = 8.7, 2.7 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 4.06 (t, J = 6.7 Hz, 4H), 3.95 (t, J = 6.6 Hz, 4H), 1.87 (p, J = 6.9 Hz, 4H), 1.78 (p, J = 6.7 Hz, 4H), 1.53–1.21 (m, 56H), 0.88 (dt, J = 12.0, 7.0 Hz, 12H). 13C NMR (126 MHz, CDCl3) δ 160.29, 158.55, 139.36, 133.04, 132.91, 124.23, 119.70, 118.85, 117.79, 115.57, 112.37, 112.09, 92.19, 87.04, 78.62, 78.56, 77.71, 76.47, 69.24, 68.59, 68.53, 64.23, 32.09, 32.05, 29.76, 29.70, 29.54, 29.52, 29.50, 29.48, 29.22, 29.08, 26.11, 26.05, 22.86, 22.85, 14.29, 14.28. HRMS (APPI+): m/z calcd for C80H96O4: 1120.7308; found: 1120.7350 [M + H]+. UV/vis (CHCl3) λmax (ε) 421 (9649) nm. Fluorescence (CHCl3) λmax 472 nm.
:1 (19 mL) were added 3 drops of water and K2CO3 (0.353 g, 2.55 mmol). The solution was stirred until the TLC showed a complete reaction. The solution was diluted in CHCl3, extracted three times with water and dried over sodium sulfate. The solvent was removed in vacuo. To a solution of CuCl (0.006 g, 0.064 mmol), TMEDA (0.03 mL, 0.192 mmol) in CHCl3 (22 mL) degassed with air for 1 h was added the unprotected alkyne. The reaction was stirred overnight. The solution was diluted in CHCl3, extracted three times with saturated aqueous NH4Cl, washed with water and brine, and dried over sodium sulfate. The crude product was recrystallized from EtOH/benzene 4:1 to afford compound 4 as a yellow solid (0.456 g, 99%). 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 2.1 Hz, 2H), 7.44 (dd, J = 8.6, 2.2 Hz, 2H), 7.39 (d, J = 8.7 Hz, 2H), 7.02 (d, J = 2.6 Hz, 2H), 6.91 (dd, J = 8.7, 2.6 Hz, 2H), 6.82 (d, J = 8.7 Hz, 2H), 3.99 (t, J = 6.2 Hz, 4H), 3.95 (t, J = 6.5 Hz, 4H), 1.84–1.73 (m, 8H), 1.48 (dd, J = 17.2, 8.2 Hz, 8H), 1.27 (d, J = 5.9 Hz, 48H), 1.14 (d, J = 1.5 Hz, 42H), 0.88 (q, J = 3.4 Hz, 12H). 13C NMR (101 MHz, CDCl3) δ 160.47, 158.48, 136.93, 133.34, 133.10, 124.06, 120.54, 118.21, 117.65, 114.90, 113.50, 111.77, 102.32, 95.41, 92.58, 86.45, 77.83, 76.81, 68.80, 68.50, 68.43, 64.24, 32.06, 29.78, 29.72, 29.71, 29.68, 29.52, 29.51, 29.48, 29.22, 26.36, 26.11, 22.85, 18.86, 14.28, 11.50. HRMS (APPI+): m/z calcd for C98H139O4Si2: 1436.0206; found: 1436.0258 [M + H]+.:3) to afford compound 5 as a light yellow solid (0.132 g, 84%). 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 2.1 Hz, 2H), 7.48 (dd, J = 8.6, 2.2 Hz, 2H), 7.39 (d, J = 8.7 Hz, 2H), 7.02 (d, J = 2.6 Hz, 2H), 6.91 (dd, J = 8.7, 2.6 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 4.03 (t, J = 6.6 Hz, 4H), 3.95 (t, J = 6.5 Hz, 4H), 3.27 (s, 2H), 1.87–1.73 (m, 8H), 1.45 (h, J = 8.3, 7.9 Hz, 8H), 1.39–1.20 (m, 48H), 0.91–0.85 (m, 12H). 13C NMR (126 MHz, CDCl3) δ 160.35, 158.56, 137.41, 133.74, 133.05, 124.17, 120.59, 118.11, 117.71, 115.18, 112.19, 112.08, 92.41, 86.71, 81.67, 79.33, 77.92, 69.07, 68.61, 68.54, 64.34, 32.06, 32.05, 29.71, 29.70, 29.50, 29.48, 29.21, 29.13, 26.10, 26.03, 22.84, 14.28. HRMS (APPI+): m/z calcd for C80H98O4: 1122.7465; found: 1122.7478 [M + H]+. UV/vis (CHCl3) λmax (ε) 436 (13323) nm. Fluorescence (CHCl3) λmax 463 nm.:1 to afford compound 6 as a yellow solid (0.040 g, 72%). 