DOI:
10.1039/C6RA26532D
(Paper)
RSC Adv., 2017,
7, 6315-6322
Structural deformation phenomenon of synthesized poly(isosorbide-1,4-cyclohexanedicarboxylate) in hot water†
Received
9th November 2016
, Accepted 27th December 2016
First published on 19th January 2017
Abstract
Previously syntheses of poly(isosorbide 1,4-cyclohexanedicarboxylate) (PICD) have overcome synthetic problems associated with the low-reactivity of isosorbide, using acetic anhydride to achieve in situ acetylation. However, this amorphous polymer exhibits unusual behavior when submerged in water at 100 °C. Severe deformation occurs, with cylindrical pellets changing into a disc-like morphology, similar to solvent-induced crystallization. The influence of water on the thermal behavior of PICD was analyzed, resulting in a mechanism analogous to solvent-induced crystallization, where the solvent functions as a plasticizer. Furthermore, the effects of acetic anhydride and open-ring isosorbide on structural deformation were investigated, revealing the occurrence of ester hydrolysis. Finally, solid state CP-MAS 13C-NMR was used to elicit the rearrangement or packing of carbons within the PICD structure.
Introduction
Biodegradable polymers have received great attention because of their environmental advantages, benefiting from reductions in fossil fuel usage, carbon footprints, and global warming potential. However, biodegradability has given rise to the fact that it has limited range in market for inexpensive short-term, causing the emergence of the “biobased” concept. Biobased polymers are polymeric materials derived from biomass resources, and are a potential replacement for conventional polymeric materials derived from fossil resources.
Research on utilizing biobased materials, such as lactic acid,1–5 citric acid,6–9 tartaric acid,10–12 and chitosan,13–15 in polymer synthesis has been conducted. These contributions are now being tested for utilization and industrialization. Therefore, understanding the nature and behavior of these synthesized polymers could further improve and expand the applications of biobased materials.
Isosorbide (ISB) is a promising biobased material, and one of three isomers of 1,4:3,6-dianhydrohexitol. ISB can easily be utilized as a monomer in polyester synthesis; however, polyester syntheses incorporating ISB have resulted in low Mw values due to its secondary alcohol nature and the uneven reactivity of the endo and exo hydroxyl groups, caused by the presence of hydrogen bonds with oxygen in the ring. Such disadvantages are compensated by unique structural properties, which cause high rigidity, superior thermal/optical properties, and nontoxicity.16–21 This sugar-derived monomer has been used to generate a variety of polymers, such as polyesters,22–24 polycarbonates,25–27 and polyurethanes,16,28–30 while little research focusing on its characteristics, such as crystallizability.
In our previous research with 4-cyclohexanedicarboxylic acid (CHDA),31 poly(isosorbide 1,4-cyclohexanedicarboxylate) (PICD) was synthesized using acetic anhydride to achieve in situ acetylation, which decreased the steric hindrance in ISB and accelerated PICD chain growth. We recently discovered that, in this particular polyester, reorientation/chain ordering can occur when it is heated in a solvent medium, particularly water. Furthermore, a phenomenon similar to solvent-induced crystallization can be observed, where solvent diffuses between the polymer chains and disrupts the free space. This removes rotation restrictions on the polymer chains and naturally lowers the polymer glass transition temperature (Tg), meaning that the solvent acts as a plasticizer. When such a restriction is removed, an energetically favorable conformation is readily adopted, resulting in crystal structure development under ambient conditions.32 Solvent-induced crystallization of polymers has already been reported in several cases, including syndiotactic polystyrene (sPS),33–35 poly(ethylene terephthalate),36 poly(ethylene-2,6-naphthalate),37 poly(ether ether ketone),38,39 polycarbonates,40–42 isotactic polypropylene,43,44 and aromatic polyimide.45
To identify the unique structural deformation behavior induced by water, we analyzed PICD polyester synthesized using various amounts of acetic anhydride. This study focuses on the deformation kinetics, and determining the effect and transition for each monomer. Therefore, the role of acetic anhydride and the open-ring ISB content were determined through a detailed analysis of PICD synthesis, which suggested two kinetic theories of deformation. Since the kinetics of these phenomena are similar to some aspects of solvent-induced crystallization, thermal analysis using DSC was carried out analogously to verify the plasticizing effect of water. Finally, solid state cross polarization-magic angle spinning (CP-MAS) 13C-NMR was conducted to describe the function of each carbon during deformation, along with theoretical confirmation.
Experimental
Materials
CHDA (99.8%) with 22 mol% trans-isomer and ISB (99.8%, Roquette Freres S.A.) were supplied by SK Chemicals. The monomers were high-purity commercial products and used as received. Solvents and chemical reagents used for acetylation and characterization, including acetic anhydride, acetic acid and chloroform, were all high-purity grade and purchased from Sigma-Aldrich. All reagents were used as received.
Synthesis of PICD polyester
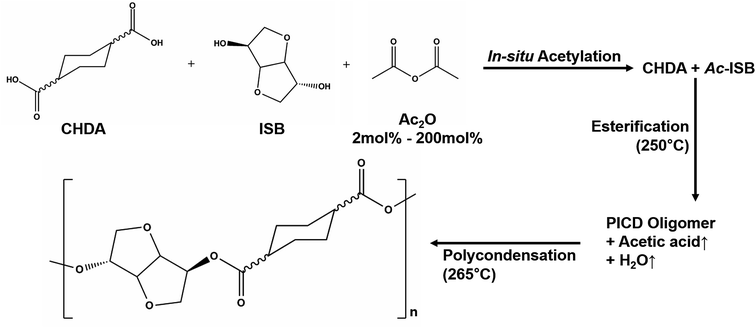
PICD samples were prepared using same method as our previous study.31 In brief, CHDA (1220 g, 7.09 mol), ISB (1040.8 g, 7.12 mol), germanium dioxide (200 ppm) and dibutyltin oxide (150 ppm) were weighed into an agitator, condenser and trap-fitted 3 L reactor in a molar ratio of CHDA/ISB = 1/1.003. To determine the effect of Ac2O, it was added to the reactor at levels of 2, 5, 50, 70 and 200 mol% relative to the amount of ISB. Esterification was performed at 250 °C for 2 h after stabilizing at 120 °C under 1 bar of nitrogen for 1 h. After esterification, the oligomer was moved to a different reactor fitted with a spiral agitator. Transesterification was performed at 265 °C and 0.4 mbar, while stirring at 98 rpm. The stirring speed was slowly decreased to 30 rpm with an increasing degree of polymerization, and was terminated when the stirrer torque reached 2 N m. The resulting polyester was removed from the reactor, quenched with liquid nitrogen and dried in a vacuum oven at 80 °C for 48 h before characterization.
