
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoenzymatic epoxidation of alkenes with Candida antarctica lipase B and hydrogen peroxide in deep eutectic solvents†
Pengfei Zhoua,
Xuping Wangb,
Bo Yanga,
Frank Hollmannc and
Yonghua Wang *b
*b
aSchool of Bioscience and Bioengineering, South China University of Technology, Guangzhou 510006, P. R. China
bSchool of Food Science and Engineering, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: yonghw@scut.edu.cn; Fax: +86 (0)20 8711 3842; Tel: +86 (0)20 8711 3842
cDepartment of Biotechnology, Delft University of Technology, Van der Maasweg 9, 2629HZ, Delft, The Netherlands
First published on 21st February 2017
Abstract
Epoxides are important synthetic intermediates for the synthesis of a broad range of industrial products. This study presents a promising solution to the current limitation of enzyme instability. By using simple deep eutectic solvents such as choline chloride/sorbitol, significant stabilization of the biocatalyst has been achieved leading to more robust reactions while using environmentally more acceptable solvents as compared to ionic liquids.
Introduction
Epoxides are important synthetic intermediates in the production of many industrial products.1 Next to established chemical syntheses such as the Prileshajev epoxidation using stoichiometric peracids2 or metal-catalyzed epoxidations,3,4 enzymatic5–9 and chemoenzymatic methods10–12 have also gained relevance. Particularly, the latter methods are attractive due to the broad availability of hydrolases. In essence, chemoenzymatic methods rely on the in situ formation of a reactive peracid making use of the ‘perhydrolase’ promiscuous activity of many lipases. The peracids formed react with the alkene forming the product of interest and re-forming the carboxylic acid, which enters a new catalytic cycle (Scheme 1).13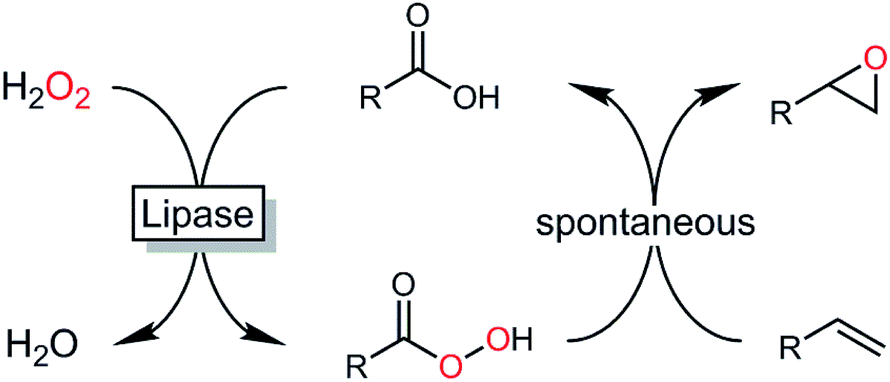 | ||
| Scheme 1 Chemoenzymatic epoxidation reaction using in situ generated peracids. In the first step, lipase-catalyzed perhydrolysis of a carboxylic acid with H2O2 forms the corresponding peracid, which then, in a non-enzymatic Prileshajev reaction forms the epoxide and the (catalytic) carboxylic acid. | ||
A broad range of hydrolases has been investigated for the perhydrolysis of carboxylic acids or their esters with hydrogen peroxide to produce epoxides.14–18 Among them the lipase B from Candida antarctica (CalB), immobilized onto an acrylic resin (tradename Novozym 435), is probably the most-widely used biocatalyst.19 Several studies dealing with lipase-catalyzed in situ peracid formation to promote the Prileshajev epoxidation underline the preparative potential of this approach.20–23
In recent years, it has become clear that CalB suffers from a pronounced instability against the harsh conditions of the chemoenzymatic epoxidation reaction as compared to the ‘natural’ esterification reactions. Arends and coworkers reported that ionic liquids (ILs) could substantially improve the performance of chemoenzymatic Baeyer–Villiger and epoxidation reactions.24 The authors suggested a two-fold effect of hydrogen-bond-donor-ILs in the reaction: first by stabilizing the biocatalyst and second, by accelerating the reaction through solvent-activation of the peracid.
Though frequently termed ‘green solvents’ (mostly due to their non-volatility), ILs are actually questionable from an environmental point-of-view.25 For example, toxicity issues can impair the environmental friendliness as well as the sometimes rather complex and resource-consuming synthesis (vide infra). Therefore, it is not astonishing that current research efforts focus on environmentally less demanding substitutes such as (natural) deep eutectic solvents (DESs).
