DOI:
10.1039/C7RA00090A
(Paper)
RSC Adv., 2017,
7, 20919-20928
Construction of the oxaphenalene skeletons of mansonone F derivatives through C–H bond functionalization and their evaluation for anti-proliferative activities†
Received
3rd January 2017
, Accepted 3rd April 2017
First published on 12th April 2017
Abstract
Novel mansonone F derivatives were conveniently synthesized via a key step of Ru(II)-catalyzed C–H functionalization to rapidly construct oxaphenalene skeletons. This synthetic procedure is sufficiently robust and flexible to offer both the generation of diverse mansonone F analogs and the scale-up synthesis of selected compounds. The structural formulas of all products were confirmed and characterized using spectral data. Most of the derivatives exhibited significant cytotoxicity against four tested human tumor cell lines in vitro.
Introduction
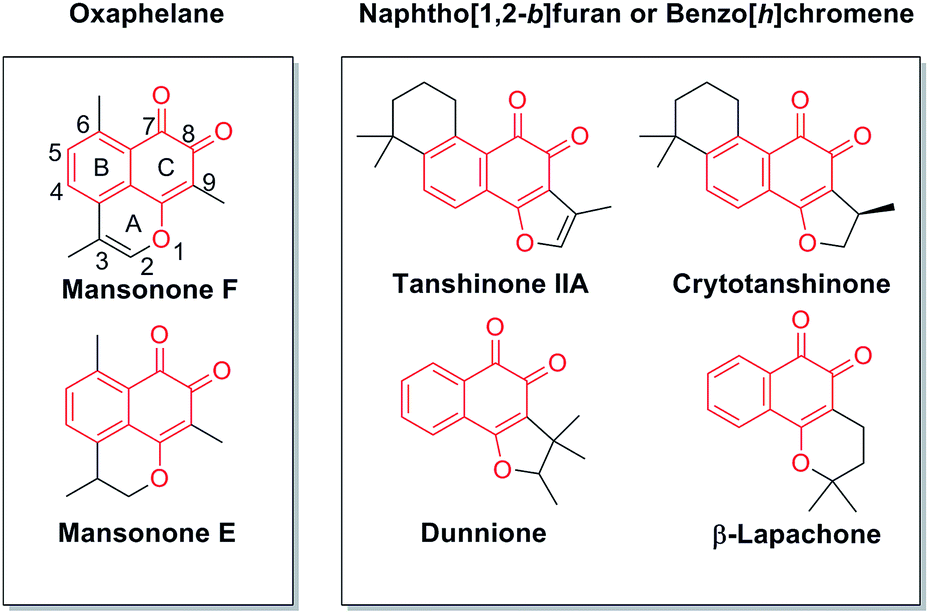
Natural products have long been an important source of drugs or drug leads. Natural products or their derivatives still comprise more than 50% of the drugs that are used for cancer chemotherapy.1 Many natural quinones and quinoid heterocycles have been reported to show antitumor effects. For example, tanshinone IIa,2,3 crytotanshinone, dunnione, β-lapachone,4,5 and mansonone E and F6,7 show great potential as antitumor agents (Fig. 1). Unlike the common o-quinone compounds with structural features of naphtho[1,2-b]furan or benzo[h]chromene, mansonone F (Fig. 1, MsF), a tricyclic sesquiterpenoid naturally occurring as ortho-naphthoquinone with an unusual oxaphelane skeleton, was first isolated from the heartwood of a west African tree Mansonia altissima A. Chev. (Sterculiaceae).8–10 MsF is a natural phytoalexin and MsF derivatives have attracted considerable attention because of their significant antimicrobial, antifungal, antioxidant, larvicidal and anticancer activities.11,12
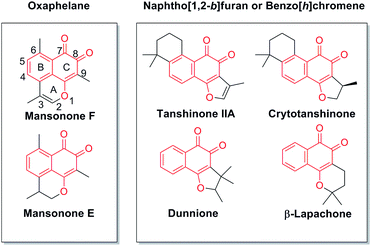 |
| | Fig. 1 The structures of natural o-quinone compounds. | |
To date, several approaches for synthesizing MsF and its derivatives have been reported. For instance, intramolecular Diels–Alder addition of benzynes to furans and intramolecular Friedel–Crafts acylation has been utilized for this purpose.13 Based on the regioselective hydrolysis of a hydroquinone diacetate and IBX (2-iodoxybenzoic acid) as oxidant for the MsF,14 the oxaphenalene skeleton of MsF has also been constructed by peri ring closure of naphthol ether.15,16 However, these multistep synthetic routes are generally time-consuming as they present inherent structural constraints that limit further optimization of their biological properties through structural modification. Furthermore, the existing routes are hampered by low efficiency in constructing the parent rings. Therefore, development of a new synthetic route for biologically active MsF derivatives is necessary. A novel synthetic route for modification via a key step of Ru(II)-catalyzed C–H functionalization has also been used to construct the oxaphenalene skeleton and shorten previously used synthetic routes.17,18 Oxidative C–H bond functionalization provides economical access to important bioactive heteroarenes and thus has been extensively used in medicinal chemistry.19
In this study, a series of highly active derivatives were successfully synthesized by establishing a novel and efficient synthetic route for MsF derivatives. The method is convenient and a one-step reaction is sufficient to construct the key tricyclic oxaphenalene intermediate using C(sp2)–H activation through Ru(II)-catalyzed cyclization. The synthesized compounds were then evaluated for their anti-proliferative activities with different cancer cell lines in vitro.
Results and discussion
Chemistry
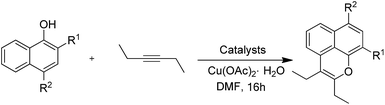
The starting material, 1-naphthol, was treated with hex-3-yne in anhydrous dimethylformamide (DMF) with [RuCl2(p-cymene)]2 as a catalyst and Cu(OAc)2·H2O to obtain the key tricyclic oxaphenalene intermediate (Table 1, entry 1). Furthermore, the reactivity of the o-substituted 1-naphthol was investigated. o-Chloro-1-naphthol has low yield of 30% when (Cp*RhCl2)2 was used as catalyst (Table 1, entry 9). We found that o-methyl, o-bromo or o-nitro naphthol could not form the oxaphenalene structure even under 150 °C and under the presence of RhCp*(CH3CN)(SbF6)2 (Table 1, entries 2–7 and 10–18). Ortho-substituents have a large steric hindrance effect on the cyclization reactivity. In addition, the reaction yield of 4-nitro-1-naphthol is higher than that of 1-naphthol (Table 1, entry 19). The reason is that the presence of electron-withdrawing groups leads to the stronger acidity of phenols and the easier dissociation of phenoxy anions. Thus, 4-nitro-1-naphthol was used as the starting substrate and [RuCl2(p-cymene)]2 as the catalyst to prepare the oxaphenalene intermediates. Introduction of substituent groups is the optimal strategy to synthesize Msn derivatives. The reactivity of different alkynes was also investigated in this cyclization reaction. The result showed that only alkyl or aromatic alkynes can participate in the reaction (Table 2, entries 1–15). Aromatic substituted alkynes illustrate an easier reaction with higher yield and faster speed than that of alkyl alkynes (Table 2, entries 4–12). Interestingly, the terminal and TMS-protected alkynes could not afford the expected product in this catalytic system (Table 2, entries 22 and 23).
