
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective hydrodeoxygenation of 5-hydroxymethylfurfural to 2,5-dimethylfuran on Ru–MoOx/C catalysts†
Yue Yang a,
Qiying Liu*b,
Dan Lib,
Jin Tanb,
Qi Zhangb,
Chenguang Wangb and
Longlong Ma*b
a,
Qiying Liu*b,
Dan Lib,
Jin Tanb,
Qi Zhangb,
Chenguang Wangb and
Longlong Ma*b
aDepartment of Chemistry, University of Science and Technology of China, Hefei 230026, P. R. China
bKey Laboratory of Renewable Energy, Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, P. R. China. E-mail: mall@ms.giec.ac.cn; liuqy@ms.giec.ac.cn; Tel: +86-20-3702-9721, +86-20-87057673
First published on 14th March 2017
Abstract
Selective hydrogenation of 5-hydroxymethylfurfural (HMF) has potential application in high quality biofuels. Herein, the catalytic hydrodeoxygenation (HDO) of HMF to 2,5-dimethylfuran (DMF) was investigated using bi-functional Ru–MoOx/C catalyst prepared by initial wetness impregnation. The high dispersion and electronic transfer between Ru and MoOx were demonstrated by a series of characterization techniques. During this HDO process, the synergy effect between metallic Ru and acidic MoOx species in the Ru–MoOx/C catalyst plays an essential role in obtaining maximized target product DMF (79.4%) via effective aldehyde group hydrogenation by Ru followed by dehydration over MoOx. This work also elucidated that DMF production proceeded through two distinct pathways: the 2,5-hydroxymethyl furan intermediate was preferable by the aldehyde group hydrogenation of HMF over the Ru–MoOx/C catalyst. Over MoOx/C catalyst, comparatively, 5-methyl furfural was the key intermediate by direct hydrogenolysis of the hydroxyl group in HMF. This kind of catalyst is stable for the first two runs by maintaining the target product yield. After the third run, the catalyst showed deactivation gradually but could be almost completely recovered after regeneration by H2 reduction.
1. Introduction
With the decrease in fossil resources, the demand for exploring new sustainable energy to produce chemicals and fuel is urgent for the future.1–3 Renewable and sustainable lignocellulose is composed of cellulose (40–50%), hemicellulose (25–35%) and lignin (15–20%), and can be used as feedstock for value-added chemicals/fuels production, which has attracted great attention during the last decade.4,5 However, the typical biomass-derived compounds usually have high oxygen-content and could not be used for fuel directly.6,7 Therefore, an additional hydrogenation step is necessary to complete or selectively remove the oxygen atoms in the compounds.8,95-Hydroxymethylfurfural (HMF), one of the important platform compounds from cellulose,10–12 can be further converted into a series of valuable high-quality chemicals/fuels, such as γ-valerolactone, levulinic acid, oxidative 2,5-furandicarboxylic, 2,5-dimethylfuran and so on.13–17 Among those chemicals/fuels, 2,5-dimethylfuran (DMF)19–21 by HMF hydrogenolysis has attracted much attention, due to the oxygen-containing DMF being an ideal renewable oxygen liquid fuel according to its higher energy density, higher octane number and higher boiling point compared to the traditional cellulose based ethanol.22,23
Catalysts played the key role in producing DMF with high yields from HMF, lignocellulosic biomass and the derivatives. In 2007, Dumesic et al.1 (Table 1, entry 1) firstly reported a catalytic strategy using a biphasic system to extract HMF, and a separated process using 6.8 bar of H2 over CuRu/C catalyst at 220 °C to produce 71% yield of DMF. Under the same reaction conditions, Binder and Raines25 (Table 1, entry 8) achieved 49% DMF yield by hydrogenolysis of crude HMF which obtained from corn stover. Chidambaram and Bell2 (Table 1, entry 7) added acetonitrile as auxiliaries and obtained 32% DMF yield over Pd/C catalyst in ionic liquids. For improving the yield of DMF, Rauchfuss et al.27 (Table 1, entry 6) utilized HCOOH as the hydrogen donor and obtained 95% DMF yield. However, HCOOH is corrosive and environment unfriendly, which leads to apply restrictedly.7 Recently, researchers reported to employ adding transitional metal additives which can form Lewis acid to obtain high DMF yield by adsorption and activation the hydroxyl and aldehyde group in HMF. And the metal sites were active for hydrogenation while the acidic sites for promoting dehydration of CH2–OH group.28,29 Zu et al.24 (Table 1, entry 4) used Ru/Co3O4 to produce 93.4% DMF yield at 130 °C after 24 h and Huang et al.26 (Table 1, entry 5) reached up to 96% DMF yield over Ni-W2C/AC at 180 °C after 3 h in THF. Judging from the results, the bi-functional catalyst was an ideal pattern for HMF hydrogenolysis, but these process involved either long reaction time or high H2 pressure and the interaction between metal and acid sites was not clearly. Therefore, there is a need to develop a new-type metal–acid catalyst to conversion of HMF to DMF in mild condition and explore the synergy effect between metal and acid sites.
| Order | Substrate | Medium | Catalyst | Condition | Yield% | References |
|---|---|---|---|---|---|---|
| 1 | HMF | 1-Butanol | Cu/Ru/C | 220 °C, 10 h | 71 | 1 |
| 2 | HMF | n-Butyl alcohol | Ru/C | 260 °C, 1.5 h | 60.3 | 18 |
| 3 | HMF | THF | Zn/Pd/C | 150 °C, 8 h | 85 | 22 |
| 4 | HMF | THF | Ru/Co3O4 | 130 °C, 24 h | 93.4 | 24 |
| 5 | HMF | THF | Ni–W2C/AC | 180 °C, 3 h | 96 | 26 |
| 6 | HMF | THF + HCOOH | H2SO4 + Pd/C | 70 °C, 15 h | 95 | 27 |
| 7 | Glucose | EMIMCl | 12-MPA + Pd/C | 120 °C, 3 h | 32 | 2 |
| 8 | Cellulose | DMA-LiCl/ EMIMCl | CrCl2 + HCl + Cu/Ru/C | 220 °C, 10 h | 49 | 25 |
On the basis of the above-mentioned research, we designed the metal–acid Ru–MoOx/C catalyst by step wetness impregnation for HMF hydrogenolysis to DMF in n-butyl alcohol. Due to the different functional groups in the HMF, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CH2–OH, furan ring, hydrogenation of HMF to DMF needs CO hydrogenation to the corresponding hydroxyl groups and subsequent C–OH groups hydrogenolysis. Under the optimum condition (180 °C, 1.5 MPa, 1 h), the selectivity of DMF was 79.8%. In addition, the characterization of catalyst was conducted to explain the interaction and electronic transfer between Ru and MoOx, and the cause of catalyst deactivation. In addition, various reaction parameters was modulated to investigate the pathway for HMF hydrogenolysis and the synergistic effect of the metal and acid sites.