The number of studies evaluating DESs as more environmentally acceptable alternatives to ILs is steadily increasing.26 For example, beneficial effects of DESs on lipase-catalysed esterification reactions have been reported.27–29 Inspired by these very promising reports, we set out to investigate the possible beneficial effects of DESs on the chemoenzymatic epoxidation reaction.
Results and discussion
In a first set of experiments, we evaluated the influence of various DESs of the efficiency of the chemoenzymatic epoxidation of 1-octadecene as a model reaction (Table 1).
|
||
|---|---|---|
| Entry | Solvents (molar ratio) | Conversion (%) |
| a Conditions: octadecene (1.6 mmol), octanoic acid (1.6 mmol) and H2O2 (1.6 mmol added as 30% w/w solution) were reacted in different DESs at 40 °C using Novozym 435 (100 mg) as catalyst for 24 h with magnetic stirring (300 rpm). | ||
| 1 | ChCl/urea (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) :2) |
30.2 |
| 2 | ChCl/acetamide (1:2) |
13.9 |
| 3 | ChCl/ethylene glycol (1:2) |
37.7 |
| 4 | ChCl/glycerol (1:1) |
56.7 |
| 5 | ChCl/xylitol (1:1) |
65.1 |
| 6 | ChCl/sorbitol (1:1) |
72.4 |
| 7 | ChCl/xylose/water (5:2:5) |
58.4 |
| 8 | ChCl/glucose/water (5:2:5) |
65.3 |
| 9 | ChCl/sucrose/water (5:2:5) |
56.2 |
| 10 | Phosphate buffer (pH6.0, 50 mM) | 62.5 |
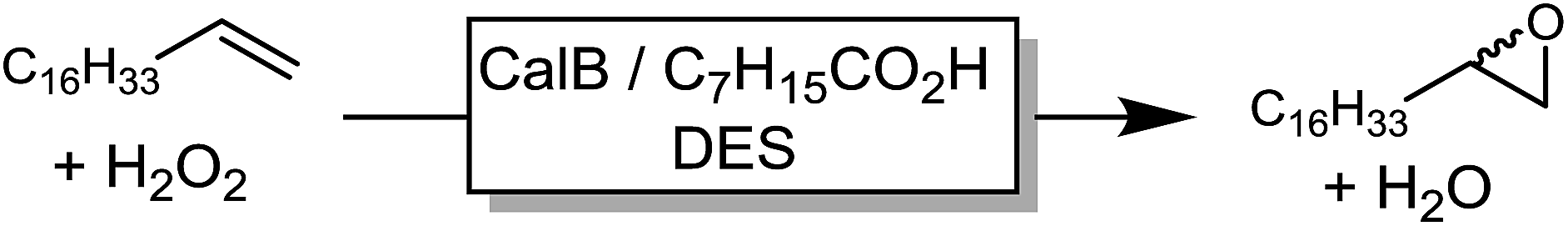
From the nine DESs chosen for this experiment, the amine-based DES gave the poorest results. Essentially product formation ceased already after approx. 6 h. Possibly, inactivation of the enzyme (maybe due to deprotonation of structurally essential amino acid residues) accounted for this behavior. Polyol-based DESs gave significantly higher robustness and consequently also higher product titers. Hence, ChCl/sorbitol as solvent resulted in more than 70% conversion of the starting material. This effect has also been observed by Arends and co-workers who have pointed out the importance on hydrogen-bond-donating cosolvents (ILs) on the stabilization of the protein structure.24 Similarly, Diego et al. have emphasized the importance of polyols such as sorbitol on protein structure.30
Encouraged by these results, we became interested in shedding some more light on the beneficial and detrimental effect of amine-based or polyol-based DESs on the stability of the biocatalyst. Therefore, the apparent half-life times of CalB in the presence of different solvents were determined (Fig. 1). In accordance to our previous observation,31 the amine-based DESs significantly reduced (as compared to buffer as incubation solvent) the stability of CalB over the entire temperature range investigated whereas the polyol-based DESs significantly increased the stability of the biocatalyst.