Table 1 Scope of the oxidative cyclization reactions using Ru(II) as catalysts
|
| Entry |
R1 |
R2 |
T (°C) |
Catalysts |
Yield [%] |
| 1 |
H |
H |
100 |
[RuCl2(p-cymene)]2 |
55 |
| 2 |
CH3 |
H |
100 |
[RuCl2(p-cymene)]2 |
Trace |
| 3 |
CH3 |
H |
150 |
[RuCl2(p-cymene)]2 |
Trace |
| 4 |
CH3 |
H |
100 |
(Cp*RhCl2)2 |
Trace |
| 5 |
CH3 |
H |
150 |
(Cp*RhCl2)2 |
Trace |
| 6 |
CH3 |
H |
100 |
RhCp*(CH3CN)(SbF6)2 |
Trace |
| 7 |
CH3 |
H |
150 |
RhCp*(CH3CN)(SbF6)2 |
Trace |
| 8 |
Cl |
H |
100 |
[RuCl2(p-cymene)]2 |
Trace |
| 9 |
Cl |
H |
100 |
(Cp*RhCl2)2 |
30 |
| 10 |
Br |
H |
100 |
[RuCl2(p-cymene)]2 |
Trace |
| 11 |
Br |
H |
150 |
[RuCl2(p-cymene)]2 |
Trace |
| 12 |
Br |
H |
100 |
(Cp*RhCl2)2 |
Trace |
| 13 |
Br |
H |
150 |
(Cp*RhCl2)2 |
Trace |
| 14 |
Br |
H |
100 |
RhCp*(CH3CN)(SbF6)2 |
Trace |
| 15 |
Br |
H |
150 |
RhCp*(CH3CN)(SbF6)2 |
Trace |
| 16 |
NO2 |
H |
100 |
[RuCl2(p-cymene)]2 |
Trace |
| 17 |
NO2 |
H |
100 |
(Cp*RhCl2)2 |
Trace |
| 18 |
NO2 |
H |
100 |
RhCp*(CH3CN)(SbF6)2 |
Trace |
| 19 |
H |
NO2 |
100 |
[RuCl2(p-cymene)]2 |
75 |
Table 2 Reaction of 4-nitronaphthalene-1-ol with alkynes
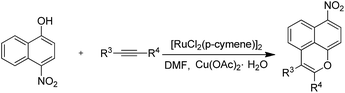
|
| Entry |
R3 |
R4 |
T (°C) |
Time (h) |
Yield (%) |
| 1 |
C2H5 |
C2H5 |
100 |
6 |
26 |
| 2 |
C2H5 |
C2H5 |
100 |
12 |
65 |
| 3 |
C2H5 |
C2H5 |
100 |
16 |
75 |
| 4 |
CH3 |
Ph |
60 |
3 |
35 |
| 5 |
CH3 |
Ph |
60 |
9 |
58 |
| 6 |
CH3 |
Ph |
100 |
3 |
76 |
| 7 |
CH3 |
Ph |
100 |
9 |
88 |
| 8 |
CH2CH3 |
Ph |
100 |
9 |
84 |
| 9 |
Ph |
Ph |
60 |
3 |
28 |
| 10 |
Ph |
Ph |
60 |
9 |
46 |
| 11 |
Ph |
Ph |
100 |
3 |
70 |
| 12 |
Ph |
Ph |
100 |
9 |
85 |
| 13 |
(CH2)2CH3 |
(CH2)2CH3 |
100 |
16 |
78 |
| 14 |
(CH2)3CH3 |
(CH2)3CH3 |
100 |
16 |
74 |
| 15 |
(CH2)4CH3 |
(CH2)4CH3 |
100 |
16 |
78 |
| 16 |
COOC2H5 |
COOC2H5 |
100 |
16 |
0 |
| 17 |
CN |
CN |
100 |
16 |
0 |
| 18 |
CH2OH |
CH2OH |
100 |
16 |
0 |
| 19 |
CH2Cl |
CH2Cl |
100 |
16 |
0 |
| 20 |
COOH |
COOH |
100 |
16 |
0 |
| 21 |
COOH |
Ph |
100 |
16 |
0 |
| 22 |
H |
Ph |
100 |
16 |
0 |
| 23 |
Si(CH3)3 |
Ph |
100 |
16 |
0 |
| 24 |
CH2Cl |
Ph |
100 |
16 |
0 |
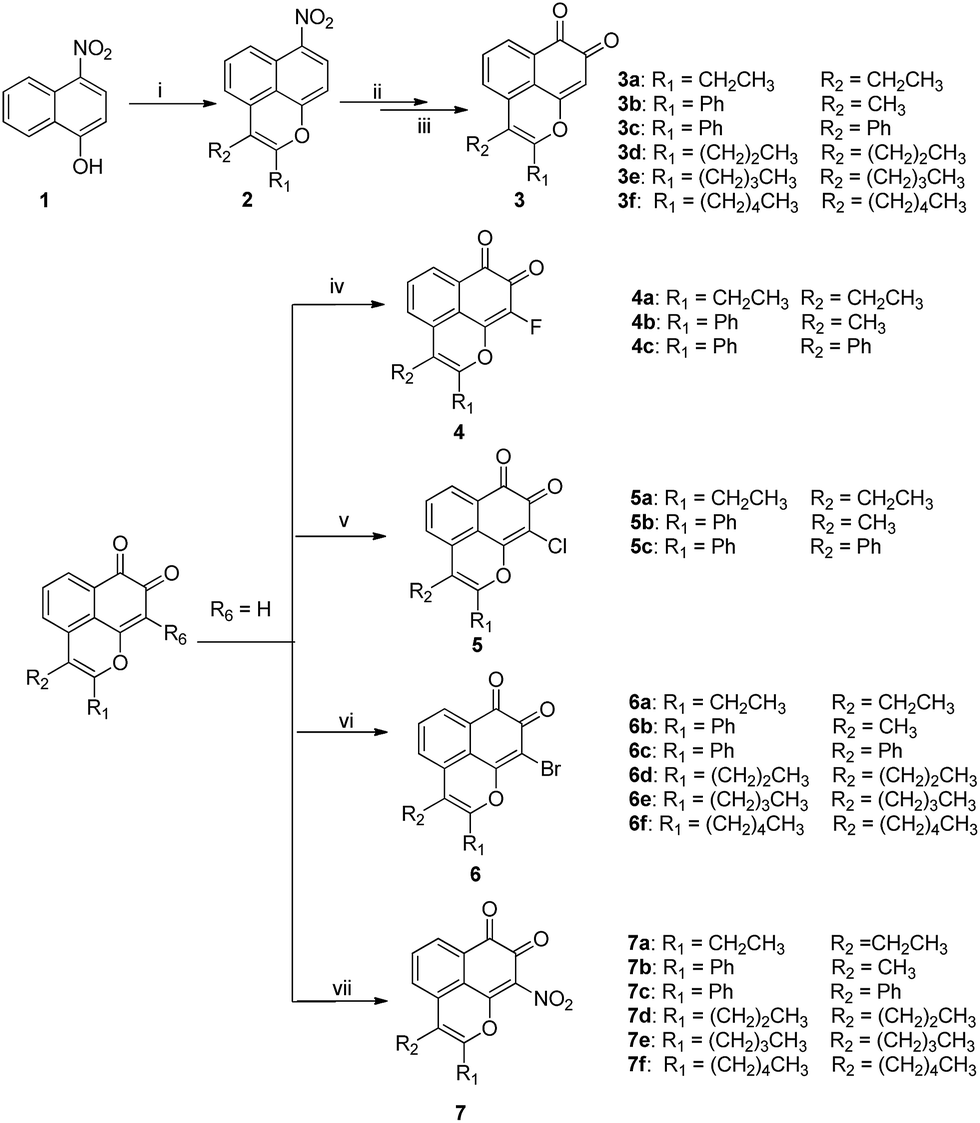
The proposed MsF derivatives were obtained as outlined in Scheme 1 and 2.20 The key tricyclic oxaphenalene intermediate 2 was constructed through a one-step Ru(II)-catalyzed cyclization reaction based on the above optimized conditions. The two subsequent steps were performed according to a previously described method with minor modification.7 The nitro group of intermediate 2 was reduced to amine by using sodium hydrosulfite as the reducing reagent. The amine was oxidized by Fremy's salt, thereby generating target compounds 3 (Scheme 1).
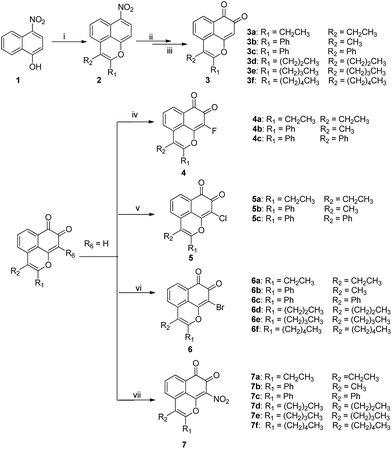 |
| | Scheme 1 Reagents and conditions: (i) alkyne, [RuCl2(p-cymene)]2, Cu(OAc)2·H2O, DMF, 80 °C, 14 h; (ii) Na2S2O4, THF/H2O, 50 °C,1 h; (iii) acetone, 0.06 M KH2PO4, Fremy's salt, 1 h; (iv) Selectfluor, MeCN, 70 °C, 4 h; (v) NCS, BPO, CCl4, 80 °C,4 h; (vi) NBS, BPO, CCl4, 80 °C, 4 h; (vii) Cu(NO3)2·3H2O, Ac2O, 20 min. | |
MsF derivatives substituted at the C-9 position were obtained from 3a to 3f via different methods. Fluoro-substituted MsF derivatives 4 were obtained by using Selectfluor®. Meanwhile, chloro- and bromo-substituted MsF derivatives 5 and 6 were obtained by using N-chlorosuccinimide and N-bromosuccinimide, respectively. Nitro-substituted MsF derivatives 7 were obtained by using cupric nitrate (Scheme 1).
Furthermore, 4,5-disubstituted MsF derivatives were synthesized from 6,7-difluoronaphthalen-1-ol 10. However, given that it is not commercially available, the starting 1-bromo-2,4,5-trifluorobenzene 8 was treated with Mg in anhydrous THF. Afterward, furan was added via Diels–Alder cyclization to obtain 6,7-difluoro-1,4-dihydro-1,4-epoxynaphthalene 9.21,22 The O-ring opening of 9 was created by adding BF3-etherate in dichloromethane obtaining 6,7-difluoronaphthalen-1-ol 10. For the compounds with 6,7-difluoro group, the yield in the C–H functional step was improved from 10% to 45% (Table 3) when m-xylene was used as solvent instead of DMF. The 5-amination products 15 were produced by treating 12 with various amine and K2CO3 in DMF. Based on the information of NOESY spectra of compound 17, both 6-H(δ = 7.79 ppm) and piperidyl α-CH2 (δ = 3.23 ppm) have a through-space correlation. Piperidyl should be considered only as a 5-position substituent group. This finding is possibly due to the C-5 position having lower steric hindrance and higher electron density than the C-4 position, which makes electrophilic substitution more likely at the C-5 position. As shown in Scheme 2, target compounds 13 and 16a–16d were obtained by using the strategy mentioned in Scheme 1, whereas 13 and 16a were brominated by using N-bromosuccinimide in carbon tetrachloride to obtain 14 and 17, respectively (Scheme 2). All structures of the novel intermediates and target compounds were confirmed by 1H NMR, 13C NMR and HRMS (ESI).
Table 3 Reaction of 6,7-difluoronaphthalen-1-ol with diethylacetylene
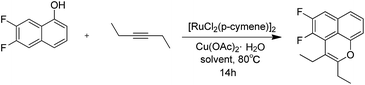
|
| Entry |
Solvent |
Yeild [%] |
| 1 |
DMF |
10 |
| 2 |
Xylene |
45 |
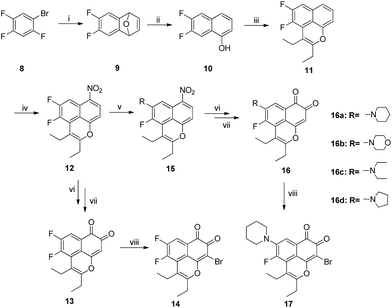 |
| | Scheme 2 Reagents and conditions: (i) furan, Mg, THF, 80 °C, 10 h; (ii) BF3·Et2O, CH2Cl2, 1 h; (iii) [RuCl2(p-cymene)]2, Cu(OAc)·H2O, 3-hexyne, m-xylene, 80 °C, 14 h; (iv) Cu(NO3)2·3H2O, Ac2O, 0.5 h; (v) Na2S2O4, THF/H2O, 1 h; (vi) acetone, Fremy's salt, KH2PO4(aq), 1 h; (vii) amine, K2CO3, DMF, 60 °C, 4–8 h; (viii) NBS, BPO, CCl4, 80 °C, 4 h. | |
Anti-proliferation activities and summary of SARs
Cytotoxicity of all the synthesized compounds was evaluated against four different human cancer cell lines. The inhibitory activities (IC50) of all of the synthesized compounds were determined (as shown in Table 4). In general, most of the compounds exhibited greater potency on suspension cell lines (HL-60, K562) than toward attached cell lines (HeLa, A549). For example, compound 6a showed excellent anti-proliferative activity with an IC50 value of 0.69 μM for the HL-60 cell line, which was equal to the IC50 value for the positive control VP-16 and far exceeded the lead compound MsF.