O, CH2–OH, furan ring, hydrogenation of HMF to DMF needs CO hydrogenation to the corresponding hydroxyl groups and subsequent C–OH groups hydrogenolysis. Under the optimum condition (180 °C, 1.5 MPa, 1 h), the selectivity of DMF was 79.8%. In addition, the characterization of catalyst was conducted to explain the interaction and electronic transfer between Ru and MoOx, and the cause of catalyst deactivation. In addition, various reaction parameters was modulated to investigate the pathway for HMF hydrogenolysis and the synergistic effect of the metal and acid sites.
2. Experimental
2.1. Materials
HMF (98%) and 5-MF (99%) was supplied by J&K chemical Co. Ltd. DMF (99%) and RuCl3·3H2O was purchased from Shanghai Aladdin Reagent Co. Ltd. 2,5-Hexanediol (98%) and 2,5-hexanedione (98%) was sourced by Macklin Reagent Co. Ltd. 2,5-dihydroxymethylfuran (95%), (5-methyl-2-furyl)methanol (97%), (cis-tetrahydrofuran-2,5-diyl)di-methanol (95%) and (NH4)6Mo7O24·4H2O, 2,5-dimethyltetrahydrofuran (98%) was obtained from Maya Reagent, Energy chemical, Shu ya Reagent, Tianjin Kaida chemical and TCI Co. Ltd respectively. All the reagents was used without further purification.2.2. Catalyst preparation
The catalyst was prepared by step-wise impregnation method. In a typical synthesis, active carbon (40–60 mesh) was chose as support materials. Firstly it was pretreated by nitric acid (0.6 mol l−1) at 110 °C for 5 h, then filtered until cooled to room temperature, and dried at 100 °C overnight. RuCl3·3H2O was dissolved in deionized water and a certain amount of active carbon was added to a desired Ru loading by weight, and stirred at 100 °C to remove excess water. The solid was dried at 100 °C overnight and calcined under a N2 atmosphere at 400 °C for 4 h. After the preparation of Ru/C catalyst, a second impregnation with (NH4)6Mo7O24 solution by the similar procedure mentioned above. Prior to the experiment, the dried sample was reduced in flowing H2 (40 ml min−1) at 350 °C for 4 h.2.3. Catalyst performance
Hydrogenolysis of HMF was carried out in a stainless steel reactor with a Teflon inner. In a typical experiment, a mixture of 0.5 g HMF dissolved in 40 ml n-butyl alcohol was loaded to the autoclave, followed by adding 0.1 g of freshly reduced 5% Ru–10% MoOx/C catalyst (reduced at 350 °C for 4 h). The reaction system was sealed, flushed by N2 to remove air residue, pressurized the H2 pressure to 1.5 MPa, and then heated to a certain temperature for a certain time. After the reaction, the reactor was cooled to room temperature immediately in a water bath. The liquid products was collected and qualitatively analyzed by a GC-MS instrument with an Agilent 7890A-5975C and quantitatively determined by GC with SHIMADZU GC-2014C gas chromatograph equipped with a FID and a HP-INNOWAX column.The product yields were calculated based on internal standard with using cyclohexanone as the internal standard. The conversion (conv.) of HMF and the selectivity of product i (Si) were defined as:
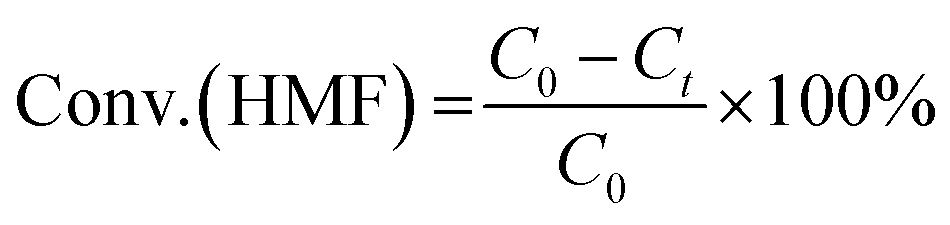
here, C0 refers to the initial concentration of HMF and Ct means the mole concentration of final HMF after reaction; Ci indicates the concentration of product i, and ai represents the carbon numbers of product i.