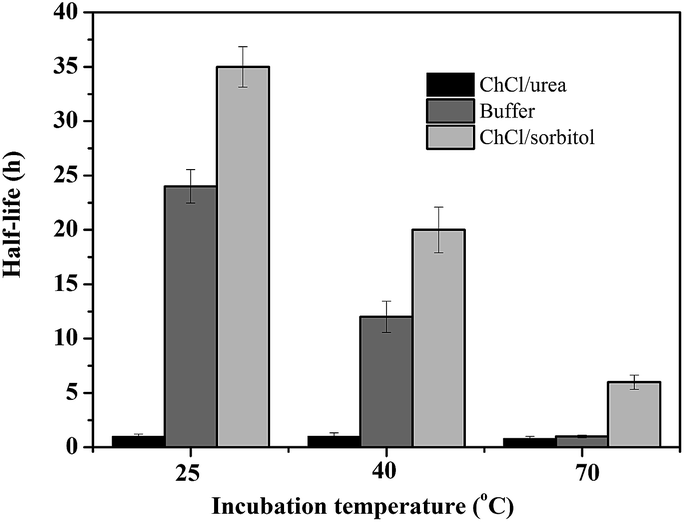 | ||
| Fig. 1 CalB stability (displayed as half life time) at different temperatures in the presence of buffer, ChCl/urea and ChCl/sorbitol. | ||
The effect of the single solvents on the structural integrity of CalB was investigated in some more detail using circular dichroism spectroscopy (CD). As shown in Table 2, the relative amount of secondary structure elements within the enzyme decreased to some extent after incubation in phosphate buffer for 24 h, while in ChCl/urea almost all secondary structural elements disappeared at the benefit of random coil. On the contrary, secondary structure elements were almost entirely preserved (as compared to the freshly dissolved enzyme) upon incubation of the enzyme in ChCl/sorbitol. This result is also in agreement with our previous study.31 Hence, we conclude that the increased stability of the enzyme was due to the preservation of its structural integrity in ChCl/sorbitol.
| Medium | α-helix [%] | β-strand [%] | Turn [%] | Random coil [%] |
|---|---|---|---|---|
| a Determined directly after complete dissolution of the enzyme.b Conditions: CalB incubated in 50 mM pH 6.0 phosphate buffer.c Conditions: CalB incubated in ChCl/urea and.d Conditions: CalB incubated in ChCl/sorbitol at 30 °C for 24 h. | ||||
| Initial CalBa | 19.8 | 13.4 | 31 | 35.8 |
| KPi bufferb | 12.3 | 17.5 | 33.5 | 36.7 |
| ChCl/ureac | 2.2 | 1.3 | 34.3 | 62.2 |
| ChCl/sorbitold | 16.6 | 19.1 | 32.5 | 32 |
Next, the key parameters effecting the chemoenzymatic epoxidation reaction; particularly the influence of the reaction temperature, the octanoic acid concentration and the hydrogen peroxide concentration were investigated (Fig. 2).
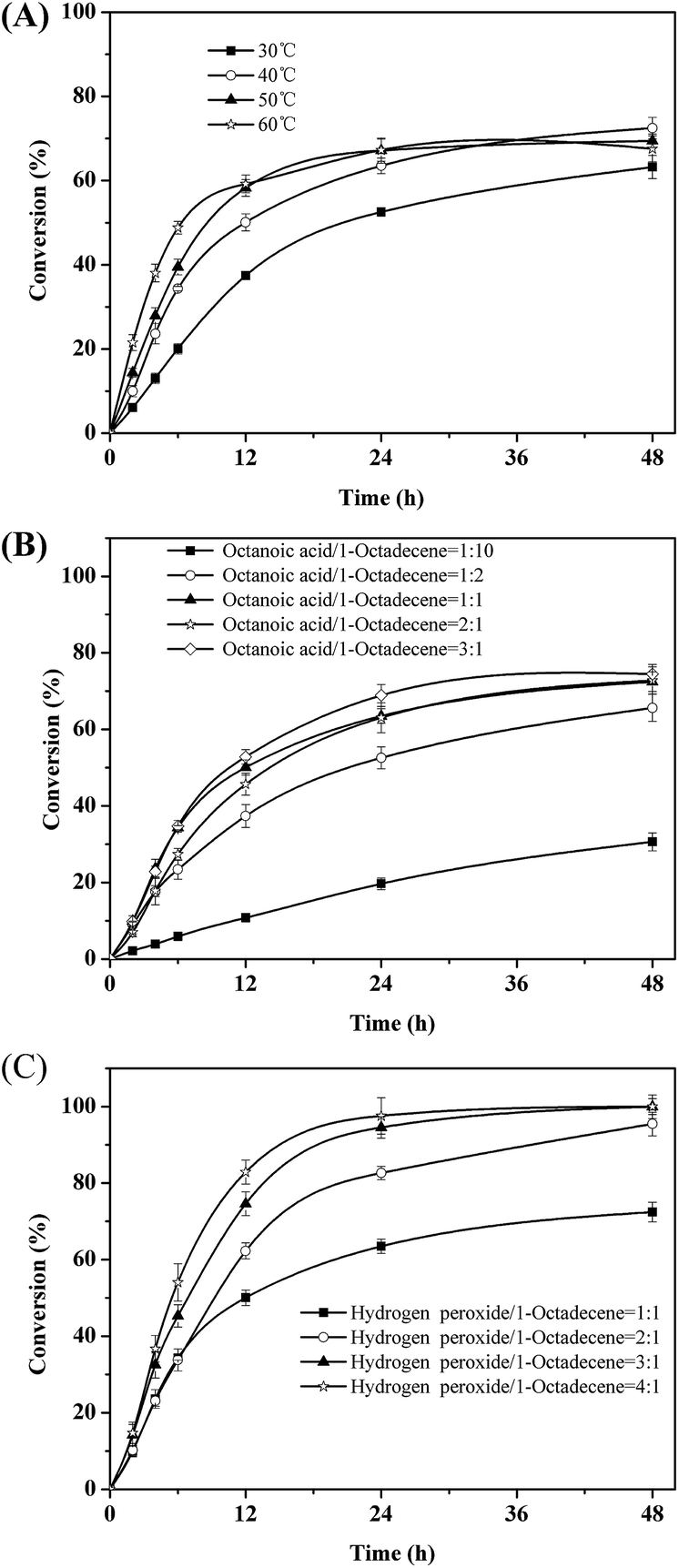 | ||
| Fig. 2 Effects of temperature (A), molar ratio of octanoic acid to 1-octadecene (B) and molar ratio of hydrogen peroxide to 1-octadecene molar ratio (C) on the epoxidation of 1-octadecene. | ||
Quite expectedly, initial this reaction rate increased linearly with temperature (Fig. 2A). Hence, the rate-limiting step may be diffusion of either the enzyme substrates (octanoic acid and/or H2O2) or products (peroctanoic acid). It is worth pointing out here that in similar studies Törnvall et al.32 reported a sharp decrease of the enzyme activity at 60 °C. This underlines the stabilizing effect of ChCl/sorbitol on the enzyme.