Preparation of deformed-PICD in water
Each PICD sample was extruded using a Thermo HAAKE™ MiniLab Rheomex CTW 5. Each sample (5 g) was weighed from each extrusion at 60 rpm and 250 °C. The direct extrusion time for each sample was controlled at 3 min. Extruded samples were quenched with liquid nitrogen and cut into 5 mm lengths. The average diameter was 2.3 mm. PICD samples were weighed at 5 g each and placed in 400 mL tube sealed with a silicon stopper. Using 100 mL of deionized water and a magnetic stirrer, the temperature was set to 100 °C and held for 48 h. Deformed samples were then dried in a vacuum oven at 60 °C for 48 h and analysed.
Characterization
Intrinsic viscosity
The intrinsic viscosity (IV) of all PICD copolymers was measured in chloroform at 25 °C and 0.01–2.5 g dL−1 using an automated Ubbelohde viscometer (Schott Co.).
Nuclear magnetic resonance (NMR)
1H-NMR spectra were obtained using a Varian VNMRS 600 MHz spectrometer operating at 600 MHz with deuterated chloroform as the solvent. Tetramethylsilane (TMS) was used as an internal standard and as a reference for the chemical shifts. Sixteen scans with 16 K data points each were acquired for each 1H-NMR spectrum. The relaxation delay was 5 s. 13C-NMR spectra were recorded at 150 MHz using the same NMR spectrometer and solvent. Each 13C-NMR spectrum comprised 8192 scans with 64 K data points. Solid state NMR spectroscopy was carried out using a Bruker AVANCE III 300 equipped with a CP-MAS accessory to determine the alteration of free space during water induced deformation.
Differential scanning calorimetry (DSC)
Thermal analysis was carried out using a TA instruments Auto-DSCQ20 System. All analyses were performed under nitrogen, and the samples each weighed 8 mg. Samples were enclosed in standard 40 μL aluminium cells, melted at 280 °C for 5 min, and then quenched to 30 °C. The temperature was raised at a scanning rate of 10 °C min−1 to 280 °C. The glass transition temperature (Tg) was taken as the onset of the change in heat capacity associated with a transition.