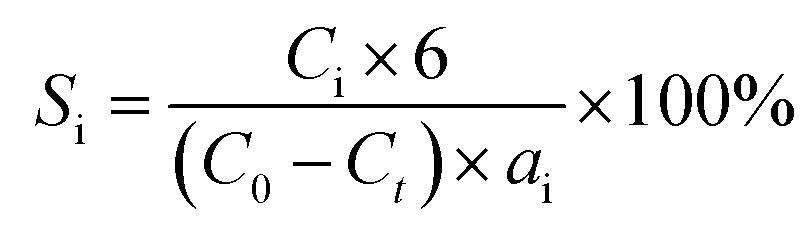
2.4. Catalyst characterization
X-ray photoelectron spectroscopy (XPS) was conducted on a Thermo Fisher Scientific Escalab 250 Xi photoelectron spectrometer, the values were revised by C 1s (284.8 eV). The specific surface area was measured by N2 isothermal (77 K) adsorption on Mesoporous ASAP-2010 automated system. The tested samples was outgassed at 200 °C for 8 h prior to analysis. The surface area was calculated by BET method. The morphology and energy spectrum analysis of catalysts were investigated by transmission electron microscopy (TEM, JEM-2100F) and scanning electron microscopy (SEM, SU-70). Temperature programmed reduction (TPR) equipped with a TCD detector was used to measure the reduction behavior under H2. For H2-TPR experiments, 100 mg sample was laid into a quartz tube and pretreated by Ar at 300 °C for 1 h, then cooling to 50 °C, the catalysts were heated to 600 °C at the rate of 5 °C min−1. Temperature programmed desorption of NH3 (NH3-TPD) experiments were conducted on AutoChem II 2920 to analyze the acid content. The catalysts were purged by He at 673 K prior to adsorption, then cooling to 373 K to saturate with NH3 and sweep by He to remove the physical absorbed NH3 for 30 min, whereafter, the samples were heated to 673 K at the speed of 10 K min−1. All of the catalysts which needed to reduce were pretreated under H2 stream for 4 h at 350 °C and cooled to room temperature, then passivated by 0.6% O2/N2 gas for 4 h.3. Results and discussion
3.1. Catalyst characterization
Hydrogen temperature-programmed reduction (H2-TPR) was conducted to investigate the reduction properties of the current Ru–MoOx/C catalysts, and the results were shown in Fig. 1. For 5% Ru/C catalyst, two reduction peaks at 100 °C and 230 °C were observed, these peaks were the reduction of RuOx species due to the different interactions with support. The first peak was assigned to the weak interaction of Ru with support, which could be easily reduce. While, the second peak was assigned to the reduction of RuOx species which was strongly interaction with support.30–32 the Ru catalytic activity can promote the break of C–C bond on the surface of active carbon, which leaded to the uphill of TCD signal at 500 °C.32,33 The profile of 10% MoOx/C catalyst presented a main peak and a shoulder peak centered at 450 °C and 550 °C, respectively. It was attributed to the reduction of Mo6+ with octahedral coordination to Mo4+ at 450 °C, while the H2 consumption peaks located at 550 °C belonged to the reduction of tetrahedrally coordinated Mo species to lower oxidation state species.34–37 Over the 5% Ru–10% MoOx/C catalyst, the small reduction peak at 180 °C was attributed to RuOx to Ru0.38 However, the main Mo reduction peak located at 350 °C and it was lower than the 10% MoOx/C catalyst (450 °C). These results suggested that Ru facilitated to the reduction of Mo species,39 in which H2 was adsorbed on Ru and dissociated to highly reactive hydrogen atoms.40 When Ru and MoOx species was close on the support, the H2 spillover facilitated the reduction of MoOx.39,41–43 This behavior increased a possible electronic transfer between Ru and MoOx.39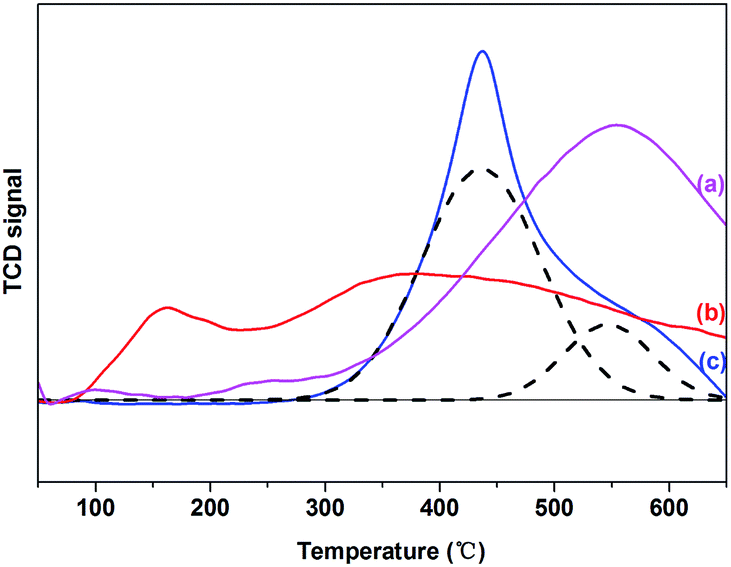 | ||
| Fig. 1 H2-TPR profiles of (a) 5% Ru/C, (b) 5% Ru–10% MoOx/C and (c) 10% MoOx/C catalysts. | ||
To verify the interaction between Ru and MoOx, the surface chemical state of 5% Ru/C, 10% MoOx/C and 5% Ru–10% MoOx/C were conducted by X-ray photoelectron spectroscopy (XPS) characterization. Because of 5% Ru/C and 5% Ru–10% MoOx/C catalyst were liable to oxidization, they were reduced by in situ technology to detect the chemical state of elements. Due to the photoemission line of Ru 3d was overlapped with C 1s,44 Ru 3p3/2 line was used to analyze the surface species. As shown in Fig. 2a, for the fresh 5% Ru–10% MoOx/C catalyst, the binding energy of 463.2 eV was ascribed to RuO2,45 and shifted to relatively low binding energy after reduction by H2 (reduced 5% Ru–10% MoOx/C catalyst), assigning to a reduction of RuO2 to metal Ru. In addition, over the reduced 5% Ru/C catalyst, the binding energy of metal Ru shifted to higher binding energy compared to the reduced 5% Ru–10% MoOx/C catalyst, which indicated that there have some electrons was transferred from MoOx to Ru.39 On the other hand, the chemical status of Mo 3d was measured and the results were shown in Fig. 2b. The fresh 5% Ru–10% MoOx/C sample presented only one doublet located at 232.6 eV which was assigned to Mo6+ species (MoO3).35 The reduced 5% Ru–10% MoOx/C catalyst revealed three doublets, and the values at 232.0 eV and 229.6 eV were contributed to the Mo5+ and Mo4+ formation during H2 reduction.46,47 The binding energy of Mo4+ over 5% Ru–10% MoOx/C reduced catalyst was shifted to left compared to 10% MoOx/C catalyst, which was attributed to the fact that some electrons were transferred from MoOx to Ru. The low chemical valence of MoOx (x < 3) could form surface detects, which promotes hydrogenolysis of hydroxyl groups as Lewis acid.46 In addition, from Fig. 2a, the value of the used 5% Ru–10% MoOx/C catalyst reflected this pattern existed two chemical state, namely Ru and RuO2.