The initial rate of the overall reaction showed a saturation-type behavior with respect to the concentration of octanoic acid used (Fig. 2B). Above 1.6 mmol of octanoic acid, no significant increase of the initial rate was observed. This may be due to the Michaelis–Menten-type kinetics of the lipase-catalyzed perhydrolysis reaction with octanoic acid. However, it should be pointed out that in these experiments we could demonstrate that octanoic acid performs as catalyst (TN = 3), pointing towards a truly catalytic system. Nevertheless, for all further experiments we used a molar ratio of 1:1 (acid:alkene) in order not to impair the overall reaction rate. Again, the maximal conversion reached in these experiments was approx. 70%.
Interestingly, the amount of hydrogen peroxide applied to the reaction system had almost no influence on the initial rate of the reaction (which may be explained by a comparably low KM value for H2O2) (Fig. 2C). But it had a very significant influence on the overall conversion reached. At a nominal molar ration of 1:1 (H2O2 to alkene) the maximal conversion observed in all experiments so far was approx. 70%. However, already at a molar ratio of 2:1 (H2O2 to alkene) full conversion was achieved. We believe that the most likely explanation for this observation may be that the active H2O2 content of the stock solution may have been significantly lower (approx. 70%) than the nominal concentration. Further experiments clarifying this issue are currently underway.
We also evaluated the substrate scope of this partially optimized reaction system. As shown in Table 3, a range of (ali)cyclic alkenes as well as styrene derivates were converted in good to excellent conversions in high selectivity as confirmed by NMR analysis of the isolated products (see ESI† for details). Only trace amounts (is any) of possible degradation (isomerization or hydrolysis) products were detectable.
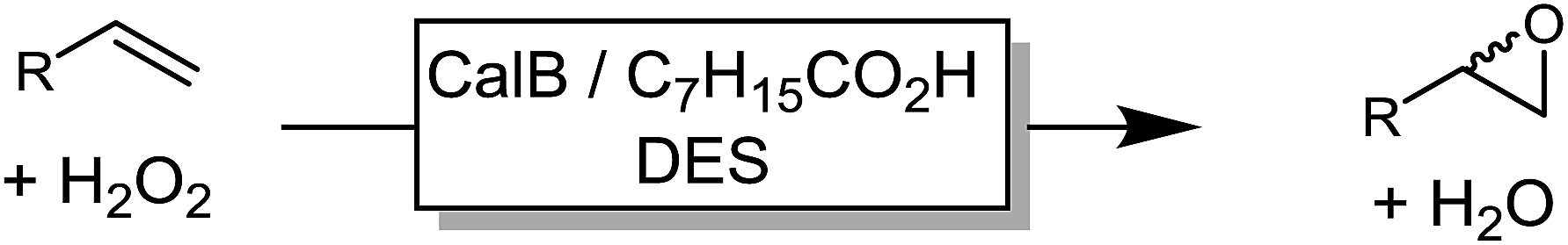
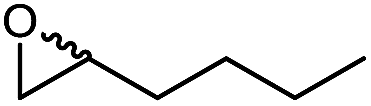
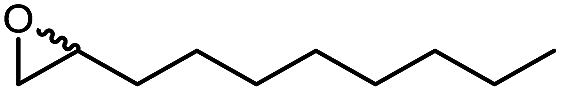
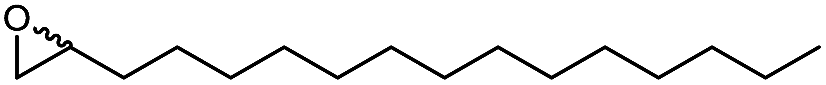
The performance of the proposed chemoenzymatic reaction system using ChCl/sorbitol as ‘performance additive’ compares well with similar reactions reported in the literature.31 For example, the conversion of 1-octadecene in aqueous reaction media was 68% or 30% using the lipase from Penicillium camemberti33 or the lipase from Malassezia globosa,34 respectively. In a three-phase system of water/methyl dichloride/ionic liquid, the conversion of cyclohexene was 41%.18 Therefore, the new oxidation system proposed here represents a promising approach for preparative epoxidation reactions.