Scanning electron microscopy (SEM)
Surface images of deformed PICD were taken with SEM (Nova Nano SEM, FEI).
Results and discussion
Synthesis and deformation of PICD homopolyester
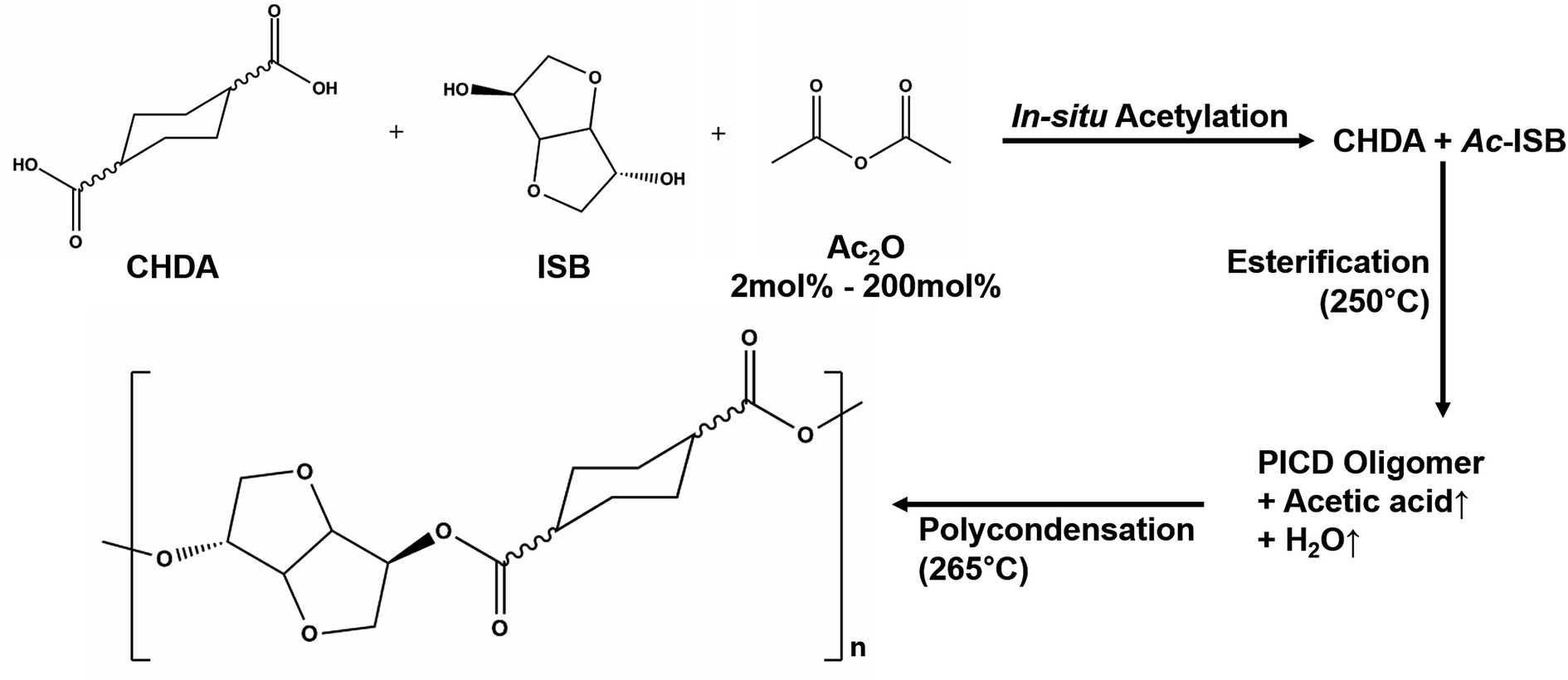
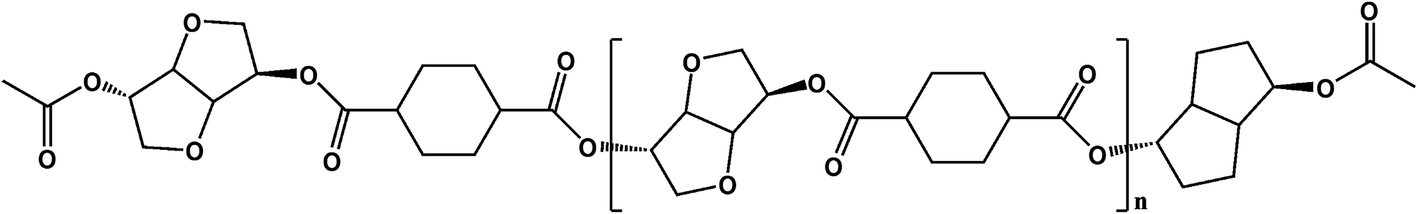
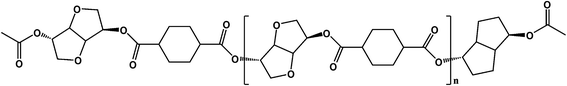
PICD homopolyester was synthesized using CHDA and ISB via acetylation, as shown in Scheme 1. To substitute the hydroxyl end group of ISB with an acetyl group using Ac2O, in situ acetylation was performed to compensate for the inferior reactivity of ISB in esterification and polycondensation. As mentioned in our previous paper, in situ acetylation is an effective method, even using small amounts of Ac2O. In this study, various contents, ranging from 2 mol% to 200 mol% Ac2O, were used to determine the effect of Ac2O on the kinetics of water-induced deformation.
 |
| | Scheme 1 Polymerization of PICD using in situ acetylation. | |
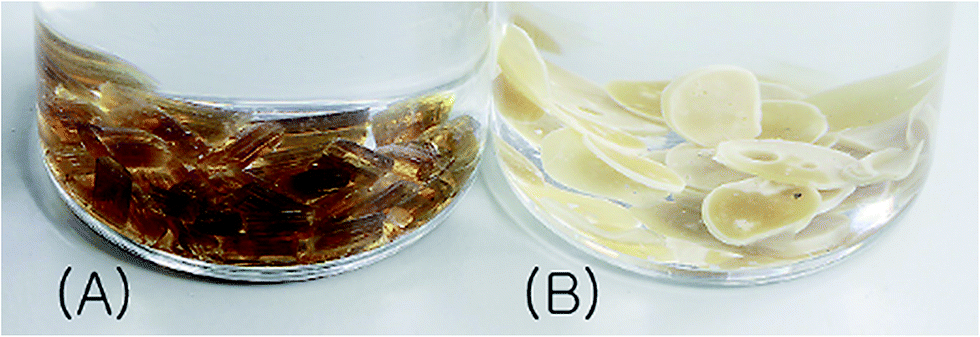
PICD deformation was discovered when a PICD pellet was submerged in water and the temperature was raised to 100 °C and held for 24 h. When PICD was submerged in water without heat, deformation did not occur. As shown in Fig. 1, the deformation was quite severe, with the length and width changing from 5 mm to 0.9 mm, and 2.3 mm to 5.3 mm, respectively. The volume decreased from 20.76 mm3 to 19.84 mm3, resulting in a 0.04% loss. Furthermore, the brownish transparent PICD had become opaque. This drastic change in length and width indicated that this deformation had occurred at the molecular level. Since the PICD chain was almost oriented with the vertical axis, which resulted from extrusion, it would have rearranged itself laterally with the help of water molecules, which seemed more thermodynamically stable. The decrease in volume signified an increased packing density, meaning that the distance between the chains had decreased. This behavior, along with the loss of transparency, was somewhat similar to that of solvent-induced crystallization.