Table 4 IC50 values (μM) of the mansonone F derivatives against cancer cells
| Compd. |
IC50a (μM) |
| A549 |
HeLa |
K562 |
HL-60 |
| The IC50 represents compound concentration giving 50% survival of each cell line. |
| 3a |
12.74 |
3.04 |
0.85 |
0.62 |
| 3b |
30.71 |
25.22 |
15.12 |
5.82 |
| 3c |
>50 |
>50 |
42.32 |
27.26 |
| 3d |
9.55 |
8.33 |
6.36 |
0.84 |
| 3e |
4.24 |
22.14 |
7.33 |
0.44 |
| 3f |
6.72 |
17.83 |
8.27 |
0.95 |
| 4a |
33.13 |
9.95 |
4.56 |
1.75 |
| 4b |
37.34 |
30.92 |
5.21 |
1.37 |
| 4c |
32.33 |
18.32 |
10.06 |
2.48 |
| 5a |
15.52 |
4.10 |
2.65 |
0.74 |
| 5b |
37.52 |
32.24 |
21.14 |
2.84 |
| 5c |
28.74 |
28.52 |
43.14 |
3.32 |
| 6a |
2.65 |
1.47 |
0.97 |
0.69 |
| 6b |
34.23 |
28.76 |
8.74 |
11.22 |
| 6c |
27.31 |
13.98 |
6.94 |
2.28 |
| 6d |
5.63 |
7.61 |
7.18 |
0.93 |
| 6e |
4.94 |
6.23 |
2.26 |
0.60 |
| 6f |
5.53 |
4.61 |
1.53 |
0.58 |
| 7a |
>50 |
42.82 |
45.24 |
6.12 |
| 7b |
>50 |
>50 |
>50 |
31.13 |
| 7c |
>50 |
>50 |
>50 |
35.43 |
| 7d |
41.17 |
38.25 |
36.02 |
12.26 |
| 7e |
38.14 |
29.71 |
15.32 |
4.41 |
| 7f |
>50 |
>50 |
20.06 |
13.96 |
| 13 |
36.36 |
2.11 |
2.21 |
0.59 |
| 14 |
20.25 |
2.56 |
2.31 |
0.58 |
| 16a |
13.14 |
9.27 |
15.42 |
2.65 |
| 16b |
14.15 |
9.67 |
11.17 |
1.87 |
| 16c |
6.07 |
3.51 |
5.21 |
1.92 |
| 16d |
5.56 |
2.26 |
3.71 |
1.14 |
| 17 |
5.06 |
2.91 |
2.91 |
0.64 |
| MsF |
9.77 |
38.88 |
10.93 |
18.44 |
| VP-16 |
26.43 |
11.32 |
1.64 |
0.49 |
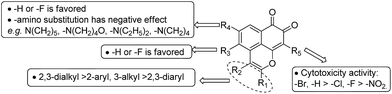
The structure-activity relationships (SARs) showed that the type and position of the substitutions play an important role in determining the anti-proliferative potency (as shown in Fig. 2). First, substitution at the C-9 position was crucial for the cytotoxicity of MsF derivatives. In general, cytotoxicity of the 9-substituted derivatives followed the order –Br ≈ –H > –Cl ≈ –F > –NO2. The 9-Br-substituted derivatives (e.g. 6a, 6c, 6d 6e and 6f) showed remarkable growth inhibitory activity against most of the tested cell lines. Second, substitution at the C-2 and C-3 positions with flexible alkyl chain (e.g., series a with 2,3-diEt, series d with 2,3-diPr, series e with 2,3-diBu, and series f with 2,3-diPent) yielded better cytotoxic activity than substitution with a rigid aromatic ring (e.g., series b with 2-Ph, 3-Me and series c with 2,3-diPh). Alkyl chain length had no influence on cytotoxicity. Finally, compared with the a series without 4,5-position substitutions (e.g., 3a–7a), 4,5-diF substituted derivatives (e.g., 13 and 14) could maintain cytotoxic activity. However, reduced cytotoxic activity was observed when the 5-fluorine atom of compound 13 was replaced by other amino groups (e.g., series 16).
 |
| | Fig. 2 Summary of SARs of MsF derivatives. | |
Conclusions
In summary, a novel and convenient synthetic route for MsF derivatives was established. The key step for synthesizing MsF derivatives involved the rapid construction of oxaphenalene structure by a one-step reaction of Ru(II)-catalyzed C–H functionalization.
This concise and practical synthetic procedure allows easy induction of a variety of substituents at the C-2, C-3, C-4, C-5 and C-9 positions, and scaled-up production of promising candidate compounds. Furthermore, the cytotoxic activity of synthesized compounds was evaluated in four human cancer cell lines. The SARs of MsF derivatives were discussed in this study and we found that 6a has excellent anti-proliferative activities which needs to be further studied.
Experimental
General materials and methods
All reactions were monitored by using TLC purchased from Qingdao Haiyang Chemical Co. Ltd. High resolution mass spectra (HRMS) were recorded on Shimadzu LCMS-IT-TOF in electrospray ionization (ESI) positive or negative modes. Melting points (Mp) were determined using a SRS-OptiMelt automated melting point instrument without correction. 1H (400 MHz), 13C NMR (100 MHz), HMBC, HMQC and NOESY were recorded on a Bruker BioSpin GmbH spectrometer with tetramethylsilane (TMS) as an internal standard. IR spectra were recorded on Bruker Equinox 55 Fourier transform spectrometer. The purification of synthesized compounds were carried out by using flash column chromatography with silica gel (200–300 mesh) purchased from Qingdao Haiyang Chemical Co. Ltd. Their purities were proved to be higher than 95% by using analytical HPLC equipped with Shimadzu LC-20AB system and an Ultimate XB-C18 column (4.6 × 250 mm, 5 μm) at a flow rate of 0.25 mL min−1. The detection wavelength was 254 nm, and the mobile phase was methanol–water (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20–60:40) containing 0.1% TFA. The main text of the article should appear here with headings as appropriate.
:20–60:40) containing 0.1% TFA. The main text of the article should appear here with headings as appropriate.
Cell culture and cell lines. The human acute promyelocytic leukemia cell HL-60, human chronic myelogenous leukemia cell K-562, human cervical cancer cell HeLa, human lung cancer cell A549 were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and preserved at our lab. The HL-60 and K-562 cells were cultured in a RPMI-1640 medium (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA), and other cell lines were grown in Dulbecco's Modified Eagle's Medium (D-MEM, Gibco Carlsbad, CA) supplemented with 10% fetal bovine serum. All cells were cultured with 5% CO2 at 37 °C.
Synthetic procedures
General procedure for synthesis of 2a–2f. A suspension of 4-nitronaphthalen-1-ol (945.0 mg, 5.00 mmol), alkyne (5 mmol), [RuCl2(p-cymene)]2 (48 mg, 1.5% mmol) and Cu(OAc)2·H2O (955 mg, 5 mmol), and dimethylformamide (8.0 mL) in ChemGlass pressure vessel was stirred at 100 °C for 9–16 h. After the starting material was consumed up, the reaction mixture was cooled to room temperature, diluted with dichloromethane, and filtered with a short column chromatography on silica gel. The organic layers were washed with water (3 × 30 mL) and dried over Na2SO4. After filtration, the filtrate was collected and the solvent was removed under reduced pressure, the crude product was purified by using column chromatography on silica gel (petroleum ether) to give the target compounds.
2,3-Diethyl-7-nitrobenzo[de]chromene (2a). Reddish yellow solid, yield 75%. 1H NMR (400 MHz, CDCl3) δ 8.59 (d, J = 8.9 Hz, 1H), 8.43 (d, J = 8.9 Hz, 1H), 7.66 (dd, J = 8.8, 7.5 Hz, 1H), 7.11 (d, J = 7.4 Hz, 1H), 6.80 (d, J = 8.9 Hz, 1H), 2.57 (m, 4H), 1.28 (t, J = 7.5 Hz, 3H), 1.20 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 158.2, 153.2, 136.9, 132.0, 130.9, 128.9, 128.5, 122.8, 119.3, 114.4, 114.2, 106.0, 23.7, 19.5, 12.7, 12.3. HRMS (ESI): calcd for C16H15NO3 [M + H]+: 270.1125; found: 270.1117.
3-Methyl-7-nitro-2-phenylbenzo[de]chromene (2b). Orange-red solid, yield 88%. 1H NMR (400 MHz, CDCl3) δ 8.68 (d, J = 8.9 Hz, 1H), 8.47 (d, J = 8.9 Hz, 1H), 7.74 (dd, J = 8.9, 7.4 Hz, 1H), 7.61–7.58 (m, 2H), 7.56–7.42 (m, 3H), 7.19 (d, J = 7.9 Hz, 1H), 6.89 (d, J = 8.9 Hz, 1H), 2.17 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 157.9, 149.7, 137.2, 133.3, 132.0, 132.0, 129.5, 129.3, 128.9, 128.4, 128.2, 122.4, 120.3, 115.6, 110.7, 106.9, 13.7. HRMS (ESI): calcd for C19H13NO3 [M + H]+: 304.0968; found: 304.0958.
7-Nitro-2,3-diphenylbenzo[de]chromene (2c). Orange-red solid, yield 85%. 1H NMR (400 MHz, CDCl3) δ 8.62 (d, J = 8.9 Hz, 1H), 8.49 (d, J = 8.8 Hz, 1H), 7.55 (dd, J = 8.8, 7.6 Hz, 1H), 7.46–7.37 (m, 3H), 7.32–7.29 (m, 2H), 7.27–7.19 (m, 5H), 6.95 (d, J = 8.8 Hz, 1H), 6.78 (d, J = 7.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 158.0, 149.3, 137.5, 134.2, 132.8, 132.3, 131.9, 130.7, 129.3, 129.1, 129.0, 128.9, 128.1, 127.9, 122.7, 120.4, 118.3, 117.9, 106.4. HRMS (ESI): calcd for C24H15NO3 [M + H]+: 366.0947; found: 366.0939.