To further understand the molecular basis of the perhydrolysis reaction we modelled octanoic acid and H2O2 into the active site of CalB (Fig. 3). Carbonyl atom of the octanoic acid is bound in the oxyanion hole of the enzyme by two hydrogen bonds to the amide nitrogens of Gln106 and Thr40. The distance between carbonyl atom of octanoic acid and Oγ of Ser105 is 2.9 Å, which is facilitate to nucleophilic attack carbonyl group by Ser105 in active site to form an acetyl-enzyme intermediate. Simultaneously, H2O2 is stabilized in the active site by Gln157, Asp134 and His224. Then, hydrogen peroxide attacked acetyl-enzyme intermediate to achieve perhydrolysis reaction. This mechanism is similar to previous reports.33–35
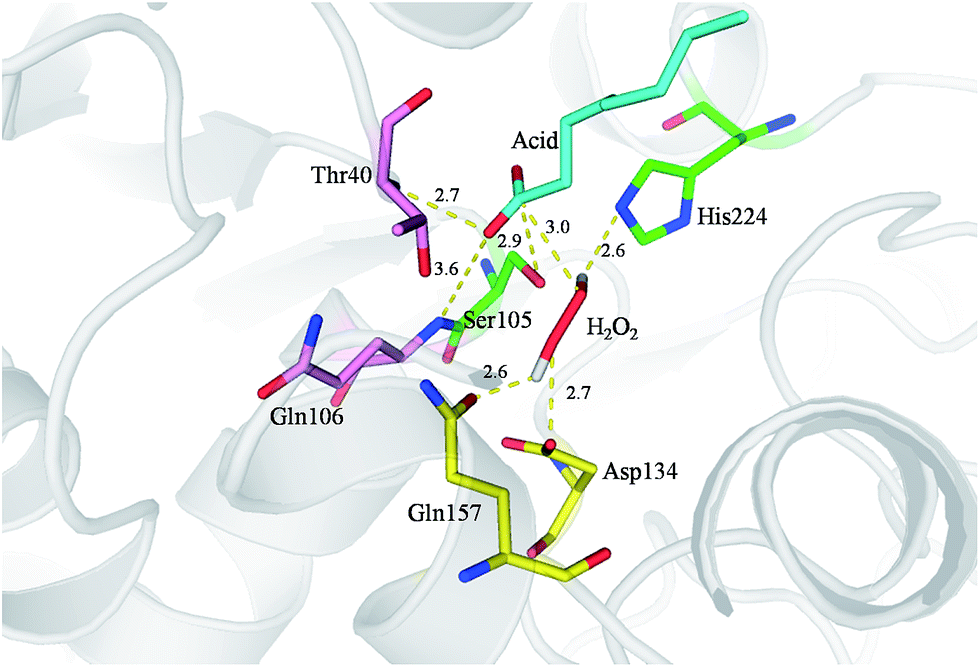 | ||
| Fig. 3 The mode of CalB (PDB ID: ILBT) catalyzed epoxidation mechanism. The atom of hydrogen, oxygen and nitrogen are shown in white, red and blue, respectively. Hydrogen bonds are shown in yellow dash with distances in angstroms. | ||
Finally, we performed a preliminary evaluation of the environmental impact of the chemoenzymatic epoxidation reaction. For this, the Sheldon's E-factor (environmental factor) analysis was used (eqn (1))36,37 and compared our chemoenzymatic epoxidation of styrene with a similar reaction reported by Arends and coworkers.24
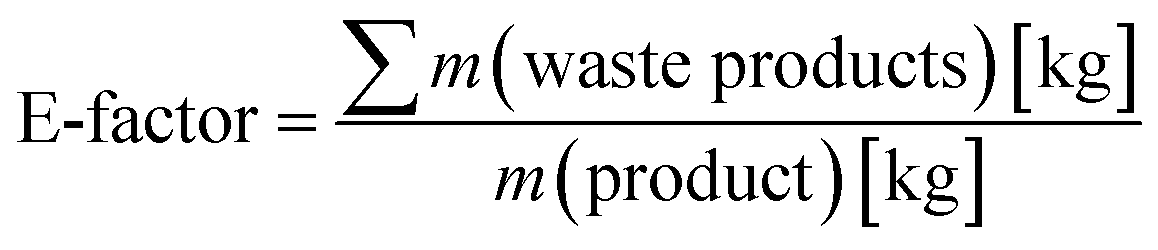 | (1) |
Eqn (1) calculation of Sheldon's E-factor.