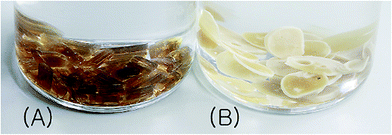 |
| | Fig. 1 PICD pellet (A) before and (B) after deformation. | |
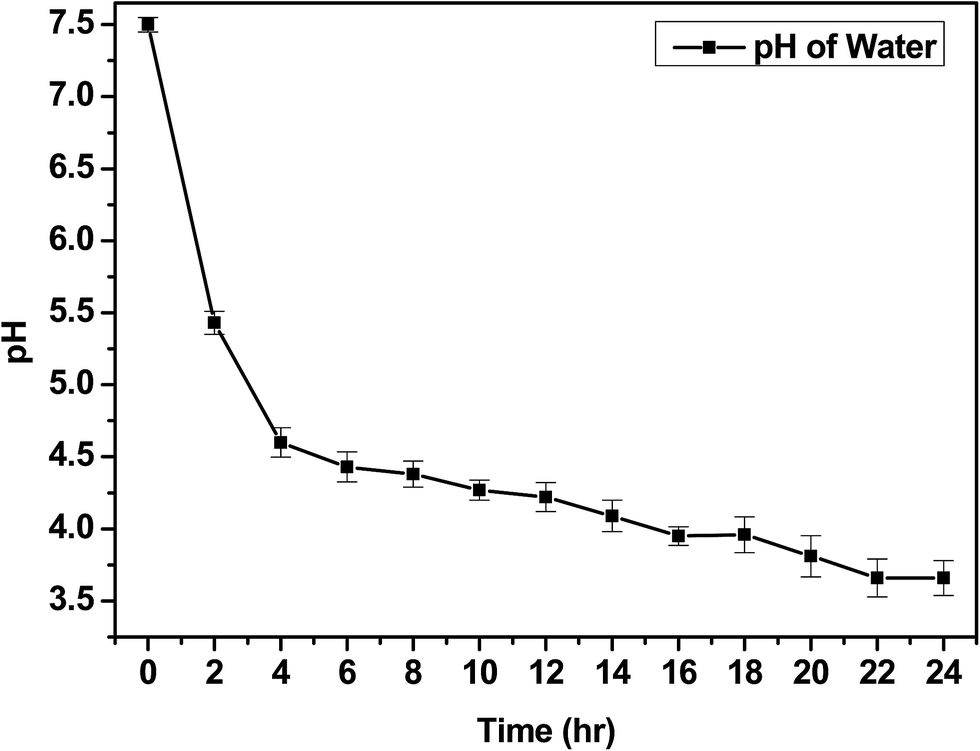
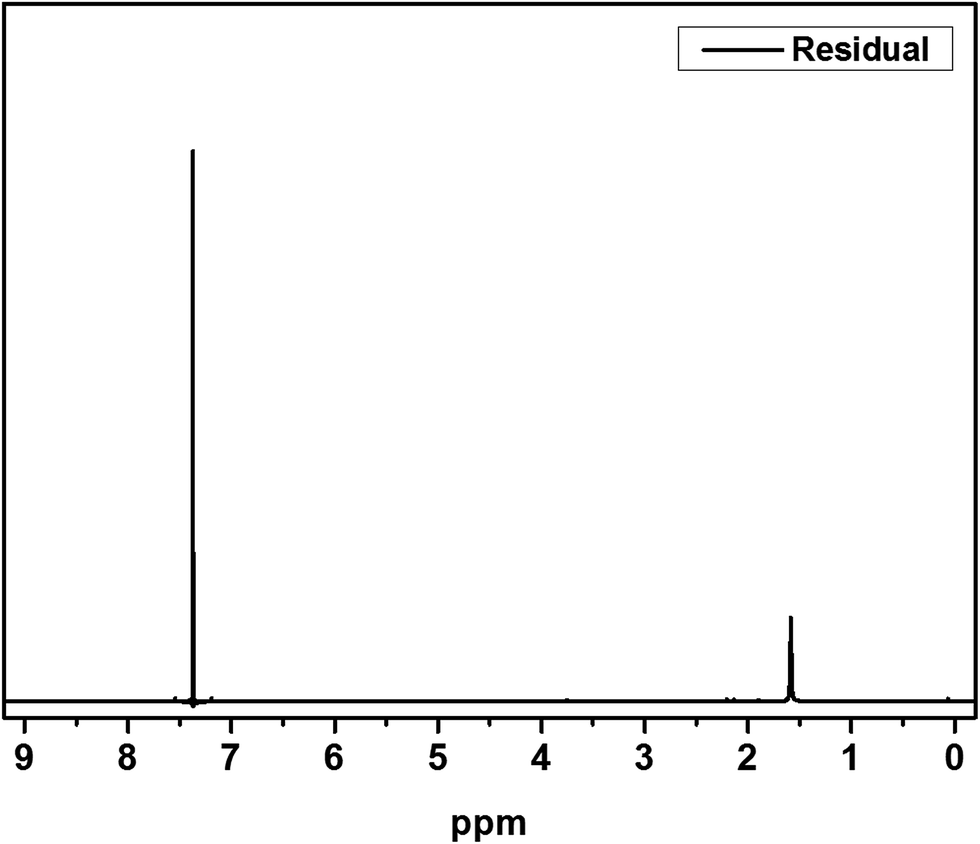
The IV values of PICD and deformed PICD are summarized in Table 1. All samples showed different IV values, decreasing from PICD to deformed PICD, with seemingly no relationship between IV difference and Ac2O content. The cause of decreased IV was most likely due to ester hydrolysis and open-ring ISB. The common foundational basis of both reactions is that the surrounding system must be acidic. To confirm the basic requirements for these reactions, the water pH was measured every 2 h throughout the deformation process, with the results shown in Fig. 2. The water pH started at 7.5, rapidly dropping to 4.6 after 4 h, followed by a gradual decrease that plateaued at 3.6 after 24 h. This increasing water acidity would affect ester hydrolysis and the open-ring ISB content. This introduction of acidity into the system was explained by analyzing the evaporated residual content of water. As shown in Fig. 3, the residual content showed a singlet at δ 1.58 ppm, matching that of acetic acid. The origin of this acetic acid can be explained using Fig. 4. As PICD synthesis progresses, acetic anhydride that had reacted with ISB produced acetyl-ISB, which can be placed at the end of PICD chains due to steric hindrance and the less reactive nature of ISB. Another possibility is entrapment of acetic acid in the PICD melt, which, given the reaction temperature and volatility of acetic acid, was highly unlikely to occur. Therefore, acetic acid can be assumed to be slowly secreted during the deformation process, which decreased the pH value.