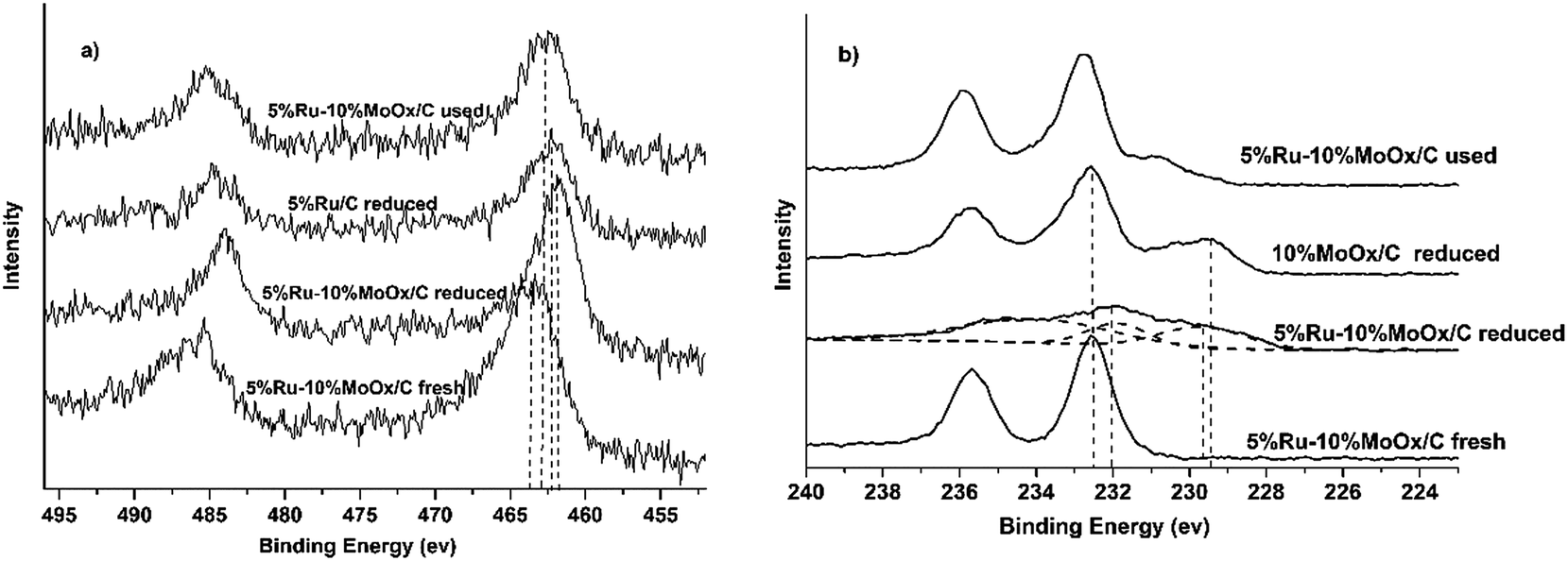 | ||
| Fig. 2 XPS spectra of Ru–MoOx/C catalysts: (a) Ru 3p and (b) Mo 3d. 5% Ru–10% MoOx/C fresh: the catalyst had not been reduced by H2; 5% Ru–10% MoOx/C, 10% MoOx/C and 5% Ru/C reduced: the catalysts were reduced by H2 under 350 °C; 5% Ru–10% MoOx/C used: the catalyst was reduced by H2 under 350 °C and then used at the reaction condition. | ||
Fig. 3 showed the TEM images of specified catalysts and the diameter of particles was calculated by the software Nano Measure. All of the particles was included to calculate the average particle size in the visible range. For the 10% MoOx/C catalyst, the MoOx particles (Fig. 3e) presented the average diameter of about 7.3 nm. The supported 5% Ru/C (Fig. 3d) catalyst showed a better dispersion by observing the smaller average diameter of about 2.6 nm. Compared to the previous two monometallic catalysts, however, the average particle size of 5% Ru–10% MoOx/C catalyst (Fig. 3a) was about 1.8 nm which was highly dispersed on the support of activation carbon, owing to the interaction between Ru and MoOx nanoparticles and facilitates the dispersion.48,49 The high dispersion was benefit to hydrogenolysis.48,50 While, the few larger particles in Fig. 3a were MoOx species (distinguished by the HRTEM on the corner), which had not been combined with Ru, and their particle size was about 17 nm, it must be pointed out that this size can not reflect the actual size of MoOx because of few statistical particles. The average size of 5% Ru–5% MoOx/C (Fig. 3b) and 5% Ru–20% MoOx/C (Fig. 3c) were 1.1 nm and 2.5 nm respectively, and the 5% Ru–20% MoOx/C was showed poor dispersion, which revealed the MoOx content could impact the size of particle and dispersion. Meanwhile, the high-resolution electron microscopy (HRTEM) image showed the morphology of the two species (Fig. 3f) of 5% Ru–10% MoOx/C catalyst. The MoOx species was close contact with the Ru moiety in this two metallic catalyst, which induces the electron transferring from MoOx to Ru. This result is well consistent with the XPS measurement.39
 | ||
| Fig. 3 TEM images of Ru–MoOx/C catalysts: (a) reduced 5% Ru–10% MoOx/C; (b) reduced 5% Ru–5% MoOx/C; (c) reduced 5% Ru–20% MoOx/C; (d) reduced 5% Ru/C; (e) reduced 10% MoOx/C and (f) HRTEM image of reduced 5% Ru–10% MoOx/C. | ||
Surface acidity and acidic strength distribution of Ru–MoOx/C with different MoOx loading was evaluated by NH3-TPD (Fig. S1†). The NH3-TPD profiles of 5% Ru/C, 5% Ru–5% MoOx/C, 5% Ru–10% MoOx/C, and 5% Ru–20% MoOx/C consisted a wider shoulder peak in the temperature range of 390–480 K, and these peaks could be attributed to NH3 adsorb on the weak acid site of the catalyst surface.51 For the Ru/C catalyst, almost no peak was observed (Table S1,† entry 4), with the increase of MoOx loading, the peak area augmentation and the strength intensified which indicated that the total acid was enhanced (Table S1†).