7-Nitro-2,3-dipropylbenzo[de]chromene (2d). Deep-red solid, yield 78%. 1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 8.8 Hz, 1H), 8.33 (d, J = 8.9 Hz, 1H), 7.56 (dd, J = 8.8, 7.6 Hz, 1H), 6.99 (d, J = 7.4 Hz, 1H), 6.68 (d, J = 8.8 Hz, 1H), 2.48–2.37 (m, 4H), 1.71–1.60 (m, 2H), 1.58–1.46 (m, 2H), 0.97 (td, J = 7.4, 5.4 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 158.1, 152.4, 136.8, 132.0, 131.1, 128.8, 128.4, 122.7, 119.3, 114.6, 113.4, 105.9, 32.3, 28.4, 21.2, 21.0, 14.2, 13.8. HRMS (ESI): calcd for C18H19NO3 [M + H]+: 298.1438; found: 298.1418.
2,3-Dibutyl-7-nitrobenzo[de]chromene (2e). Deep-red oli, yield 74%. 1H NMR (400 MHz, CDCl3) δ 8.58 (d, J = 8.8 Hz, 1H), 8.41 (d, J = 8.9 Hz, 1H), 7.65 (dd, J = 8.8, 7.5 Hz, 1H), 7.07 (d, J = 7.4 Hz, 1H), 6.78 (d, J = 8.9 Hz, 1H), 2.56–2.47 (m, 4H), 1.68 (m, 2H), 1.54–1.42 (m, 6H), 0.99 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 158.1, 152.5, 136.7, 132.0, 131.1, 128.8, 128.4, 122.6, 119.2, 114.5, 113.4, 105.9, 30.2, 30.1, 29.8, 26.1, 22.9, 22.5, 13.9, 13.8. HRMS (ESI): calcd for C20H23NO3 [M + H]+: 326.1751; found: 326.1728.
7-Nitro-2,3-dipentylbenzo[de]chromene (2f). Deep-red oil, yield 78%. 1H NMR (400 MHz, CDCl3) δ 8.60 (d, J = 8.8 Hz, 1H), 8.44 (d, J = 8.9 Hz, 1H), 7.67 (dd, J = 8.8, 7.5 Hz, 1H), 7.09 (d, J = 7.4 Hz, 1H), 6.80 (d, J = 8.9 Hz, 1H), 2.58–2.48 (m, 4H), 1.77–1.67 (m, 2H), 1.60–1.57 (m, 2H), 1.47–1.36 (m, 8H), 0.99–0.92 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 158.1, 152.5, 136.7, 132.0, 131.1, 128.8, 128.4, 122.6, 119.2, 114.5, 113.4, 105.9, 32.0, 31.5, 30.4, 27.6, 27.4, 26.4, 22.5, 22.4, 14.0, 13.9. HRMS (ESI): calcd for C22H27NO3 [M + H]+: 354.2064; found: 354.2056.
General procedure for synthesis of 3a–3f. Sodium hyposulfite (1.74 g, 10 mmol, 5 equiv.) was added to a solution of nitro-substituted products (2a–2f, 2 mmol) in THF/H2O (120 mL, 3:1). The reaction mixture was heated to 50 °C and further stirred for 2 h at this temperature. After the starting material was consumed up, the reaction mixture was concentrated to remove the THF under reduced pressure and then the residue was extracted with dichloromethane (30 mL × 3). The combined extracts were evaporated to remove the solvent. The solid residue was dissolved in acetone (60 mL) and a solution of Fremy's salt (1.34 g, 5 mmol, 2.5 equiv.) in 0.06 M KH2PO4 solution (68 mL) was added with vigorous stirring. After stirring for 1 h, the mixture was extracted with dichloromethane (30 mL × 3). The combined extracts were washed with brine (30 mL), dried over Na2SO4, filtered and evaporated to dryness. The solid residue was purified by using flash column chromatography (silica gel), eluting with 5% ethyl acetate in dichloromethane to give the target compounds.
2,3-Diethylbenzo[de]chromene-7,8-dione (3a). Reddish brown solid, yield 65%. Mp: 200.2–203.5 °C; IR νmax (KBr) cm−1: 1639, 1695. 1H NMR (400 MHz, CDCl3) δ 8.11 (dd, J = 5.8, 2.8 Hz, 1H), 7.86–7.50 (m, 2H), 6.07 (s, 1H), 2.64 (qd, J = 7.5, 2.4 Hz, 4H), 1.30 (t, J = 7.5 Hz, 3H), 1.22 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 180.3, 178.1, 166.3, 155.1, 133.2, 131.7, 130.0, 129.9, 129.0, 121.5, 113.1, 104.1, 23.9, 19.3, 14.4, 12.4. HRMS (ESI): calcd for C16H14O3 [M + Na]+: 277.0835; found: 277.0829. HPLC purity: 99.9%.
3-Methyl-2-phenylbenzo[de]chromene-7,8-dione (3b). Reddish brown solid, yield 71%. Mp: 215.6–218.3 °C; IR νmax (KBr) cm−1: 1633, 1696. 1H NMR (400 MHz, CDCl3) δ 8.17 (dd, J = 7.3, 1.3 Hz, 1H), 7.80 (dd, J = 7.3, 1.2 Hz, 1H), 7.75 (t, J = 7.2 Hz, 1H), 7.56–7.50 (m, 5H), 6.11 (s, 1H), 2.30 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 180.1, 178.2, 166.0, 151.1, 133.3, 132.8, 132.3, 130.6, 130.0, 129.8, 129.6, 129.3, 128.5, 121.7, 109.1, 104.5, 13.5. HRMS (ESI): calcd for C19H12O3 [M + H]+: 289.0859; found: 289.0864. HPLC purity: 99.3%.
2,3-Diphenylbenzo[de]chromene-7,8-dione (3c). Reddish brown solid, yield 55%. Mp: 294.1–296.3 °C; IR νmax (KBr) cm−1: 1627, 1689. 1H NMR (400 MHz, CDCl3) δ 8.15 (dd, J = 7.5, 1.0 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.45–7.40 (m, 3H), 7.36 (dd, J = 8.1, 1.0 Hz, 1H), 7.32–7.21 (m, 7H), 6.24 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 180.0, 178.3, 165.8, 150.9, 133.1, 132.7, 132.0, 131.7, 131.0, 130.9, 129.6, 129.5, 129.2, 129.0, 128.5, 128.1, 121.3, 116.5, 104.7. HRMS (ESI): calcd for C24H14O3 [M + H]+: 351.1016; found: 351.1016. HPLC purity: 99.7%.
2,3-Dipropylbenzo[de]chromene-7,8-dione (3d). Reddish brown solid, yield 65%. Mp: 165.6–168.4 °C; IR νmax (KBr) cm−1: 1631, 1696. 1H NMR (400 MHz, CDCl3) δ 8.11 (dd, J = 4.6, 4.0 Hz, 1H), 7.70 (m, 2H), 6.06 (s, 1H), 2.64–2.54 (m, 4H), 1.75 (m, 2H), 1.61 (m, 2H), 1.07 (m, 6H). 13C NMR (400 MHz, CDCl3) δ 180.3, 178.1, 166.3, 154.3, 133.1, 132.0, 130.0, 129.94, 129.2, 121.5, 112.3, 104.1, 32.4, 28.1, 23.1, 21.1, 14.1, 13.8. HRMS (ESI): calcd for C18H18O3 [M + H]+: 283.1329; found: 283.1325. HPLC purity: 97.9%.
2,3-Dibutylbenzo[de]chromene-7,8-dione (3e). Reddish brown solid, yield 60%. Mp: 160.7–162.6 °C; IR νmax (KBr) cm−1: 1629, 1699. 1H NMR (400 MHz, CDCl3) δ 8.10 (dd, J = 5.2, 3.4 Hz, 1H), 7.77–7.46 (m, 2H), 6.05 (s, 1H), 2.67–2.35 (m, 4H), 1.80–1.59 (m, 2H), 1.59–1.27 (m, 6H), 0.99 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 180.4, 178.1, 166.3, 154.4, 133.1, 132.0, 130.0, 129.9, 129.1, 121.5, 112.2, 104.1, 32.0, 30.3, 29.9, 25.8, 22.7, 22.4, 13.8, 13.8. HRMS (ESI): calcd for C20H22O3 [M + H]+: 311.1642; found: 311.1637. HPLC purity: 95.3%.
2,3-Dipentylbenzo[de]chromene-7,8-dione (3f). Reddish brown solid, yield 66%. Mp: 153.6–155.1 °C; IR νmax (KBr) cm−1: 1627, 1699.1H NMR (400 MHz, CDCl3) δ 8.07 (dd, J = 5.3, 3.3 Hz, 1H), 7.73–7.52 (m, 2H), 6.03 (s, 1H), 2.61–2.43 (m, 4H), 1.73–1.65 (m, 2H), 1.61–1.48 (m, 2H), 1.47–1.32 (m, 8H), 0.95–0.92 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 180.3, 178.1, 166.3, 154.4, 133.1, 132.0, 130.0, 129.9, 129.2, 121.4, 112.3, 104.1, 31.8, 31.4, 30.5, 29.6, 27.5, 26.1, 22.4, 22.3, 13.9, 13.9. HRMS (ESI): calcd for C22H26O3 [M + H]+: 339.1955; found: 339.1954. HPLC purity: 97.1%.
General procedure for synthesis of 4a–4c. Selectfluor (1 mmol, 2 equiv.) was added to a solution of o-quinone products (3a–3c, 0.5 mmol) dissolved in anhydrous acetonitrile. The reaction mixture was heated at 80 °C with stirring for 4 h. After the starting material was consumed up, the reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The solid residue was purified by using flash column chromatography (silica gel), eluting with dichloromethane (DCM) to give the target compounds.
2,3-Diethyl-9-fluorobenzo[de]chromene-7,8-dione (4a). Reddish brown solid, yield 19%. Mp: 212.4–213.8 °C; IR νmax (KBr) cm−1: 1634, 1695. 1H NMR (400 MHz, CDCl3) δ 8.06 (dd, J = 7.2, 1.3 Hz, 1H), 7.70–7.59 (m, 2H), 2.68 (q, J = 7.6, 2H), 2.64 (q, J = 7.6, 2H), 1.32 (t, J = 7.5 Hz, 3H), 1.22 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 179.1 (d, J = 6.5 Hz), 169.4 (d, J = 9.0 Hz), 154.9, 151.8 (d, J = 8.4 Hz), 142.1 (d, J = 253.4 Hz), 132.8, 131.6 (d, J = 6.7 Hz), 130.7, 129.7, 127.8, 119.3, 113.6, 23.9, 19.3, 14.4, 12.2. HRMS (ESI): calcd for C16H13O3F [M + Na]+: 295.0741; found: 295.0743. HPLC purity: 95.4%.