Table 1 Intrinsic viscosity of PICD before and after deformation
| Sample codea |
Ac2O contents (mol%) |
IV of PICDb (dL g−1) |
IV of deformed PICD (dL g−1) |
ΔIVc |
| Sample codes denote the mol% of Ac2O added relative to ISB. Intrinsic viscosity (dL g−1) measured in chloroform at 25 °C. IV difference between amorphous PICD and crystallized PICD. |
| PICD-2 |
2 mol% |
0.52 |
0.50 |
0.02 |
| PICD-5 |
5 mol% |
0.62 |
0.53 |
0.09 |
| PICD-50 |
50 mol% |
0.64 |
0.58 |
0.06 |
| PICD-70 |
70 mol% |
0.55 |
0.52 |
0.03 |
| PICD-200 |
200 mol% |
0.65 |
0.62 |
0.03 |
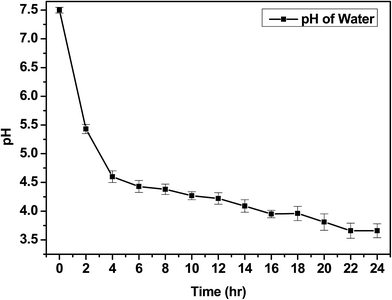 |
| | Fig. 2 Change in water pH during deformation. | |
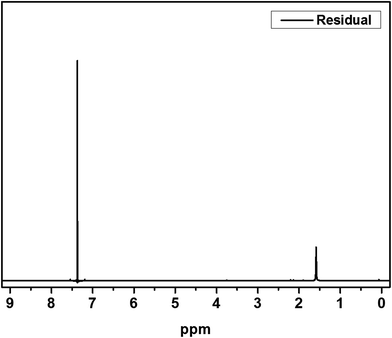 |
| | Fig. 3 1H-NMR spectrum of residual. | |
 |
| | Fig. 4 Probable end group structure of PICD. | |
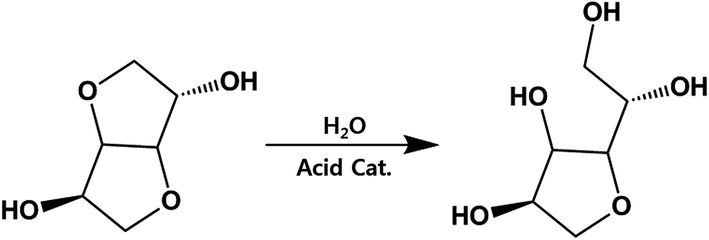
Considering that residual analysis showed only acetic acid, ester hydrolysis would conclusively occur in sizable portions, not just the end-group diol, which would lead to a slight decrease in IV. As shown in Fig. 5, ISB has been shown to undergo ring-opening reactions when the internal ether is exposed to acid catalyst. As a result, it can induce increased flexibility and higher mobility, which decreases the IV value. Table 2 summarizes the open-ring ISB contents in both PICD and deformed-PICD. PICD-2 showed no difference in open-ring ISB content. Considering that 2 mol% is a very low amount of Ac2O, it would have little effect on the acidity of PICD synthesis, hence the lack of increase or decrease in open-ring ISB content, even after deformation. The open-ring content decreased in PICD-5 and PICD-50, which can be explained by ester hydrolysis, as discussed earlier. As hydrolysis would occur at the ether moiety in ISB, causing decreased IV, it would also decrease the open-ring ISB content. PICD-70 and PICD-200 showed increased open-ring ISB contents. As 70 and 200 mol% were large amounts of Ac2O, it was clear that abundant acetic acid would be produced by the PICD end group, as discussed above. This acetic acid would be released into the solvent medium (water) faster, actively triggering the ring-opening reaction and resulting in a higher open-ring ISB content.
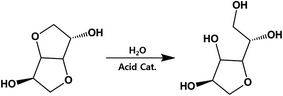 |
| | Fig. 5 Open-ring ISB and the possible reaction outcome. | |
Table 2 Open-ring ISB contents in PICD and deformed PICD
| Sample codea |
Open-ring ISB content of PICDa (mol%) |
Open-ring ISB content of deformed PICD (mol%) |
| Open-ring ISB content was measured using the relative integrated area of the methylene proton in 1H-NMR. |
| PICD-2 |
10.92 mol% |
10.92 mol% |
| PICD-5 |
18.33 mol% |
17.55 mol% |
| PICD-50 |
6.63 mol% |
6.59 mol% |
| PICD-70 |
10.86 mol% |
10.92 mol% |
| PICD-200 |
8.82 mol% |
9.30 mol% |
Thermal properties of PICD homopolyester
Normally, the kinetics of crystallization induced by solvent are derived from their solvent diffusion coefficients, which have the mobile strength to remove any restriction placed upon the polymer chain, functioning as a plasticizer. Therefore, solvent-induced crystallization does not require any external source of energy, such as heat, for crystallization.
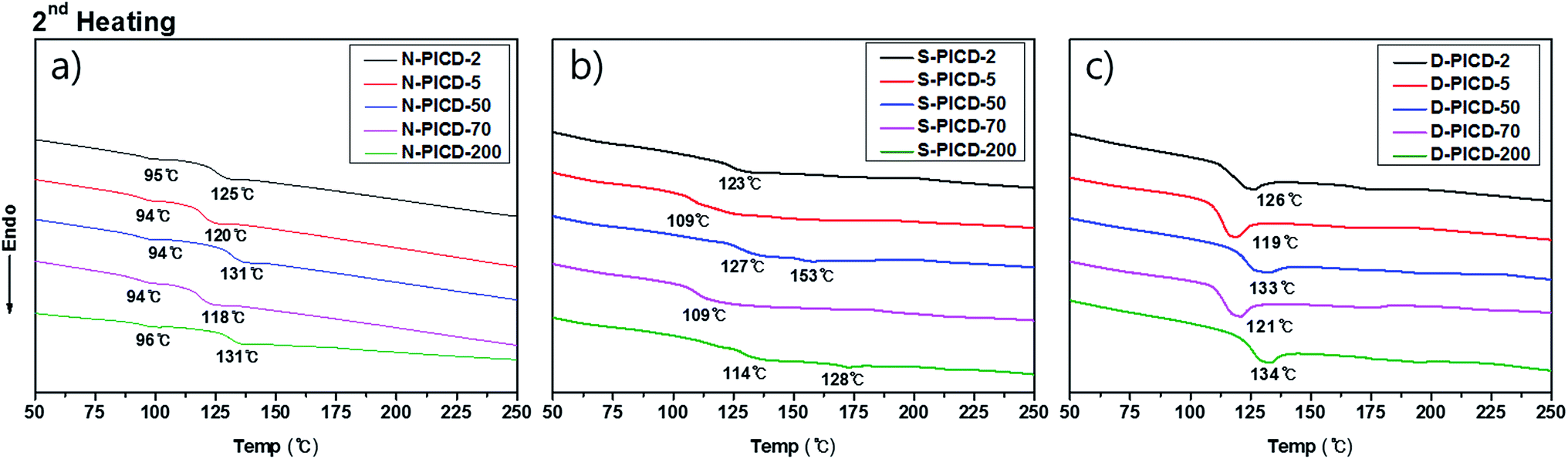
However, for PICD, water alone was not able to cause crystallization or deformation. Since water molecules have low diffusion coefficients (self-diffusion coefficient: 0.0012 cm2 s−1, diffusion coefficient of water to polymer: ∼10−6 cm2 s−1),46,47 they cannot diffuse into the core of the PICD structure. PICD only deformed when exposed to 100 °C heat in water for a long period, preferably more than 24 h, suggesting that vibrant water molecules induce PICD structure deformation with an external energy source. To ensure that the deformation kinetics resembled solvent-induced crystallization with water functioning as a plasticizer, the thermal properties of PICD and PICD were analyzed under various conditions. The thermal behaviors of neat PICD, PICD submerged in water, and deformed PICD are summarized in Fig. 6 and Table 3. Neat PICD showed two glass transition temperatures, denoted T1 and T2. As two glass transition temperatures represent a polymer having two phases, PICD can be thought as having two different phases in the homopolyester chain. The first phase was in the presence of open-ring ISB, whose flexible nature induced localized reordering at its branches and loose sites, giving a lower transition temperature, T1. The secondary phase was main chain PICD, where whole-bodied long-range segmental motion resulted in a higher transition temperature, T2.