3.2. HMF hydrogenolysis to DMF
The catalyst was tested in HMF hydrogenolysis to produce DMF. During the reaction process, 2,5-dihydroxymethylfuran (DHMF) and 5-methylfurfural (MF) were the main intermediates along with different reaction pathways. Meanwhile, the byproducts mainly contained the O-containing intermediates (e.g. 5-methyl-2-furanmethanol (MFA), 2,5-dihydroxymethyl tetrahydrofuran (DHMTHF)) and the over-hydrogenated products such as 2,5-dimethyl tetrahydrofuran (DMTHF), 2,5-hexanedione and 2,5-hexanediol by furan ring opening. The carbon balance of all detected products was about 65–86% maybe due to the formation of undetected molecules7,24,52 and polymers and/or cokes.53 In the studied condition, the n-butyl alcohol had no reactivity over catalysts.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | HMF conv.% | DMF sel.% | DMTHF sel.% | 5-MF sel.% | MFA sel.% | DHMF sel.% | DHMTHF sel.% | 2,5-Hexanedione sel.% | 2,5-Hexanediol sel.% |
| a Reaction condition: 180 °C, 1.5 MPa of H2, 1 h, HMF 0.5 g, n-butyl alcohol 40 ml and 0.1 g catalyst; the Ru and Mo loading were defined by the mass fraction on support. | ||||||||||
| 1 | 2% Ru–10% MoOx/C | 100 | 60.8 | 3.6 | — | — | — | 0.2 | 1.4 | 0.7 |
| 2 | 5% Ru–10% MoOx/C | 100 | 79.8 | 3.4 | — | — | — | 0.4 | 0.7 | 1.0 |
| 3 | 5% Ru–5% MoOx/C | 100 | 71.8 | 2.6 | — | — | — | 1.1 | 0.4 | 0.9 |
| 4 | 5% Ru–20% MoOx/C | 100 | 72.6 | 3.1 | — | — | — | 0.2 | 1.9 | 0.9 |
| 5 | 5% Ru/C | 100 | 54.1 | 6.7 | — | — | — | 10.1 | 2.2 | 0.5 |
| 6 | 10% MoOx/C | 39.9 | 3.9 | 8.5 | 37.5 | 0.4 | 2.6 | — | 6.9 | 11.6 |
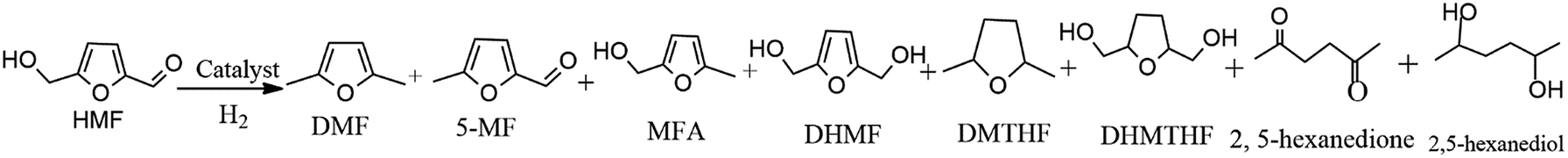
In order to understand the role of Ru and MoOx in this hydrogenolysis reaction, 5% Ru/C and 10% MoOx/C were prepared and tested under the same reaction condition. Over 5% Ru/C catalyst (Table 2, entry 5), the target DMF selectivity was 54.1%, in addition, over-hydrogenated DMTHF and DHMTHF selectivity was 6.7% and 10.1% respectively, which indicated the good hydrogenation and medium DMF selectivity for the sole Ru particle. However, if 10% MoOx/C (Table 2, entry 6) was introduced to the reaction, the HMF conversion was only 39.8% and the DMF selectivity was 3.9%. Besides the target DMF, 37.5% of the main intermediate 5-MF was detected. Apparently, compared to Ru particle, MoOx/C exhibited a good deoxygenation activity to crack the CH2–OH bond in HMF, while its hydrogenation activity to activate aldehyde group (–CHO) was rather limited. Based on the above results, the synergy of Ru and MoOx with proper ratios played a key role in obtaining high DMF yield in hydrogenolysis of HMF.
This catalytic performance could also be explained by the electronic modification between Ru and MoOx. Compared the results of 5% Ru/C (Table 2, entry 5) with 5% Ru–10% MoOx/C (Table 2, entry 2), the selectivity of DMTHF and DHMTHF were 6.7% and 10.1% over the 5% Ru/C catalyst, while the selectivities were decreased to 3.4% and 0.4% over 5% Ru–10% MoOx/C catalyst. These results reflected that the hydrogenolysis ability on Ru particle was restrained when added MoOx, which leaded to obtain more target DMF over Ru–MoOx/C catalyst. The relatively week hydrogenolysis for furan ring was attributed to the electron enrichment of Ru, which was similar to the result of hydrogenolysis of n-butane by Scott et al.54 In the characterization section, we had interpreted the increase in the Ru electronic density due to the electronic transfer from partially reduced MoOx to Ru. A higher electronic density over Ru increased the hydrogenolysis of the minimum electron density C–OH bond since Ru electron-donating properties.39 On the other hand, the mutual electronic impact maybe was attributed to the synergistic activity. Considering the highest DMF yield obtained, the 5–10% RuMo/C catalyst was selected for the following controlled experiments.
As shown in Table 3 (entry 1–5), when the temperature was 120 °C, only 21.7% of DMF selectivity was observed even though at the 97.1% of HMF conversion. The intermediates (5-MF, MFA and DHMF) and byproducts (DMTHF, DHMTHF, 2,5-hexanedione and 2,5-hexanediol) were detected with DHMF (36.3%) and MFA (11.2%) as the largest two. With increasing the reaction temperature from 120 °C to 180 °C, the HMF conversion and DMF selectivity were correspondingly increased from 97.1% to 100% and from 21.7% to 79.8%, respectively, which was attributed to that the intermediate products was converted to the target DMF gradually. Additionally, the over-hydrogenated byproducts were also risen but with the total amounts below 6%. However, when the temperature was further increased to 200 °C, the selectivity of DMF was decreased to 69.0%. Meanwhile, the byproducts (DMTHF, 2,5-hexanedione and 2,5-hexanediol) were significantly increased to 12.5%.