9-Fluoro-3-methyl-2-phenylbenzo[de]chromene-7,8-dione (4b). Reddish brown solid, yield 16%. Mp: 216.7–219.3 °C; IR νmax (KBr) cm−1: 1632, 1699. 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 7.4 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.68 (t, J = 7.8 Hz, 1H), 7.63–7.56 (m, 2H), 7.54–7.47 (m, 3H), 2.31 (s, 3H). 13C NMR (101 MHz, DMSO) δ 178.1 (d, J = 6.8 Hz), 169.5 (d, J = 9.4 Hz), 150.2 (d, J = 8.2 Hz), 149.2, 140.0 (d, J = 249.4 Hz), 133.0, 131.8, 131.6 (d, J = 6.4 Hz), 130.4, 130.2, 130.0, 129.2, 128.6, 127.7, 118.9, 109.6, 13.2. HRMS (ESI): calcd for C19H11O3F [M + H]+: 307.0765; found: 307.0763. HPLC purity: 96.2%.
9-Fluoro-2,3-diphenylbenzo[de]chromene-7,8-dione (4c). Reddish brown solid, yield 20%. Mp: 242.3–245.7 °C; IR νmax (KBr) cm−1: 1630, 1708. 1H NMR (400 MHz, CDCl3) δ 8.12 (dd, J = 7.5, 1.1 Hz, 1H), 7.54 (t, J = 7.8 Hz, 1H), 7.46 (dd, J = 6.5, 3.9 Hz, 3H), 7.38–7.30 (m, 6H), 7.26 (d, J = 2.0 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 178.9 (d, J = 6.3 Hz), 170.0 (d, J = 9.7 Hz), 151.2 (d, J = 8.3 Hz), 150.4, 140.8 (d, J = 255.6 Hz), 132.8, 132.6, 132.3, 131.5, 131.0, 129.8, 129.4 (d, J = 7.9 Hz), 129.3, 129.0, 128.7, 128.3, 128.2, 127.4, 119.1, 116.7. HRMS (ESI): calcd for C24H13O3F [M + H]+: 369.0921; found: 369.0929. HPLC purity: 96.0%.
General procedure for synthesis of 5a–5c. N-Chlorosuccinimide (0.6 mmol, 1.2 equiv.) and benzoyl peroxide (12 mg) were added to a solution of o-quinone products (3a–3c, 0.5 mmol) in carbon tetrachloride. The reaction mixture was heated at 75 °C with stirring. After the starting material was consumed up, the reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The solid residue was purified by using flash column chromatography (silica gel), eluting with dichloromethane to give the target compounds.
9-Chloro-2,3-diethylbenzo[de]chromene-7,8-dione (5a). Reddish brown solid, yield 55%. Mp: 209.3–211.4 °C; IR νmax (KBr) cm−1: 1634, 1695. 1H NMR (400 MHz, CDCl3) δ 8.12 (dd, J = 7.1, 1.3 Hz, 1H), 7.82–7.64 (m, 2H), 2.74 (q, J = 7.5 Hz, 2H), 2.67 (q, J = 7.5 Hz, 2H), 1.36 (t, J = 7.5 Hz, 3H), 1.22 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 178.3, 172.4, 160.8, 155.4, 133.2, 131.5, 130.8, 130.7, 129.5, 128.6, 121.0, 113.9, 23.9, 19.3, 14.3, 11.9. HRMS (ESI): calcd for C16H13O3Cl [M + H]+: 289.0626; found: 289.0627. HPLC purity: 98.8%.
9-Chloro-3-methyl-2-phenylbenzo[de]chromene-7,8-dione (5b). Reddish brown solid, yield 47%. Mp: 223.4–225.6 °C; IR νmax (KBr) cm−1: 1638, 1693. 1H NMR (400 MHz, CDCl3) δ 8.18 (dd, J = 7.4, 1.1 Hz, 1H), 7.84 (dd, J = 8.1, 1.1 Hz, 1H), 7.75 (t, J = 7.6 Hz, 1H), 7.68–7.64 (m, 2H), 7.56–7.51 (m, 3H), 2.38 (s, 3H). 13C NMR (101 MHz, Pyr) δ 178.2, 172.9, 160.1, 150.5, 133.1, 132.5, 132.4, 130.3, 130.1, 129.5, 129.2, 128.7, 121.3, 110.0, 65.5, 13.1. HRMS (ESI): calcd for C19H11O3Cl [M + H]+: 323.0469; found: 323.0461. HPLC purity: 98.9%.
9-Chloro-2,3-diphenylbenzo[de]chromene-7,8-dione (5c). Reddish brown solid, yield 52%. Mp: 243.6–245.1 °C; IR νmax (KBr) cm−1: 1638, 1692. 1H NMR (400 MHz, CDCl3) δ 8.17 (dd, J = 7.4, 0.8 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.50–7.46 (m, 3H), 7.42–7.36 (m, 3H), 7.32 (d, J = 7.2 Hz, 1H), 7.28–7.24 (m, 4H), 7.24 (d, J = 1.6 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 178.2, 172.7, 160.4, 150.6, 133.1, 132.8, 132.7, 132.3, 131.5, 131.4, 130.9, 129.9, 129.4, 128.9, 128.8, 128.2, 120.7, 117.0. HRMS (ESI): calcd for C24H13O3Cl [M + H]+: 385.0626; found: 385.0637. HPLC purity: 99.7%.
General procedure for the preparation of 6a–6f. N-Bromosuccinimide (0.6 mmol, 1.2 equiv.) and benzoyl peroxide (12 mg) were added to a solution of o-quinone products (3a–3f, 0.5 mmol) dissolved in carbon tetrachloride. The reaction mixture was heated at 75 °C with stirring. After the starting material was consumed up, the reaction mixture was filtered and concentrated under reduced pressure. The solid residue was purified by using flash column chromatography (silica gel), eluting with dichloromethane to give the target compounds.
9-Bromo-2,3-diethylbenzo[de]chromene-7,8-dione (6a). Reddish brown solid, yield 48%. Mp: 242.8–244.7 °C; IR νmax (KBr) cm−1: 1632, 1695. 1H NMR (400 MHz, CDCl3) δ 8.13 (dd, J = 6.5, 2.0 Hz, 1H), 7.78–7.62 (m, 2H), 2.74 (q, J = 7.5 Hz, 2H), 2.67 (q, J = 7.6 Hz, 2H), 1.37 (t, J = 7.5 Hz, 3H), 1.22 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 177.9, 172.6, 162.3, 155.5, 133.4, 131.5, 130.7, 129.6, 128.6, 121.2, 114.1, 99.6, 23.9, 19.3, 14.3, 11.9. HRMS (ESI): calcd for C16H13O3Br [M + H]+: 333.0121; found: 333.0124. HPLC purity: 99.2%.
9-Bromo-3-methyl-2-phenylbenzo[de]chromene-7,8-dione (6b). Reddish brown solid, yield 50%. Mp: 245.4–247.3 °C; IR νmax (KBr) cm−1: 1633, 1690. 1H NMR (400 MHz, CDCl3) δ 8.19 (dd, J = 7.4, 1.1 Hz, 1H), 7.84 (dd, J = 8.1, 1.1 Hz, 1H), 7.77 (t, J = 7.6 Hz, 1H), 7.71–7.66 (m, 2H), 7.57–7.51 (m, 3H), 2.39 (s, 3H). 13C NMR (101 MHz, Pyr) δ 177.6, 173.2, 161.6, 150.3, 133.2, 132.4, 130.2, 130.1, 129.5, 129.2, 128.7, 123.7, 122.7, 121.3, 110.1, 100.0, 13.2. HRMS (ESI): calcd for C19H11O3Br [M + H]+: 366.9964; found: 366.9961. HPLC purity: 98.0%.
9-Bromo-2,3-diphenylbenzo[de]chromene-7,8-dione (6c). Reddish brown solid, yield 45%. Mp: 271.4–273.3 °C; IR νmax (KBr) cm−1: 1641, 1699. 1H NMR (400 MHz, CDCl3) δ 8.17 (dd, J = 7.5, 1.1 Hz, 1H), 7.60 (m, J = 7.6 Hz, 1H), 7.48 (m, 3H), 7.44–7.40 (m, 2H), 7.38 (dd, J = 8.2, 1.1 Hz, 1H), 7.33–7.27 (m, 5H). 13C NMR (101 MHz, CDCl3) δ 178.2, 177.7, 172.9, 162.0, 150.8, 133.2, 132.8, 132.82, 132.3, 131.5, 130.9, 129.9, 129.5, 129.0, 128.9, 128.2, 121.0, 117.1, 100.1, 100.0. HRMS (ESI): calcd for C24H13O3Br [M + H]+: 429.0121; found: 429.0120. HPLC purity: 96.8%.
9-Bromo-2,3-dipropylbenzo[de]chromene-7,8-dione (6d). Reddish brown solid, yield 46%. Mp: 256.4–257.7 °C; IR νmax (KBr) cm−1: 1633, 1693. 1H NMR (400 MHz, CDCl3) δ 8.11 (dd, J = 5.3, 3.2 Hz, 1H), 7.78–7.65 (m, 2H), 2.69 (t, J = 7.2 Hz, 2H), 2.61 (t, J = 7.2 Hz, 2H), 1.93–1.75 (m, 2H), 1.60 (m, 2H), 1.07 (t, J = 7.4 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 177.9, 172.6, 162.3, 154.8, 133.3, 131.8, 130.7, 129.7, 128.7, 121.2, 113.2, 99.6, 32.4, 28.0, 23.0, 20.7, 14.0, 13.8. HRMS (ESI): calcd for C18H17O3Br [M + H]+: 361.0434; found: 361.0432. HPLC purity: 99.2%.
9-Bromo-2,3-dibutylbenzo[de]chromene-7,8-dione (6e). Reddish brown solid, yield 80%. Mp: 274.3–275.7 °C; IR νmax (KBr) cm−1: 1635, 1696. 1H NMR (400 MHz, CDCl3) δ 8.04 (t, J = 4.8, 1H), 7.69–7.56 (m, 2H), 2.63 (t, J = 7.2, 2H), 2.55 (t, J = 7.2, 2H), 1.74–1.66 (m, 2H), 1.55–1.31 (m, 6H), 0.94–0.90 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 177.9, 172.6, 162.3, 154.9, 133.3, 131.8, 130.7, 129.8, 128.6, 121.0, 113.3, 99.5, 31.9, 30.2, 29.4, 25.8, 22.7, 22.4, 13.8, 13.8. HRMS (ESI): calcd for C20H21O3Br [M + H]+: 389.0747; found: 389.0738. HPLC purity: 98.8%.