1H NMR (500 MHz, CDCl3) δ 7.73 (d, J = 2.2 Hz, 2H), 7.41 (dd, J = 8.6, 2.1 Hz, 2H), 7.38 (d, J = 8.6 Hz, 2H), 7.07 (d, J = 2.6 Hz, 2H), 6.90 (dd, J = 8.7, 2.7 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 4.06 (t, J = 6.7 Hz, 4H), 3.95 (t, J = 6.6 Hz, 4H), 1.87 (p, J = 6.9 Hz, 4H), 1.78 (p, J = 6.7 Hz, 4H), 1.53–1.21 (m, 56H), 0.88 (dt, J = 12.0, 7.0 Hz, 12H). 13C NMR (126 MHz, CDCl3) δ 160.29, 158.55, 139.36, 133.04, 132.91, 124.23, 119.70, 118.85, 117.79, 115.57, 112.37, 112.09, 92.19, 87.04, 78.62, 78.56, 77.71, 76.47, 69.24, 68.59, 68.53, 64.23, 32.09, 32.05, 29.76, 29.70, 29.54, 29.52, 29.50, 29.48, 29.22, 29.08, 26.11, 26.05, 22.86, 22.85, 14.29, 14.28. HRMS (APPI+): m/z calcd for C80H96O4: 1120.7308; found: 1120.7350 [M + H]+. UV/vis (CHCl3) λmax (ε) 421 (9649) nm. Fluorescence (CHCl3) λmax 472 nm.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) through a Discovery Grant. M. D. thanks the NSERC for a PhD scholarship. We acknowledge Compute Canada for computation time.References
- T. N. Hoheisel, S. Schrettl, R. Szilluweit and H. Frauenrath, Angew. Chem., Int. Ed., 2010, 49, 6496–6515 CrossRef CAS PubMed.
- A. Sarkar, J. J. Pak, G. W. Rayfield and M. M. Haley, J. Mater. Chem., 2001, 11, 2943–2945 RSC.
- T. Naddo, Y. Che, W. Zhang, K. Balakrishnan, X. Yang, M. Yen, J. Zhao, J. S. Moore and L. Zang, J. Am. Chem. Soc., 2007, 129, 6978–6979 CrossRef CAS PubMed.
- N. Weibel, A. Mishchenko, T. Wandlowski, M. Neuburger, Y. Leroux and M. Mayor, Eur. J. Org. Chem., 2009, 6140–6150 CrossRef CAS.
- Q. Zhou, P. J. Carroll and T. M. Swager, J. Org. Chem., 1994, 59, 1294–1301 CrossRef CAS.
- S. Rondeau-Gagne and J.-F. Morin, Chem. Soc. Rev., 2014, 43, 85–98 RSC.
- S. Rondeau-Gagné, J. R. Néabo, M. Desroches, J. Larouche, J. Brisson and J. F. Morin, J. Am. Chem. Soc., 2013, 135, 110–113 CrossRef PubMed.
- S. Rondeau-Gagné, J. R. Néabo, M. Desroches, K. Cantin, A. Soldera and J.-F. Morin, J. Mater. Chem. C, 2013, 1, 2680 RSC.
- M. Desroches, M.-A. Courtemanche, G. Rioux and J.-F. Morin, J. Org. Chem., 2015, 80, 10634–10642 CrossRef CAS PubMed.
- K. Tahara, T. Yoshimura, M. Ohno, M. Sonoda and Y. Tobe, Chem. Lett., 2007, 36, 838–839 CrossRef CAS.
- J. A. Marsden, J. J. Miller and M. M. Haley, Angew. Chem., Int. Ed., 2004, 43, 1694–1697 CrossRef CAS PubMed.
- W. A. Chalifoux, R. McDonald, M. J. Ferguson and R. R. Tykwinski, Angew. Chem., Int. Ed., 2009, 48, 7915–7919 CrossRef CAS PubMed.
- S. Szafert and J. A. Gladysz, Chem. Rev., 2003, 103, 4175–4205 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- M. M. Haley, M. L. Bell, S. C. Brand, D. B. Kimball, J. J. Pak and W. B. Wan, Tetrahedron Lett., 1997, 38, 7483–7486 CrossRef CAS.
- G. Huang, B. Ma, J. Chen, Q. Peng, G. Zhang, Q. Fan and D. Zhang, Chem.–Eur. J., 2012, 18, 3886–3892 CrossRef CAS PubMed.
- S. Kato, N. Takahashi and Y. Nakamura, J. Org. Chem., 2013, 78, 7658–7663 CrossRef CAS PubMed.
- A. D. Slepkov, F. A. Hegmann, S. Eisler, E. Elliott and R. R. Tykwinski, J. Chem. Phys., 2004, 120, 6807–6810 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1531790. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01721a |
| This journal is © The Royal Society of Chemistry 2017 |