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | PH2 (MPa) | T (°C) | t (h) | HMF conv.% | DMF sel.% | DMTHF sel.% | 5-MF sel.% | MFA sel.% | DHMF sel.% | DHMTHF sel.% | 2,5-Hexanedione sel.% | 2,5-Hexanediol sel.% |
| a Reaction condition: HMF 0.5 g, n-butyl alcohol 40 ml and 0.1 g 5% Ru–0% MoOx/C catalyst; the Ru and Mo loading were defined by the mass fraction on support. | ||||||||||||
| 1 | 1.5 | 120 | 1 | 97.1 | 21.7 | 0.2 | 3.6 | 11.2 | 36.3 | 0.2 | 0.5 | 0.4 |
| 2 | 1.5 | 140 | 1 | 99.8 | 47.9 | 0.8 | 1.0 | 3.5 | 5.0 | 0.4 | 1.5 | 0.6 |
| 3 | 1.5 | 160 | 1 | 100 | 74.4 | 2.1 | 0.1 | 0.8 | 0.1 | 0.6 | 0.8 | 0.8 |
| 4 | 1.5 | 180 | 1 | 100 | 79.8 | 3.4 | — | — | — | 0.4 | 0.7 | 1.0 |
| 5 | 1.5 | 200 | 1 | 100 | 69.0 | 9.5 | — | — | — | 0.4 | 0.3 | 2.3 |
| 6 | 1.5 | 120 | 0.25 | 93.2 | 17.8 | 0.8 | 2.2 | 5.4 | 29.4 | 0.1 | 0.3 | 0.3 |
| 7 | 1.5 | 120 | 0.5 | 95.6 | 17.4 | 0.5 | 3.4 | 11.3 | 46.6 | 0.3 | 0.4 | 0.4 |
| 8 | 1.5 | 120 | 2 | 99.6 | 46.4 | 0.6 | 1.5 | 5.3 | 13.2 | 0.4 | 1.3 | 0.6 |
| 9 | 1.5 | 120 | 4 | 99.7 | 56.1 | 0.6 | 0.6 | 4.7 | 6.8 | 0.5 | 0.6 | 0.7 |
| 10 | 1.5 | 120 | 6 | 99.8 | 50.9 | 0.9 | 0.9 | 5.4 | 6.9 | 1.0 | 0.3 | 0.8 |
The influence of reaction time was listed in Table 3, entry 6–10. At the initial time of 0.25 h, the conversion of HMF was reached to 93.2%, nearly completely converted. However, the selectivity of the target DMF was only 17.8% and DHMF (29.4%) and MFA (5.4%) were presented as the main products. Prolonging the reaction time to 0.5 h, the conversion of HMF was increased to 95.6% and the selectivity of DMF was almost constant. While the selectivity of DHMF (46.6%) and MFA (11.3%) reached to the maximum. As the reaction time was further extended to 4 h, 56.1% of DMF was obtained while the slightly reduced MFA and DHMF intermediates were obtained. When the time was exceeded 4 h, the yield of DMF was slightly decreased and some byproducts including DHMTHF and DMTHF were produced, which is generated by furan ring saturation via over-hydrogenation. The Fig. S2† showed the concentration of the products as a function of time.
Based on the above results, the aldehyde group in HMF was firstly attacked to form DHMF over the bimetallic Ru–MoOx/C catalyst. Since DHMF could be converted to MFA and further be hydrogenated to DMF through cleavage of CH2–OH bond, MFA was a key precursor to be converted into DMF. Interestingly, over 10% MoOx/C catalyst (Table 2, entry 6), the result revealed that the main intermediate was 5-MF and the selectivity was 37.5%, which indicates that a different reaction pathway was observed via the first step of dehydroxylation in the presence of aldehyde group, comparing to the bimetallic Ru–MoOx/C catalyst. The proposed reaction pathway was shown in Scheme 1.
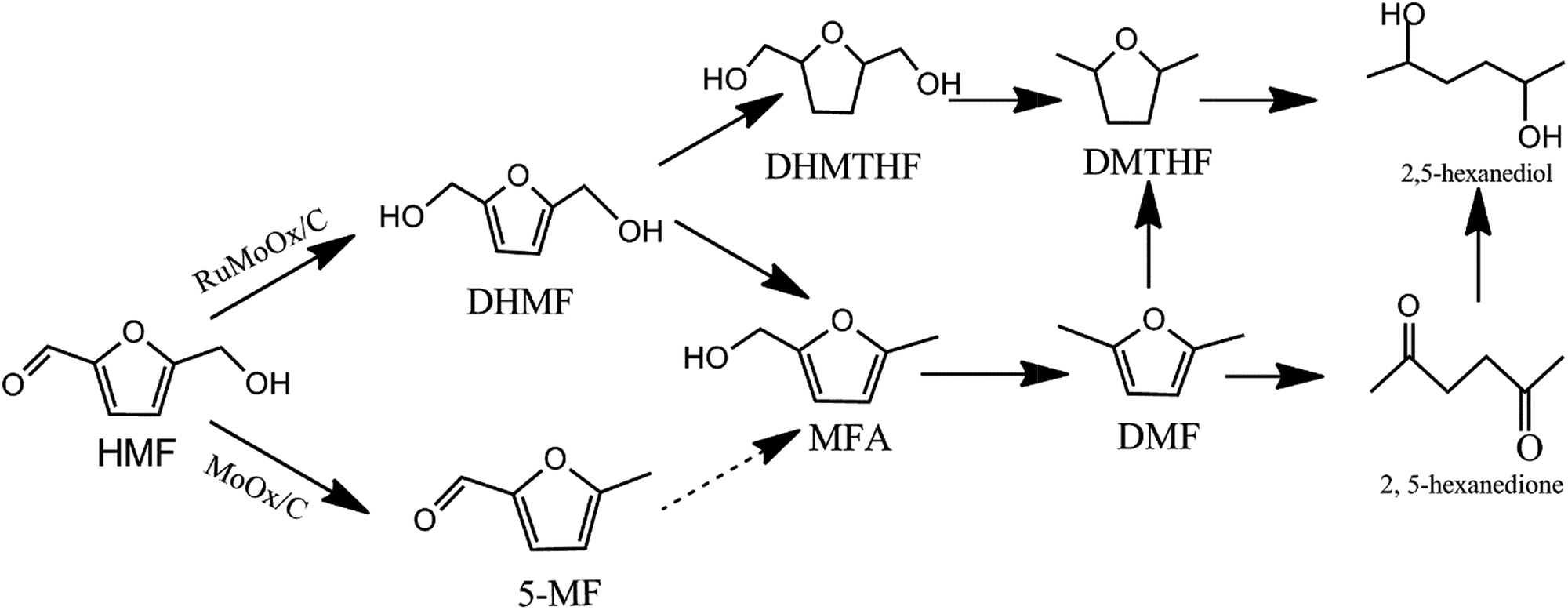 | ||
| Scheme 1 Proposed reaction network for hydrogenolysis of HMF to DMF. | ||
According to the rules of concentration intermediates change, the catalytic step can be described as follows. Initially, Ru metal site could activated H2 and further dissociative absorbed on the metal site. Meanwhile, the O atom of aldehyde group in HMF was activated by the MoOx vacant orbital through Mo–O bond.55 Firstly, activated H attacked the aldehyde group to produce DHMF, which was a key intermediate in HMF hydrolysis. Then, the MoOx acid site accelerated dehydroxylation to generate DMF via MFA intermediate. After that, the DMF was desorbed and catalyst was used into the next round.19
 | ||
| Fig. 4 Reusability of the Ru–MoOx/C catalyst. Reaction condition: 180 °C, 1.5 MPa of H2, 1 h, HMF 0.5 g, n-butyl alcohol 40 ml and 5% Ru–10% MoOx/C catalyst 0.1 g. | ||
 | ||
| Fig. 5 SEM images of (a) the fresh 5% Ru–10% MoOx/C catalyst, (b) the used of 5% Ru–10% MoOx/C catalyst after first run, (c) the used of 5% Ru–10% MoOx/C catalyst after five runs. And TEM image of (d) the used of 5% Ru–10% MoOx/C catalyst after first run, (e) the used of 5% Ru–10% MoOx/C catalyst after five runs and (f) the regenerated of 5% Ru–10% MoOx/C catalyst after five runs. | ||