9-Bromo-2,3-dibutylbenzo[de]chromene-7,8-dione (6f). Reddish brown solid, yield 52%. Mp: 273.4–275.7 °C; IR νmax (KBr) cm−1: 1632, 1689. 1H NMR (400 MHz, CDCl3) δ 8.05 (t, J = 4.3 Hz, 1H), 7.64 (d, J = 4.5 Hz, 2H), 2.63 (t, J = 7.2 Hz, 2H), 2.54 (t, J = 7.2 Hz, 2H), 1.78–1.68 (m, 2H), 1.50–1.48 (m, 4H), 1.36–1.28 (m, 6H), 0.88–0.84 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 178.0, 172.7, 162.3, 154.9, 133.3, 131.8, 130.7, 129.7, 128.7, 121.2, 113.3, 99.6, 31.8, 31.3, 30.4, 29.6, 26.9, 26.1, 22.4, 22.3, 13.9, 13.8. HRMS (ESI): calcd for C22H25O3Br [M + H]+: 417.1060; found: 417.1065. HPLC purity: 96.9%.
General procedure for synthesis of 7a–7f. O-Quinone product (3a–3f, 0.5 mmol) was dissolved in acetic anhydride (25 mL) and cooled to 0 °C in an ice bath. Cupric nitrate (0.6 mmol, 1.2 equiv.) was added under stirring. The reaction mixture was warmed up to room temperature, and stirring was continued until the reaction was completed. The mixture was slowly poured into chopped ice, and the resulting mixture was extracted with DCM. The combined organic layer was washed with water (3 times) and concentrated to give a crude product under reduced pressure. The crude product was purified by using column chromatography (silica gel), eluting with dichloromethane to give the target compound.
2,3-Diethyl-9-nitrobenzo[de]chromene-7,8-dione (7a). Deep brown solid, yield 65%. Mp: 198.4–200.1 °C; IR νmax (KBr) cm−1: 1643, 1698. 1H NMR (400 MHz, DMSO) δ 8.20 (d, J = 7.7 Hz, 2H), 8.09 (t, J = 7.2 Hz, 1H), 2.89–2.80 (m, 4H), 1.28 (t, J = 7.5 Hz, 3H), 1.22 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 176.7, 167.9, 158.9, 155.1, 135.6, 132.7, 130.2, 130.0, 129.5, 129.4, 116.8, 116.0, 23.0, 18.5, 14.3, 11.9. HRMS (ESI): calcd for C16H13NO5 [M + H]+: 300.0866; found: 300.0872. HPLC purity: 99.4%.
3-Methyl-9-nitro-2-phenylbenzo[de]chromene-7,8-dione (7b). Deep brown solid, yield 64%. Mp: 275.4–276.3 °C; IR νmax (KBr) cm−1: 1658, 1702. 1H NMR (400 MHz, DMSO) δ 8.23 (d, J = 8.0 Hz, 1H), 8.22 (d, J = 7.6 Hz, 1H), 8.10 (t, J = 7.8 Hz, 1H), 7.69 (dd, J = 7.8, 1.8 Hz, 2H), 7.63–7.59 (m, 3H), 2.43 (s, 3H). 13C NMR (101 MHz, DMSO) δ 177.5, 168.6, 159.3, 150.8, 136.1, 134.5, 131.7, 131.2, 131.1, 130.8, 130.3, 129.8, 129.6, 129.2, 117.7, 112.8, 13.7. HRMS (ESI): calcd for C19H11NO5 [M + H]+: 334.0710; found: 334.0736. HPLC purity: 95.2%.
9-Nitro-2,3-diphenylbenzo[de]chromene-7,8-dione (7c). Deep brown solid, yield 71%. Mp: 284.5–286.7 °C; IR νmax (KBr) cm−1: 1657, 1696. 1H NMR (400 MHz, CDCl3) δ 8.33 (dd, J = 7.5, 1.0 Hz, 1H), 7.82 (t, J = 7.6 Hz, 1H), 7.59 (dd, J = 8.3, 1.0 Hz, 1H), 7.55–7.49 (m, 3H), 7.41–7.28 (m, 7H). 13C NMR (101 MHz, CDCl3) δ 181.7, 173.6, 163.7, 155.2, 140.9, 139.0, 137.3, 136.35, 136.2, 136.0, 135.3, 135.2, 134.8, 134.5, 134.2, 134.1, 133.6, 123.6, 122.1. HRMS (ESI): calcd for C24H13NO5 [M + H]+: 396.0866; found: 396.0885. HPLC purity: 95.4%.
9-Nitro-2,3-dipropylbenzo[de]chromene-7,8-dione (7d). Deep brown solid, yield 67%. Mp: 196.5–197.4 °C; IR νmax (KBr) cm−1: 1647, 1699. 1H NMR (400 MHz, DMSO) δ 8.14 (d, J = 7.3 Hz, 2H), 8.02 (t, J = 7.2 Hz, 1H), 2.79–2.70 (m, 4H), 1.79–1.62 (m, 2H), 1.55 (m, 2H), 0.96 (m, 6H). 13C NMR (101 MHz, DMSO) δ 176.8, 167.9, 158.9, 154.3, 135.5, 132.9, 130.2, 130.0, 129.8, 129.4, 116.8, 115.1, 31.3, 27.0, 22.7, 20.4, 13.6, 13.2. HRMS (ESI): calcd for C18H17NO5 [M + H]+: 328.1179; found: 328.1182. HPLC purity: 99.3%.
2,3-Dibutyl-9-nitrobenzo[de]chromene-7,8-dione (7e). Deep brown solid, yield 70%. Mp: 159.8–161.4 °C; IR νmax (KBr) cm−1: 1650, 1705. 1H NMR (400 MHz, DMSO) δ 8.13 (d, J = 7.6 Hz, 1H), 8.11 (d, J = 8.8 Hz, 1H), 8.03 (t, J = 7.6 Hz, 1H), 2.78–2.73 (m, 4H), 1.71–1.56 (m, 2H), 1.50–1.40 (m, 4H), 1.38–1.34 (m, 2H), 0.95–0.91 (m, 6H). 13C NMR (101 MHz, DMSO) δ 176.7, 167.9, 158.8, 154.4, 135.6, 132.9, 130.1, 129.7, 129.4, 128.5, 116.7, 115.2, 31.6, 29.2, 29.0, 24.9, 21.9, 21.4, 13.7, 13.6. HRMS (ESI): calcd for C20H21NO5 [M + H]+: 356.1492; found: 356.1491. HPLC purity: 99.6%.
9-Bromo-2,3-dibutylbenzo[de]chromene-7,8-dione (7f). Deep brown solid, yield 68%. Mp: 149.7–152.8 °C; IR νmax (KBr) cm−1: 1644, 1692. 1H NMR (400 MHz, DMSO) δ 8.16–8.09 (m, 2H), 8.03 (t, J = 7.6 Hz, 1H), 2.75 (t, J = 7.3 Hz, 4H), 1.72–1.61 (m, 2H), 1.57–1.46 (m, 2H), 1.43–1.30 (m, 8H), 0.88 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, DMSO) δ 176.7, 167.9, 158.8, 154.4, 135.6, 132.9, 130.0, 129.7, 129.4, 128.5, 116.7, 115.2, 30.9, 30.4, 29.4, 29.1, 26.5, 25.1, 21.9, 21.7, 13.8, 13.6. HRMS (ESI): calcd for C22H25NO5 [M + H]+: 384.1805; found: 384.1791. HPLC purity: 99.2%.
6,7-Difluoro-1,4-dihydro-1,4-epoxynaphthalene (9). A 500 mL, 3-neck, round-bottom flask, which was equipped with a condenser, rubber seal, gas adapter under N2, was charged 50 mL of THF, 150 mmol furan and 1.44 g of Mg (60 mmol, 1.2 equiv.). The solution which was heated to 75 °C, was added 8 (10.55 g, 50 mmol) in 30 mL of THF via gastight syringe over a 1 h period at such a rate to keep the reaction mixture. The reaction mixture was then stirred for 8 h at 75 °C. Then 20 mL of water was poured into the reaction mixture to halt the reaction, and the aqueous phase was washed (3 × 50 mL) with ether. The combined organic phases were dried over Na2SO4, vacuum filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 20:1, 10:1, 8:1, 5:1) to afford 9 (4.89 g, 31%) as yellow oily liquid. 1H NMR (400 MHz, CDCl3) δ 7.07 (t, J = 7.7 Hz, 2H), 7.03 (t, J = 1 Hz, 2H), 5.69 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 148.5 (d, J = 14.9), 146.0 (d, J = 14.9), 145.2 (t, J = 4.6 HZ), 143.1, 110.9 (m), 82.1.
6,7-Difluoronaphthalen-1-ol (10). To a stirred solution of 9 (5.76 g, 1.5 mmol) in CH2Cl2 (30 mL), diluted BF3·Et2O (4.84 mL) was added slowly and stirred for 1 h at 0 °C. After the starting material was consumed up, the solution was washed with water (40 mL) and dried over Na2SO4, and then the solvent was removed under educed pressure. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 10:1) to afford 10 (4.89 g, 84.9%) as white solid. 1H NMR (400 MHz, CDCl3) δ 7.94 (t, J = 9.5 Hz, 1H), 7.52 (t, J = 9.0 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.32–7.27 (m, 2H) 6.80 (d, J = 7.1 Hz, 1H), 5.36 (bs, 1H). 13C NMR (101 MHz, CDCl3) δ 150.9 (d, J = 5.1 Hz), 150.5 (dd, J = 249.6, 14.7 Hz), 149.6 (dd, J = 248.5, 15.2 Hz), 131.8 (d, J = 7.7 Hz), 126.4 (d, J = 2.3 Hz), 121.1 (d, J = 6.8 Hz), 119.9 (dd, J = 4.7, 1.6 Hz), 113.3 (d, J = 16.77 Hz), 108.7 (d, J = 5.8 Hz), 106.6 (d, J = 10.3 Hz).