It becomes clear from Table 4, that the solvent (either ChCl/sorbitol or [BMIM][BF4]) has a very significant impact on the E-factor of the reaction contributing 31% or even 75% to the total value, respectively.
| Entry | DESs systema,b | ILs systema24 |
|---|---|---|
| a Numbers in parentheses represent the individual E-factor contributions (i.e. g component × g product − 1).b Results taken from Table 3. | ||
| Non-converted (styrene) conversion (%) | 0.016 g (0.09) 90% | 0.08 g (1) 40% |
| CalB (biocatalyst) | 0.1 g (0.58) | 0.01 g (0.13) |
| Octanoic acid | 0.23 g (1.35) | 0.029 g (0.2) |
| Solvent | 0.4 g (ChCl/sorbitol) (2.35) | 1 g ([BMIM][BF4]) (12.5) |
| H2O/H2O2 | 0.56 g (3.29) | 0.14 g (1.75) |
| Product | 0.17 g | 0.08 g |
| E-factor | 7.7 | 16.7 |
Given this importance of the solvents, also their upstream processing should be considered. Following Jessop's approach of evaluating the environmental impact of solvent synthesis based on the synthesis trees38 we compared those for [BMIM][BF4] and ChCl/sorbitol (Fig. S1†). This comparison reveals that compared to [BMIM][BF4] (30 steps) the synthesis of ChCl/sorbitol with only 10 steps is significantly less complex. Furthermore, most of the solvent (55% w/w) of ChCl/sorbitol originates from renewable resources. Of course, only a full life cycle assessment (LCA) will be suitable to holistically compare the environmental impact of the solvent syntheses and the chemoenzymatic reactions, but we hold the results from this preliminary analysis to be very encouraging.
Conclusions
In the present study, it has been demonstrated that DESs represent a true alternative to the commonly used ILs to facilitate the chemoenzymatic epoxidation of alkenes. The system excels particularly by the lower environmental impact of the cosolvents (DESs) used as compared to ILs. This advantage, however, does not come with an impaired performance compared to classical ILs. The practical applicability and efficiency of the current system is at least as good as the one of the systems of the state-of-the-art. Therefore, we are convinced that exciting future developments will arise from the application of DESs to chemoenzymatic epoxidation reactions.Experimental sections
Materials
Candida antarctica lipase B (CalB, Novozym 435-immobilized on a polyacrylate resin) was obtained from Novo Nordisk (Denmark). The 30% (w/w) hydrogen peroxide, choline chloride (98%), urea (99%), acetamide (99%), ethylene glycol (98%), glycerol (99%), xylitol (98%), D-sorbitol (98%), D-xylose (98%), D-(+)-glucose (99.5%), sucrose (ACS grade), 1-hexene (99%), cyclohexene (99%), 1-decene (99.5%), 1-octadecene (95%), styrene (99%), α-methylstyrene (99%), cyclohexene oxide (98%) and styrene oxide (98%) were bought from Aladdin Chemistry Co., Ltd (Shanghai, China). 1,2-Hexylene oxide (98%), decene oxide (98%), octadecene oxide (99%) and α-methylstyrene oxide (97%) were obtained from TCI (Japan). All other reagents were of analytical grade.Preparation DESs
Deep eutectic solvents were formulated in different molar ratios of quaternary ammonium salt (choline chloride) to hydrogen bond donors. The eutectic mixtures were prepared by rotary evaporation of the two components at 80 °C in water bath until a homogeneous transparent liquid was formed.Stability of enzyme
Purified CalB enzyme was added into 50 mM phosphate buffer pH 6.0, DESs ChCl/urea or ChCl/sorbitol incubated at temperature from 25 °C to 70 °C for 12 h. The samples were extracted from reaction mixture at periodically, and then the perhydrolysis activity was followed spectrophotometrically at 290 nm as described by Peter Bernhardt.39 The tests were carried out in 96-well microtiter plates with a reaction of volume 100 μL (80 μL assays solution to 10 μL samples and 10 μL hydrogen peroxide in 0.1 M pentanoic acid buffer pH 5.0) at 40 °C for 5 minutes using microplate reader Cytation™ 5 (BioTek, USA). The blank experiments were carried out under the identical conditions adding inactived enzymes. All tests were performed in triplicate.Epoxidation reaction