| Catalyst | BET surface areaa (m2 g−1) | Total pore volumea (cm3 g−1) | Mean particle size diameterb (nm) |
|---|---|---|---|
| a The BET surface area, pore volume were determined by N2 physical adsorption.b Particle size was observed by TEM. | |||
| Fresh 5% Ru–10% MoOx/C | 447.9 | 0.26 | 1.5 |
| Used 5% Ru–10% MoOx/C after first run | 455.6 | 0.29 | 1.7 |
| Used 5% Ru–10% MoOx/C after five run | 194.9 | 0.15 | 2.6 |
| Regenerated after five run | 384.0 | 0.23 | 2.2 |
The above characterization results suggested that the deactivation of our sample maybe due to the agglomeration of carbon support and the decrease of surface area. Then the used five times catalyst was regenerated at 350 °C and the flowing H2 (40 ml min−1) for 4 h. As depicted by Fig. 4, the HMF conversion and DMF yield was regained 99.7% and 79%, indicating this catalyst could be completely recovered after regeneration. Table 4 also revealed that the surface area and total pore volume was increased after regenerated, which was well consistent with the reaction results.
4. Conclusion
The bi-functional Ru–MoOx/C catalysts were prepared by initial wetness impregnation and their catalytic performance was tested in DMF production from hydrogenolysis of biomass-derived HMF by finely modulating the metal–acid properties of catalysts and reaction parameters. The synergy effect between metal and acid sites plays a key role in obtaining high selectivity of DMF (79.8%). The characterizations of catalyst were interpreted the interaction and electron transfer between Ru and MoOx and the cause of deactivation, which was attributed to the agglomeration of carbon support and the shrink of surface area of catalyst. Hydrogenation of HMF were proceeded through two pathways: for the Ru–MoOx/C catalyst, hydrogenation of CO group primarily occurred, followed by hydrogenolysis of two CH2–OH bonds in DHMF to obtained the target DMF; comparatively, hydrogenolysis of CH2–OH bonds in HMF was first taken place to 5-MF over MoOx/C catalyst.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 51576199, 50536009 and 51376185).References
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef CAS PubMed.
- M. Chidambaram and A. T. Bell, Green Chem., 2010, 12, 1253 RSC.
- Y. B. Yi, J. W. Lee and C. H. Chung, Environ. Chem. Lett., 2015, 13, 173–190 CrossRef CAS.
- X. L. Tong, Y. Ma and Y. D. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS.
- G. Bottari, A. J. Kumalaputri, K. K. Krawczyk, B. L. Feringa, H. J. Heeres and K. Barta, ChemSusChem, 2015, 8, 1323–1327 CrossRef CAS PubMed.
- M. Y. Chen, C. B. Chen, B. Zada and Y. Fu, Green Chem., 2016, 18, 3858–3866 RSC.
- A. S. Nagpure, N. Lucas and S. V. Chilukuri, ACS Sustainable Chem. Eng., 2015, 3, 2909–2916 CrossRef CAS.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- N. Shi, Q. Y. Liu, T. J. Wang, Q. Zhang, J. L. Tu and L. L. Ma, Chinese J. Phys. Chem., 2014, 27, 711–717 CrossRef CAS.
- A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Commun., 2009, 41, 6276–6278 RSC.
- J. B. Binder, A. V. Cefali, J. J. Blank and R. T. Raines, Energy Environ. Sci., 2010, 3, 765–771 CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- I. T. Horvath, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC.
- L. Hu, X. Tang, J. Xu, Z. Wu, L. Lin and S. Liu, Ind. Eng. Chem. Res., 2014, 53, 3056–3064 CrossRef CAS.
- A. Villa, M. Schiavoni, S. Campisi, G. M. Veith and L. Prati, ChemSusChem, 2013, 6, 609–612 CrossRef CAS PubMed.
- Y. L. Zhang, Z. M. Xue, J. F. Wang, X. H. Zhao, Y. H. Deng, W. C. Zhao and T. C. Mu, RSC Adv., 2016, 6, 51229–51237 RSC.
- J. H. Zhang, L. Lin and S. J. Liu, Energy Fuels, 2012, 26, 4560–4567 CrossRef CAS.
- A. B. Gawade, M. S. Tiwari and G. D. Yadav, ACS Sustainable Chem. Eng., 2016, 4, 4113–4123 CrossRef CAS.
- T. S. Hansen, K. Barta, P. T. Anastas, P. C. Ford and A. Riisager, Green Chem., 2012, 14, 2457 RSC.
- J. Jae, W. Zheng, A. M. Karim, W. Guo, R. F. Lobo and D. G. Vlachos, ChemCatChem, 2014, 6, 848–856 CrossRef CAS.
- B. Saha, C. M. Bohn and M. M. Abu-Omar, ChemSusChem, 2014, 7, 3095–3101 CrossRef CAS PubMed.