2,3-Diethyl-4,5-difluorobenzo[de]chromene (11). A suspension of 10 (900 mg, 5.00 mmol), 3-hexyne (5 mmol), [RuCl2(p-cymene)]2 (48 mg, 1.5% mmol) and Cu(OAc)2·H2O (955 mg, 5 mmol), and m-xylene (8.0 mL) in ChemGlass pressure vessel was stirred at 80–100 °C overnight. After the starting material was consumed up, the reaction mixture was cooled to room temperature, diluted with dichloromethane, and filtered with a short column chromatography on silica gel. The organic layers were washed with water (3 × 30 mL) and dried over Na2SO4. After filtration, the filtrate was collected and the solvent was removed under reduced pressure, the crude product was purified by using column chromatography on silica gel (petroleum ether) to give 11 (54%) as pale yellow oily liquid. 1H NMR (400 MHz, CDCl3) δ 7.24 (t, J = 8.0 Hz, 1H), 7.10 (dd, J = 8.1, 0.6 Hz, 1H), 7.03 (dd, J = 11.1, 7.2 Hz, 1H), 6.71 (d, J = 7.7 Hz, 1H), 2.59 (qd, J = 7.4, 2.2 Hz, 2H), 2.46 (q, J = 7.5 Hz, 2H), 1.26 (t, J = 7.2 Hz, 3H), 1.20 (td, J = 7.4, 1.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 154.7 (d, J = 2.2 Hz), 152.3 (dd, J = 248.2, 17.4 Hz), 152.1 (dd, J = 4.7, 1.9 Hz), 140.8 (dd, J = 247.3, 17.5 Hz), 131.57 (d, J = 5.5 Hz), 129.0, 127.4 (d, J = 1.6 Hz), 126.3, 120.2 (d, J = 5.1 Hz), 118.4 (dd, J = 5.0, 1.7 Hz), 110.0 (dd, J = 5.4, 3.2 Hz), 107.8 (d, J = 18.2 Hz), 106.7 (d, J = 2.1 Hz), 23.5, 21.4 (d, J = 9.6 Hz), 14.5 (d, J = 4.2 Hz), 12.4.
2,3-Diethyl-4,5-difluoro-7-nitrobenzo[de]chromene (12). 5 mmol (1.3 g) of 11 was dissolved in acetic anhydride (40 mL) and cooled to 0 °C in an ice bath. 1.21 g of cupric nitrate (5 mmol, 1 equiv.) was added slowly with vigorous stirring. The reaction was warmed to room temperature, and stirring was continued until the reaction was completed. The mixture was diluted with brash ice (300 g), stirred for another 2 h, and filtered to collect the residue. The residue was subjected to chromatography column (silica gel), eluting with 30% dichloromethane in petroleum to give 12 (4.7 g, 73.09%) as yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.47 (m, 1H), 8.43 (d, J = 8.9 Hz, 1H), 6.79 (d, J = 8.9 Hz, 1H), 2.69–2.65 (m, 2H), 2.56 (q, J = 7.6 Hz, 2H), 1.28 (t, J = 7.6 Hz, 3H), 1.22 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 157.2 (d, J = 2.7 Hz), 155.0 (d, J = 2.2 Hz), 154.6 (dd, J = 251.8, 16.2 Hz), 141.4 (dd, J = 251.9, 16.9 Hz), 136.5 (d, J = 5.5 Hz), 129.2, 125.8 (dd, J = 11.1, 1.8 Hz), 123.6 (d, J = 2.2 Hz), 120.4 (d, J = 4.7 Hz), 119.6 (dd, J = 7.3, 2.4 Hz), 11.6 (dd, J = 5.7, 3.5 Hz), 109.2 (d, J = 18.9 Hz), 106.8 (d, J = 24.0 Hz), 106.5, 23.5, 21.4 (d, J = 10.4 Hz), 14.5 (d, J = 4.5 Hz). HRMS (ESI): calcd for C16H13NO3F2, [M + H]+: 306.0936; found: 306.0940.
2,3-Diethyl-4,5-difluorobenzo[de]chromene-7,8-dione (13). Sodium hyposulfite (1.74 g, 10 mmol, 5 equiv.) was added to a solution of 12 (2 mmol) in THF/H2O (120 mL, 3:1). The reaction mixture was heated to 50 °C, and further stirred for 2 h at this temperature. After the starting material was consumed up, the reaction mixture was concentrated to remove the THF under reduced pressure and then the residue was extracted with dichloromethane (30 mL × 3). The combined extracts were evaporated to remove the solvent. The solid residue was then dissolved in acetone (60 mL) and a solution of Fremy's salt (1.34 g, 5 mmol, 2.5 equiv.) in 0.06 M KH2PO4 solution (68 mL) was added with vigorous stirring. After stirring for 1 h, the mixture was extracted with dichloromethane (30 mL × 3). The combined extracts were washed with brine (30 mL), dried over Na2SO4, filtered and evaporated to dryness. The solid residue was purified by using flash column chromatography (silica gel), eluting with 5% ethyl acetate in dichloromethane to give 13 (45.19%) as purple black solid. Mp: 145.2–148.5 °C. IR νmax (KBr) cm−1: 1641, 1699. 1H NMR (400 MHz, CDCl3) δ 7.92 (t, J = 7.6 Hz, 1H), 6.06 (s, 1H), 2.71 (dq, J = 7.2, 2.4 Hz, 2H), 2.64 (q, J = 7.5 Hz, 2H), 1.29 (t, J = 7.5 Hz, 3H), 1.22 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 178.1, 177.6, 164.3, 156.6, 153.8 (dd, J = 258.9, 148 Hz), 150.5 (dd, J = 267.0, 14.7 Hz), 127.1 (d, J = 5.1 Hz), 123.2 (d, J = 8.9 Hz), 119.8 (d, J = 5.2 Hz), 119.6 (d, J = 20.3 Hz), 110.7 (d, J = 5.2 Hz), 104.3, 23.8, 21.1 (d, J = 10.9 Hz), 14.9 (d, J = 4.0 Hz), 12.5. HRMS (ESI): calcd for C16H12O3F2 [M + H]+: 291.0827; found: 291.0832. HPLC purity: 99.9%.
9-Bromo-2,3-diethyl-4,5-difluorobenzo[de]chromene-7,8-dione (14). N-Bromosuccinimide (0.6 mmol, 1.2 equiv.) and benzoyl peroxide (12 mg) were added to a solution of 13 (0.5 mmol) dissolved in carbon tetrachloride. The reaction mixture was heated at 75 °C with stirring. After the starting material was consumed up, the reaction mixture was filtered and concentrated under reduced pressure. The solid residue was purified by using flash column chromatography (silica gel), eluting with dichloromethane to give 14 (74%) as purple black solid. Mp: 304.9–307.4 °C. IR νmax (KBr) cm−1: 1646, 1697. 1H NMR (400 MHz, CDCl3) δ 7.94 (t, J = 8.0 Hz, 1H), 2.72–2.78 (m, 4H), 1.37 (t, J = 7.4 Hz, 3H), 1.23 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 175.8, 172.2, 160.3, 156.8, 153.7 (d, J = 259.1, 15.7 Hz), 150.8 (dd, J = 268.2, 14.7 Hz), 125.8 (m)1, 123.1 (d, J = 9.9 Hz), 120.2 (d, J = 20.0 Hz), 119.6 (m), 11.7 (m), 100.1, 23.8, 21.2 (d, J = 11.1 Hz), 14.8 (d, J = 3.4 Hz), 11.9. HRMS (ESI): calcd for C16H11O3F2Br [M + H]+: 368.9932; found: 368.9944. HPLC purity: 99.1%.
General procedure for synthesis of compounds 16. A solution of K2CO3 (552 mg, 4 mmol), 12 (2 mmol), different amine (6 mmol) in DMF (20 mL) was stirred in 60 °C for 12 h. Then H2O (40 mL) was added and extracted with CH2Cl2 (3 × 30 mL). The combined organic phases were dried over Na2SO4, and the solvent was removed under reduced pressure to get the crude product. The next step was similar to the synthesis of 13.
2,3-Diethyl-4-fluoro-5-(piperidin-1-yl)benzo[de]chromene-7,8-dione (16a). 16a was prepared from K2CO3 (552 mg, 4 mmol), 12 (2 mmol), piperidin (6 mmol), purple black solid, yield 65%. Mp: 167.9–169.4 °C. IR νmax (KBr) cm−1: 1630, 1696. 1H NMR (400 MHz, CDCl3) δ 7.76 (t, J = 5.4 Hz, 1H), 5.94 (d, J = 6.1 Hz, 1H), 3.20 (d, J = 4.2 Hz, 4H), 2.70 (s, 2H), 2.64–2.58 (m, 2H), 1.75 (s, 4H), 1.65 (s, 2H), 1.29–1.24 (m, 3H), 1.20 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 179.9, 177.9, 165.7, 155.5, 153.4 (d, J = 262.9 Hz), 145.4 (d, J = 10.1 Hz), 126.6 (d, J = 3.9 Hz), 122.6 (d, J = 6.0 Hz), 121.3 (d, J = 12 Hz), 115.5 (d, J = 5.7 Hz), 111.4 (d, J = 4.3 Hz), 102.4, 51.6 (d, J = 4.4 Hz), 25.9, 24.0, 23.9, 21.6 (d, J = 12.1 Hz), 15.1 (d, J = 4.4 Hz), 12.5. HRMS (ESI): calcd for C20H20NO4F [M + H]+: 358.1449; found: 358.1465. HPLC purity: 98.6%.
2,3-Diethyl-4-fluoro-5-morpholinobenzo[de]chromene-7,8-dione (16b). 16b was prepared from K2CO3 (552 mg, 4 mmol), 12 (2 mmol), morpholine (6 mmol). Deep green solid, yield 36%. Mp: 315.4–317.2 °C. IR νmax (KBr) cm−1: 1631, 1695. 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 7.6 Hz, 1H), 5.97 (s, 1H), 3.90 (d, J = 3.3 Hz, 4H), 3.26 (d, J = 2.9 Hz, 4H), 2.77–2.66 (m, 2H), 2.65–2.57 (m, 2H), 1.27 (t, J = 7.6 Hz, 3H), 1.20 (t, J = 7.1 Hz, 3H)13C NMR (101 MHz, CDCl3) δ 179.7, 177.9, 165.5, 155.7, 153.7 (d, J = 263.6 Hz), 144.3 (d, J = 10.2 Hz), 126.7 (d, J = 4.02 Hz), 121.9 (d, J = 5.6 Hz), 121.5 (d, J = 11.9 Hz), 116.7 (d, J = 6.0 Hz), 111.2 (d, J = 4.4 Hz), 102.8, 66.7, 50.4, 23.8, 21.5 (d, J = 12.1 Hz), 15.1 (d, J = 4.4 Hz), 12.6. HRMS (ESI): calcd for C20H20NO4F [M + Na]+: 380.1269; found: 380.1254. HPLC purity: 99.8%.