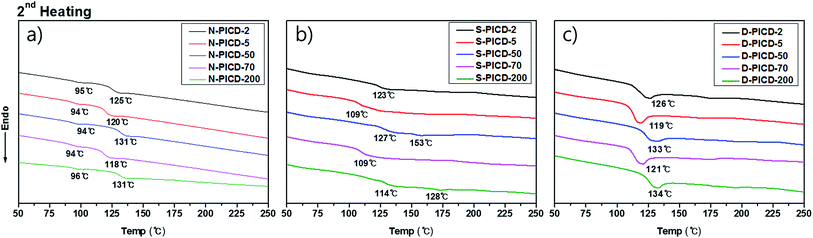 |
| | Fig. 6 Thermal analysis of (a) neat PICD, (b) PICD submerged in water, and (c) deformed PICD. | |
Table 3 Thermal properties of PICD under various conditions
| Sample codea |
T1 and T2 of PICDb (°C) |
T3 of submerged PICD (°C) |
T4 of deformed PICD (°C) |
| Sample codes denote the mol% of Ac2O added relative to ISB. Glass transition temperatures measured by DSC. |
| PICD-2 |
95/125 |
123 |
126 |
| PICD-5 |
94/120 |
109 |
119 |
| PICD-50 |
94/131 |
127/153 |
133 |
| PICD-70 |
94/118 |
109 |
121 |
| PICD-200 |
96/131 |
114/128 |
134 |
Although submergence did not induce deformation, it proved that water was capable of diffusing into the PICD chain and disrupting its original thermal behavior during the heating process. As the results show, PICD-2, PICD-5, and PICD-70 show a single T3 between T1 and T2, whereas PICD-50 and PICD-70 showed two increased T3 values. These two samples contained less than 10 mol% open-ring ISB, meaning a smaller effect on prior segmental motion. As thermal analysis progressed, the increasing temperature would begin affecting the PICD chain containing water-induced reordering. When the temperature reached the boiling point of water, which, coincidently, was close to T1, energized and vibrant water molecules forcefully removed PICD chains from the restriction. However, the duration of heating was too short in the thermal analysis of PICD-50 and PICD-70, such that they were unable to undergo reordering. Therefore, PICD treated with enough heat and water for a suitably long time would receive enough energy for water to have a plasticizing effect, inducing deformation and reordering. Conclusively, deformed PICD, which was treated at 100 °C in water for 24 h, had a single T4 at a higher value.
Kinetics of PICD deformation
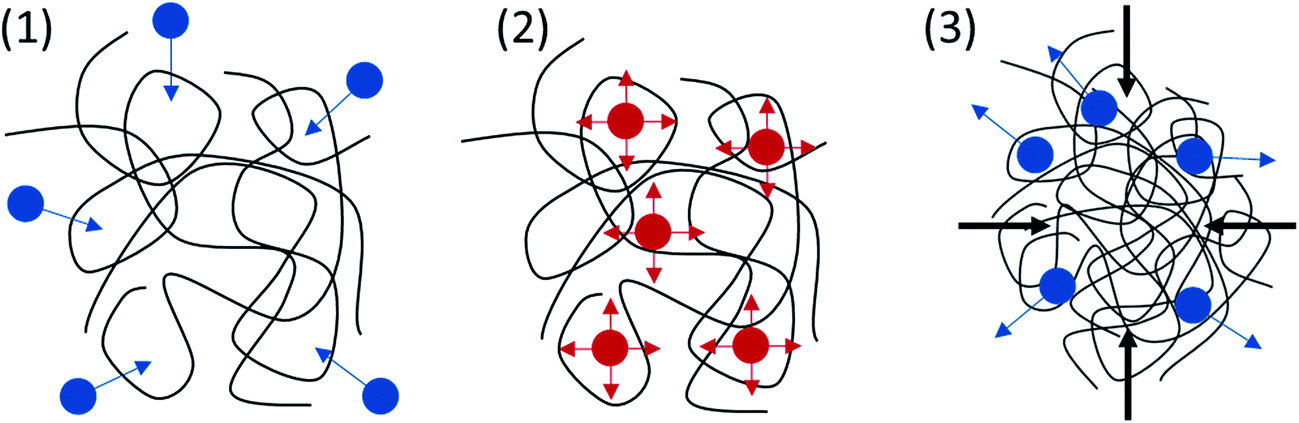
Thermal analysis proved that PICD deformation shared similar kinetics with solvent-induced crystallization, in which solvent diffusion and solvent functioning as plasticizer removed the restriction on the PICD chain, allowing it to reorder itself in favor of entropy. To better understand the movement of water, the surface of deformed PICD was analyzed. Fig. 7(A) shows the surface of deformed PICD, while Fig. 7(B) shows an internal view of the surface holes, which were numerous and variously sized, showing no regularity. However, the outer rim of these holes was white, meaning that it was higher than the grey surface. This indicated that an outward eruption had occurred. Such a phenomenon is explained in Scheme 2. Firstly, water diffusion occurs (1); as the two angled furan rings of ISB create enough free volume between the chains, water, aided by heat, can diffuse into the PICD chains. When the temperature reaches 100 °C, water starts to vibrate vigorously (2). At this stage, vibrant and diffused water molecules, both inside and outside, remove restrictions on the PICD chain and start to function as plasticizers, increasing the mobility of the PICD structure. When deformation is complete, during which the chains are brought closer to each other, the free space between the chains shrinks. Therefore, water that had diffused inside is forced to erupt out of the PICD structure, leaving holes on the deformed surface.