- S. H. Zhong, R. Daniel, H. Xu, J. Zhang, D. Turner, M. L. Wyszynski and P. Richards, Energy Fuels, 2010, 24, 2891–2899 CrossRef CAS.
- Y. H. Zu, P. P. Yang, J. J. Wang, X. H. Liu, J. W. Ren, G. Z. Lu and Y. Q. Wang, Appl. Catal., B, 2014, 146, 244–248 CrossRef CAS.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS PubMed.
- Y. B. Huang, M. Y. Chen, L. Yan, Q. X. Guo and Y. Fu, ChemSusChem, 2014, 7, 1068–1072 CrossRef CAS PubMed.
- T. Thananatthanachon and T. B. Rauchfuss, Angew. Chem., 2010, 49, 6616–6618 CrossRef CAS PubMed.
- Y. Kusunoki, T. Miyazawa, K. Kunimori and K. Tomishige, Catal. Commun., 2005, 6, 645–649 CrossRef CAS.
- X. Kong, R. X. Zheng, Y. F. Zhu, G. Q. Ding, Y. L. Zhu and Y. W. Li, Green Chem., 2015, 17, 2504–2514 RSC.
- A. S. Nagpure, A. K. Venugopal, N. Lucas, M. Manikandan, R. Thirumalaiswamy and S. Chilukuri, Catal. Sci. Technol., 2015, 5, 1463–1472 CAS.
- S. Z. Zheng, X. Y. Cao, Q. Zhou, S. H. Wang, G. S. Hu, J. Q. Lu, M. F. Luo and Y. J. Wang, J. Fluorine Chem., 2013, 145, 132–135 CrossRef CAS.
- I. Rossetti, N. Pernicone and L. Forni, Appl. Catal., A, 2003, 248, 97–103 CrossRef CAS.
- L. A. Chen, Y. L. Zhu, H. Y. Zheng, C. H. Zhang, B. Zhang and Y. W. Li, J. Mol. Catal. A: Chem., 2011, 351, 217–227 CrossRef CAS.
- T. Bhaskar, K. R. Reddy, C. P. Kumar, M. R. V. S. Murthy and K. V. R. Chary, Appl. Catal., A, 2001, 211, 189–201 CrossRef CAS.
- J. Chang, T. Danuthai, S. Dewiyanti, C. Wang and A. Borgna, ChemCatChem, 2013, 5, 3041–3049 CrossRef CAS.
- L. J. Feng, X. G. Li, D. B. Dadyburjor and E. L. Kugler, J. Catal., 2000, 190, 1–13 CrossRef CAS.
- V. R. Surisetty, I. Eswaramoorthi and A. K. Dalai, Fuel, 2012, 96, 77–84 CrossRef CAS.
- P. G. J. Koopman, A. P. G. Kieboom and H. Vanbekkum, J. Catal., 1981, 69, 172–179 CrossRef CAS.
- C. E. Scott, P. Betancourt, M. J. P. Zurita, C. Bolivar and J. Goldwasser, Appl. Catal., A, 2000, 197, 23–29 CrossRef CAS.
- H. Cheng, M. Wen, X. Ma, Y. Kuwahara, K. Mori, Y. Dai, B. Huang and H. Yamashita, J. Am. Chem. Soc., 2016, 138, 9316–9324 CrossRef CAS PubMed.
- P. A. Sermon and G. C. Bond, Catal. Rev., 1973, 8, 211–239 CAS.
- K. Fabicovicova, M. Lucas and P. Claus, Green Chem., 2015, 17, 3075–3083 RSC.
- N. Li, Y. Zheng, L. Wei, H. Teng and J. Zhou, Green Chem., 2016, 18, 1821–2242 RSC.
- J. L. Fu, K. X. Yang, C. J. Ma, N. W. Zhang, H. J. Gai, J. B. Zheng and B. H. Chen, Appl. Catal., B, 2016, 184, 216–222 CrossRef CAS.
- Y. J. Weng, S. B. Qiu, C. G. Wang, L. G. Chen, Z. Q. Yuan, M. Y. Ding, Q. Zhang, L. L. Ma and T. J. Wang, Fuel, 2016, 170, 77–83 CrossRef CAS.
- V. M. L. Whiffen and K. J. Smith, Energy Fuels, 2010, 24, 4728–4737 CrossRef CAS.
- M. Shetty, K. Murugappan, T. Prasomsri, W. H. Green and Y. Román-Leshkov, J. Catal., 2015, 331, 86–97 CrossRef CAS.
- P. P. Yang, Q. N. Xia, X. H. Liu and Y. Q. Wang, J. Energy Chem., 2016, 25, 1015–1020 CrossRef.
- W. T. Luo, Y. Lyu, L. F. Gong, H. Du, M. Jiang and Y. J. Ding, Chin. J. Catal., 2016, 37, 2009–2017 CrossRef CAS.
- X. B. Fu, H. Yu, F. Peng, H. J. Wang and Y. Qian, Appl. Catal., A, 2007, 321, 190–197 CrossRef CAS.
- Y. H. Chu, T. T. Zhang, J. X. Guo, C. Liu, H. Q. Yin, X. F. Zhu and Y. J. Liu, J. Rare Earths, 2015, 33, 371–381 CrossRef CAS.
- A. Iriondo, A. Mendiguren, M. B. Güemez, J. Requies and J. F. Cambra, Catal. Today, 2017, 279, 286–295 CrossRef CAS.
- J. J. Shi, Y. Y. Wang, X. N. Yu, W. C. Du and Z. Y. Hou, Fuel, 2016, 163, 74–79 CrossRef CAS.
- C. E. Scott, T. Romero, E. Lepore, M. Arruebarrena, P. Betancourt, C. Bolivar, M. J. Perezzurita, P. Marcano and J. Goldwasser, Appl. Catal., A, 1995, 125, 71–79 CrossRef CAS.
- C. M. Andrade, N. M. Lima, S. J. Melo and C. A. M. de Abreu, Fuel, 2015, 147, 125–132 CrossRef CAS.
- S. Nishimura, N. Ikeda and K. Ebitani, Catal. Today, 2014, 232, 89–98 CrossRef CAS.
- J. Z. Chen, F. Lu, J. J. Zhang, W. Q. Yu, F. Wang, J. Gao and J. Xu, Chemcatchem, 2013, 5, 2822–2826 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00605e |
| This journal is © The Royal Society of Chemistry 2017 |