In a 10 mL Erlenmeyer flask were added 1.6 mmol of 1-octadecene, 1.6 mmol of octanoic acid, 1.6 mmol and 1.6 mmol of 30% hydrogen peroxide, 0.4 g of DESs and 100 mg Novozym 435 at 40 °C stirring with 300 rpm. The control without DESs was performed in the phosphate buffer. The 30% hydrogen peroxide was added in 3 portions in 1 h intervals. Samples were withdrawn in different time-points. 20 μL of solution was added into 980 μL methanol and then for gas chromatograph analysis.Analytical methods
Analysis of olefins and corresponding epoxides were determined using an Agilent Technology model 7890 GC (Agilent, USA), equipped with an HP-5 column (30.0 m × 0.25 mm, 0.25 μm, Macherey Nagel, Germany). Peak of olefins and corresponding epoxides in GC were identified and calculated by the calibration curves which were made with the reference standard compounds.Circular dichroism spectroscopy
Circular dichroism (CD) spectroscopy measurements of protein were carried out with a Chirascan spectropolarimeter (Applied Photophysics, Surrey, UK), using 0.2 mg mL−1 of purified CalB enzyme in different medium. The path length was 0.5 mm. Spectra covering the wavelength range of 190–260 nm were scanned at 25 °C. The scanning rate is 1 nm per 0.5 s. Each spectrum was the average of three successive scans. Under the same conditions, the corresponding medium solution was recorded as control and subtracted from the sample spectra. The secondary structure percentages were analyzed by using the CDNN tool.Identification of epoxide
Epoxidized alkenes was identified by nuclear magnetic resonance (NMR) spectra (Bruker Avance 600 MHz). Samples were dissolved in 600 μL deuterated chloroform in a NMR tube and subjected to 1H and 13C NMR analysis.Structure analysis
The modeling of the ligand carboxylic acid in the binding site and complex energy minimization has been performed with MODELLER package (Discovery Studio 3.5). The structure 1LBT and the model of its open conformation40 has been used for analysis.Acknowledgements
This work was supported by National Natural science foundation of China (31471690). National High Technology Research and Development Program of China (863 program, 2014AA093514, 2014AA093601) and Science and Technology Planning project of Guangdong province (2014B020204003, 2015B020231006).Notes and references
- E. J. De Vries and D. B. Janssen, Curr. Opin. Biotechnol., 2003, 14, 414–420 CrossRef CAS PubMed.
- C. Orellana-Coca, S. Camocho, D. Adlercreutz, B. Mattiasson and R. Hatti-Kaul, Eur. J. Lipid Sci. Technol., 2005, 107, 864–870 CrossRef CAS.
- X. Li, Q. Shen, G. Zhang, D. Zhang, A. Zheng, F. Guan and Y. Sun, Catal. Commun., 2013, 41, 126–131 CrossRef.
- N. Scotti, N. Ravasio, R. Psaro, C. Evangelisti, S. Dworakowska, D. Bogdal and F. Zaccheria, Catal. Commun., 2015, 64, 80–85 CrossRef CAS.
- J. B. Park, B. Bühler, T. Habicher, B. Hauer, S. Panke, B. Witholt and A. Schmid, Biotechnol. Bioeng., 2006, 95, 503–505 CrossRef PubMed.
- H. Lin, J. Qiao, Y. Liu and Z. L. Wu, J. Mol. Catal. B: Enzym., 2010, 67, 236–241 CrossRef CAS.
- S. Peter, M. Kinne, R. Ullrich, G. Kayser and M. Hofrichter, Enzyme Microb. Technol., 2013, 52, 370–376 CrossRef CAS PubMed.
- C. E. Paul, D. Tischler, A. Riedel, T. Heine, N. Itoh and F. Hollmann, ACS Catal., 2015, 5, 2961–2965 CrossRef CAS.
- C. Zhang, P. X. Liu, L. Y. Huang, S. P. Wei, L. Wang, S. Y. Yang, X. Q. Yu, L. Pu and Q. Wang, Chem.–Eur. J., 2016, 22, 10969–10975 CrossRef CAS PubMed.