 |
| | Fig. 7 (A) Surface of deformed PICD, and (B) inside the rupture. | |
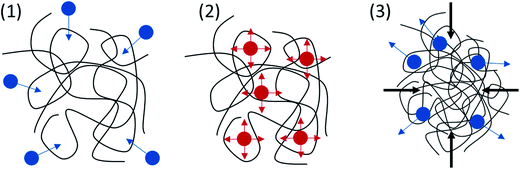 |
| | Scheme 2 Paths of movement for plasticizing water. | |
Structural analysis of PICD
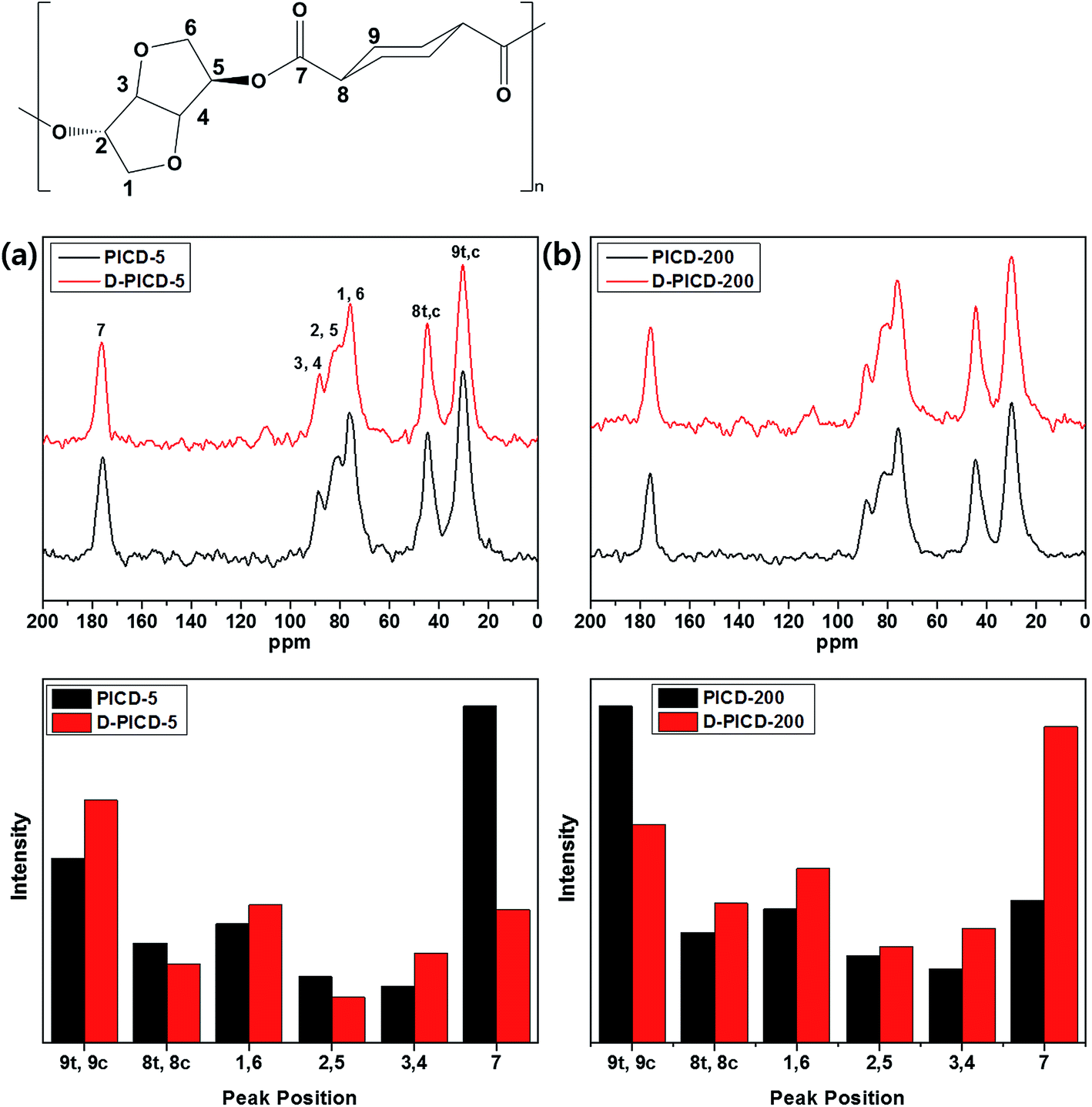
Solid-state NMR (SSNMR) offers great insight into polymer structure and free space with the help of CP-MAS. CP-MAS combines cross polarization and magic angle spinning techniques to narrow down broad peaks by spinning the sample at 54.74° (the magic angle) with respect to the direction of the magnetic field. This can obtain a clear spectrum, which is more suitable for assigning peaks according to the structure. Conventional solution NMR removes restrictions caused by structure and allows all carbons and hydrogens to freely rotate according to the applied magnetic field, resulting in instant relaxation times. However, in SSNMR, the motional freedom of carbons and hydrogens is effected and restricted by the neighboring structure because of the solid nature. Therefore, SSNMR gives longer relaxation times, resulting in altered peak intensities. This alteration signifies the transition in free space state of the corresponding carbon. If a carbon is positioned in a crystalline region, it would have little free space, hindering rotation when a magnetic field is applied. Therefore, the relaxation time would be longer, giving the peak a higher intensity. This can be applied to analyze the deformation of PICD and elicit which carbons have moved or become more closely packed, as in solution-induced crystallization, during deformation. Fig. 8 shows the solid state CP-MAS 13C-NMR spectra of PICD-5 and PICD-200, along with the intensity differences of each peak. PICD and D-PICD represent amorphous PICD and deformed PICD, respectively. The peaks at δ 30.2, 44.6, 76.1, 80.7, 88.8, and 175.8 ppm were assigned to 9 (trans/cis), 8 (trans/cis), 1/6, 2/5, 3/4, and 7, respectively. All peaks were assigned in comparison with the results of solution 13C-NMR [S1]. All peaks, except peak 7, were broad enough to cover the range of two peaks, which were closely positioned due to their symmetry. Therefore, it was reasonable to conclude that these peaks shared the same structural environment. PICD-5 and PICD-200 represented two different possible deformation kinetics. For PICD-5, the IV value and open-ring ISB content decreased after deformation, suggesting an ester hydrolysis effect. As shown by the peak intensity differences, peaks far from the breakage point, such as the ester functional group, 9, 1/6, and 3/4, showed increased intensity, whereas those directly affected by the ester functional group, 7 and 8, showed decreased intensities. Therefore, the hydrolysis effect resulted in fewer ester functional groups, and hence fewer effected carbons, to take part in deformation. However, PICD-200 presented another kinetic route to deformation. Compared with PICD-5, the IV value of PICD-200 decreased less and, since excess Ac2O induces an acidic environment during polymerization, the open-ring ISB content increased. With less hydrolysis occurring, ISB and ester functional groups were brought closer together forcefully during deformation, resulting in the exclusion of a cyclohexane group (peak 9). In conclusion, it is reasonable to state that open-ring ISB content is the key factor in water-induced deformation.