5-(Diethylamino)-2,3-diethyl-4-fluorobenzo[de]chromene-7,8-dione (16c). 16c was prepared from K2CO3 (552 mg, 4 mmol), 12 (2 mmol), diethylamine (6 mmol). Reddish black, yield 30%. Mp: 245.4–246.8 °C. IR νmax (KBr) cm−1: 1624, 1694. 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 7.9 Hz, 1H), 5.91 (s, 1H), 3.42 (m, 4H), 2.70 (m, 2H), 2.65–2.59 (m, 2H), 1.27 (t. J = 7.6 Hz, 3H), 1.21 (dd, J = 13.7, 6.8 Hz, 12H). 1.20 (t, J = 7.2 Hz, 3H)·13C NMR (101 MHz, CDCl3) δ 180.6, 177.8, 166.2, 155.5, 151.2 (d, J = 259.1 Hz), 142.6 (d, J = 10.1 Hz), 126.7 (d, J = 2.8 Hz), 121.8 (d, J = 20.0 Hz), 121.8 (d, J = 7.8 Hz), 112.9 (d, J = 5.1 Hz), 111.59 (d, J = 4.5 Hz), 101.7, 46.4 (d, J = 5.2 Hz), 23.9, 21.6 (d, J = 12.6 Hz), 15.1 (d, J = 4.6 Hz), 13.3, 12.6. HRMS (ESI): calcd for C20 H22NO3 F [M + H]+: 344.1656; found: 344.1671 HPLC purity: 99.8%.
2,3-Diethyl-4-fluoro-5-(pyrrolidin-1-yl)benzo[de]chromene-7,8-dione (16d). 16d was prepared from K2CO3 (552 mg, 4 mmol), 12 (2 mmol), pyrrolidin (6 mmol). Dark green solid, yield 50%. Mp: 163.5–166.4 °C. IR νmax (KBr) cm−1: 1626, 1695. 1H NMR (400 MHz, CDCl3) δ 8.61 (d, J = 6.3 Hz, 1H), 8.00 (s, 1H), 7.76 (s, 1H), 6.50 (s, 1H), 6.01 (s, 1H), 2.67 (s, 2H), 2.58 (d, J = 7.1 Hz, 2H), 1.23 (d, J = 7.3 Hz, 4H), 1.18 (s, 7H). 13C NMR (101 MHz, CDCl3) δ 180.8, 177.7, 166.5, 155.3, 148.4 (d, J = 256.0 Hz), 141.2 (d, J = 11.3 Hz), 127.1 (d, J = 2.1 Hz), 121.2 (d, J = 12.3 Hz), 119.5 (d, J = 8.6 Hz), 111.6 (d, J = 4.2 Hz), 110.9 (d, J = 5.4 Hz), 101.1, 50.5, 25.4, 23.9, 21.7 (d, J = 21.7 Hz), 14.9 (d, J = 4.8 Hz), 12.55. HRMS (ESI): calcd for C20H20NO3F [M + H]+: 342.1500; found: 342.1511 HPLC purity: 99.6%.
9-Bromo-2,3-diethyl-4-fluoro-5-(piperidin-1-yl)benzo[de]chromene-7,8-dione (17). N-Bromosuccinimide (0.6 mmol, 1.2 equiv.) and benzoyl peroxide (12 mg) were added to a solution of 16a (0.5 mmol) dissolved in carbon tetrachloride. The reaction mixture was heated at 75 °C with stirring. After the starting material was consumed up, the reaction mixture was filtered and concentrated under reduced pressure. The solid residue was purified by using flash column chromatography (silica gel), eluting with dichloromethane to give 17 (56%) as black solid. Mp:307.9–310.1 °C. IR νmax (KBr) cm−1: 1636, 1689. 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 7.7 Hz, 1H), 3.23 (m, J = 4.1 Hz, 4H), 2.71–2.76 (m, 4H), 1.77 (s, 4H), 1.66 (s, 2H), 1.35 (t, J = 7.4 Hz, 3H), 1.23 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 177.6, 172.5, 161.6, 155.7, 153.26 (d, J = 263.7 Hz), 145.2 (d, J = 9.8 Hz), 130.1, 128.4, 125.5 (d, J = 3.6 Hz), 123.2 (d, J = 6.3 Hz), 121.4 (d, J = 12.2 Hz), 115.0 (d, J = 5.8 Hz), 112.4 (d, J = 4.3 Hz), 97.6, 51.6, 29.8, 25.9, 24.0, 23.9, 21.6 (d, J = 12.7 Hz), 14.9 (d, J = 44 Hz), 12.1. HRMS (ESI): calcd for C21H21NO3FBr [M + H]+: 434.0762; found: 434.0770. HPLC purity: 98.3%.
MTT assay
Four different cancer cell lines, including HeLa, A549, K562 and HL-60 were used to evaluate the growth inhibitory effect of MsF derivatives by using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium-bromide) assay as described by Mosmann with modifications.23 The cells were plated at a density of 1 × 104 per well in 96-well microplates, and allowed to incubate overnight in a 5% CO2 incubator at 37 °C. MsF derivatives were added to the wells at increasing concentrations (0–50 μM). After 48 h, each well was treated with 20 mL 2.5 mg mL−1 MTT solution, and the cells were further incubated at 37 °C for 4 h. At the end of the incubation, the untransformed MTT was removed, and 100 μL of DMSO was added for HeLa and A549 cell lines. K562, and HL-60 cell lines were treated with three linked lysis solution, and then incubated at 37 °C overnight. The microplates were well shaken to dissolve the formazan dye, and the absorbance at 570 nm was measured using a microplate-reader (Bio-Tek). VP-16 were commercially available and used as positive controls.
Acknowledgements
We thank the Natural Science Foundation of China (21272288), the Natural Science Foundation of Guangdong Province (2015A030313120), and the Fundamental Research Funds for the Central Universities (13ykpy09) for financial support of this study.
Notes and references
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed.
- K. Zhang, J. Li, W. Meng, H. Xing and Y. Yang, Blood Cells, Mol., Dis., 2016, 56, 46–52 CrossRef CAS PubMed.
- J. Xie, J. Liu, H. Liu, S. Liang, M. Lin, Y. Gu, T. Liu, D. Wang, H. Ge and S. L. Mo, Acta Pharm. Sin. B, 2015, 5, 554–563 CrossRef PubMed.
- S. M. Planchon, S. Wuerzberger, B. Frydman, D. T. Witiak, P. Hutson, D. R. Church, G. Wilding and D. A. Boothman, Cancer Res., 1995, 55, 3706–3711 CAS.
- C. J. Li, C. Wang and A. B. Pardee, Cancer Res., 1995, 55, 3712–3715 CAS.
- W. B. Wu, J. B. Ou, Z. H. Huang, S. B. Chen, T. M. Ou, J. H. Tan, D. Li, L. L. Shen, S. L. Huang, L. Q. Gu and Z. S. Huang, Eur. J. Med. Chem., 2011, 46, 3339–3347 CrossRef CAS PubMed.
- Z. H. Huang, S. T. Zhuo, C. Y. Li, H. T. Xie, D. Li, J. H. Tan, T. M. Ou, Z. S. Huang, L. Q. Gu and S. L. Huang, Eur. J. Med. Chem., 2013, 68, 58–71 CrossRef CAS PubMed.
- D. Wang, M. Y. Xia, Z. Cui, S. Tashiro, S. Onodera and T. Ikejma, Biol. Pharm. Bull., 2004, 27, 1025–1030 CAS.
- J. P. Kim, W. G. Kim, H. Koshino, J. Jung and I. D. Yoo, Phytochemistry, 1996, 43, 425–430 CrossRef CAS PubMed.
- R. S. Burden and M. S. Kemp, Phytochemistry, 1984, 23, 383–385 CrossRef CAS.
- D. Y. Shin, S. N. Kim, J. H. Chae, S. S. Hyun, S. Y. Seo, Y. S. Lee, K. O. Lee, S. H. Kim, Y. S. Lee, J. M. Jeong, N. S. Choi and Y. G. Suh, Bioorg. Med. Chem. Lett., 2004, 14, 4519–4523 CrossRef CAS PubMed.
- Y. G. Suh, S. N. Kim, D. Y. Shin, S. S. Hyun, D. S. Lee, K. H. Min, S. M. Han, F. Li, E. C. Choi and S. H. Choi, Bioorg. Med. Chem. Lett., 2006, 16, 142–145 CrossRef CAS PubMed.
- P. S. Ng and A. K. Banerjee, Nat. Prod. Res., 2006, 20, 629–635 CrossRef PubMed.
- R. L. Nunes, L. W. Bieber and R. L. Longo, J. Nat. Prod., 1999, 62, 1643–1645 CrossRef CAS.
- W. Best and D. Wege, Aust. J. Chem., 1986, 39, 647–666 CrossRef CAS.
- Y. G. Suh, D. Y. Shin, K. H. Min, S. S. Hyun, J. K. Jung and S. Y. Seo, Chem. Commun., 2000, 1203–1204 RSC.
- S. Mochida, M. Shimizu, K. Hirano, T. Satoh and M. Miura, Chem.–Asian J., 2010, 5, 847–851 CrossRef CAS PubMed.
- V. S. Thirunavukkarasu, M. Donati and L. Ackermann, Org. Lett., 2012, 14, 3416–3419 CrossRef CAS PubMed.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- S. L. Huang, Z. S. Huang, L. Q. Gu and H. T. Xie, CN. Patent, CN 104744421, 2015.
- K. C. Caster, C. G. Keck and R. D. Walls, J. Org. Chem., 2001, 66, 2932–2936 CrossRef CAS PubMed.
- J. T. Repine, D. S. Johnson, A. D. White, D. A. Favor, M. A. Stier, J. Yip, T. Rankin, Q. Ding and S. N. Maiti, Tetrahedron Lett., 2007, 48, 5539–5541 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00090a |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Hong-Gen Wang
*,
Hong-Gen Wang