- E. G. Ankudey, H. F. Olivo and T. L. Peeples, Green Chem., 2006, 8, 923–926 RSC.
- M. Svedendahl, P. Carlqvist, C. Branneby, O. Allnér, A. Frise, K. Hult, P. Berglund and T. Brinck, ChemBioChem, 2008, 9, 2442–2451 CrossRef PubMed.
- D. Méndez-Sánchez, N. Ríos-Lombardía, V. Gotor and V. Gotor-Fernández, Tetrahedron, 2014, 70, 1144–1148 CrossRef.
- F. Björkling, S. E. Godtfredsen and O. Kirk, J. Chem. Soc., Chem. Commun., 1990, 1, 1301–1303 RSC.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed.
- J. Littlechild, Curr. Opin. Chem. Biol., 1999, 3, 28–34 CrossRef CAS PubMed.
- A. Li, S. Wu, J. P. Adams, R. Snajdrova and Z. Li, Chem. Commun., 2014, 450, 8771–8774 RSC.
- J. Wu, C. Liu, Y. Jiang, M. Hu, S. Li and Q. Zhai, Catal. Commun., 2010, 11, 727–731 CrossRef CAS.
- M. A. Moreira, T. B. Bitencourt, M. Graça, M. A. Moreira and T. B. Bitencourt, Synth. Commun., 2005, 35, 2107–2114 CrossRef CAS.
- E. Milchert, K. Malarczyk and M. Kłos, Molecules, 2015, 20, 21481–21493 CrossRef CAS PubMed.
- T. B. Bitencourt and M. da Graça Nascimento, Green Chem., 2009, 11, 209–214 RSC.
- Y. Xu, N. R. B. J. Khaw and Z. Li, Green Chem., 2009, 11, 2047–2051 RSC.
- C. Aouf, J. Lecomte, P. Villeneuve, E. Dubreucq and H. Fulcrand, Green Chem., 2012, 14, 2328–2336 RSC.
- C. Aouf, E. Durand, J. Lecomte, M. C. Figueroa-Espinoza, E. Dubreucq, H. Fulcrand and P. Villeneuve, Green Chem., 2014, 16, 1740–1754 RSC.
- A. J. Kotlewska, F. van Rantwijk, R. A. Sheldon and I. W. C. E. Arends, Green Chem., 2011, 13, 2154–2160 RSC.
- T. Welton, Green Chem., 2011, 13, 225 RSC.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- K. Xu, Y. Wang, Y. Huang, N. Li and Q. Wen, Anal. Chim. Acta, 2015, 864, 9–20 CrossRef CAS PubMed.
- C. X. Zeng, S. J. Qi, R. P. Xin, B. Yang and Y. H. Wang, Bioprocess Biosyst. Eng., 2015, 38, 2053–2061 CrossRef CAS PubMed.
- T. De Diego, P. Lozano, S. Gmouh, M. Vaultier and J. L. Iborra, Biotechnol. Bioeng., 2004, 88, 916–924 CrossRef CAS PubMed.
- P. Zhou, X. Wang, C. Zeng, W. Wang, B. Yang, F. Hollmann and Y. Wang, ChemCatChem, 2016 DOI:10.1002/cctc.201601483.
- U. Törnvall, C. Orellana-Coca, R. Hatti-Kaul and D. Adlercreutz, Enzyme Microb. Technol., 2007, 40, 447–451 CrossRef.
- Q. Tang, G. M. Popowicz, X. Wang, J. Liu, I. V. Pavlidis and Y. Wang, ChemistrySelect, 2016, 1, 836–839 CrossRef CAS.
- X. Wang, Q. Tang, G. M. Popowicz, B. Yang and Y. Wang, Biochem. Biophys. Res. Commun., 2015, 460, 392–396 CrossRef CAS PubMed.
- D. L. Yin, P. Bernhardt, K. L. Morley, Y. Jiang, J. D. Cheeseman, V. Purpero, J. D. Schrag and R. J. Kazlauskas, Biochemistry, 2010, 49, 1931–1942 CrossRef CAS PubMed.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- P. Bernhardt, K. Hult and R. J. Kazlauskas, Angew. Chem., Int. Ed., 2005, 44, 2742–2746 CrossRef CAS PubMed.
- J. Uppenberg, N. Ohrner, M. Norin, K. Hult, G. J. Kleywegt, S. Patkar, V. Waagen, T. Anthonsen and T. Jones, Biochemistry, 1995, 34, 16838–16851 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00805h |
| This journal is © The Royal Society of Chemistry 2017 |