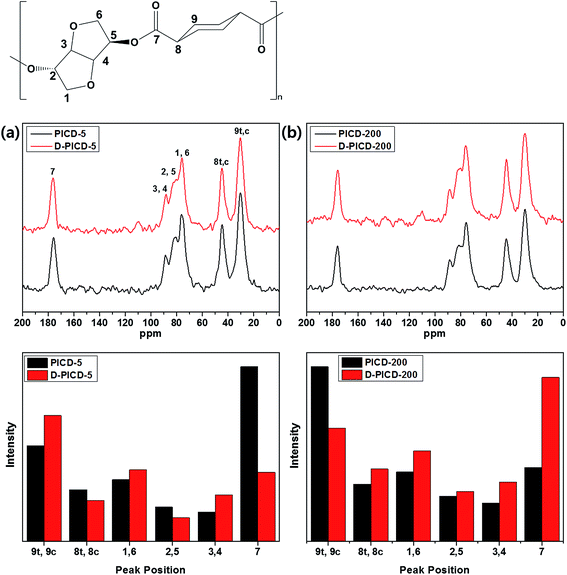 |
| | Fig. 8 Solid state CP-MAS 13C-NMR spectra of (a) PICD-5 and (b) PICD-200 with peak intensity differences. | |
Conclusion
The abnormal deformation behavior in heat and water has been discussed. Acetic anhydride has proven to be an attractive reagent for accelerating polymerization with less reactive ISB, while the open-ring ISB content it induces has a significant effect on polymer deformation. Although this phenomenon shares kinetics analogous to solvent-induced crystallization, the amorphous nature of the polymer chain and fully intact ISB prevent continuous ordering, which agrees with our previous research48 showing complete ISB exclusion during crystallization. Interestingly, open-ring ISB shows the possibility to reduce packing distance and cause severe structural/apparent deformation using an exterior energy source. This phenomenon could be utilized by controlling the open-ring ISB content in future studies.
Acknowledgements
This work was supported and funded by the Chemical R&D institute of SK Chemicals. The authors would also like to thank Professor Young-il Lee, University of Ulsan.
References
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- A. Hinoki, A. Saito, M. Kinoshita, J. Yamamoto, D. Saitoh and S. Takeoka, Br. J. Surg., 2016, 103, 692–700 CrossRef CAS PubMed.
- R. E. Lee, Y. Guo, H. Tamber, M. Planeta and S. N. S. Leung, Ind. Eng. Chem. Res., 2016, 55, 560–567 CrossRef CAS.
- K. Khodabakhshi and M. Ehsani, Handbook of Sustainable Polymers: Processing and Applications, 2016, p. 397 Search PubMed.
- K. Masutani and Y. Kimura, Polym. Int., 2016, 66(2), 260–270 CrossRef.
- J. Yang, A. R. Webb and G. A. Ameer, Adv. Mater., 2004, 16, 511–516 CrossRef CAS.
- I. Djordjevic, N. R. Choudhury, N. K. Dutta and S. Kumar, Polymer, 2009, 50, 1682–1691 CrossRef CAS.
- R. Shi, Z. Zhang, Q. Liu, Y. Han, L. Zhang, D. Chen and W. Tian, Carbohydr. Polym., 2007, 69, 748–755 CrossRef CAS.
- D. Gyawali, P. Nair, Y. Zhang, R. T. Tran, C. Zhang, M. Samchukov, M. Makarov, H. K. Kim and J. Yang, Biomaterials, 2010, 31, 9092–9105 CrossRef CAS PubMed.
- M. Pawar, A. Kadam, O. Yemul, V. Thamke and K. Kodam, Ind. Crops Prod., 2016, 89, 434–447 CrossRef CAS.
- K. Kizuka and S.-I. Inoue, Open J. Org. Polym. Mater., 2016, 6, 38 CrossRef CAS.
- S. Dhamaniya and J. Jacob, Polymer, 2010, 51, 5392–5399 CrossRef CAS.
- R. Porras, D. Bavykin, J. Zekonyte, F. C. Walsh and R. J. Wood, Nanotechnology, 2016, 27, 195706 CrossRef CAS PubMed.
- H. Gao, W. Zhou, J. H. Jang and J. B. Goodenough, Adv. Energy Mater., 2016, 6(6), 1502130 CrossRef.
- J.-S. Ahn, H.-K. Choi and C.-S. Cho, Biomaterials, 2001, 22, 923–928 CrossRef CAS PubMed.
- E. C. Varkey and K. Sreekumar, J. Mater. Sci., 2010, 45, 1912–1920 CrossRef.
- B. Philip and K. Sreekumar, J. Mater. Sci., 2003, 38, 1573–1577 CrossRef CAS.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J.-P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS.
- J. F. Treon, L. E. Gongwer and W. H. Rueggeberg, Exp. Biol. Med., 1965, 119, 39–42 CrossRef CAS.
- C. H. Lee, H. Takagi, H. Okamoto and M. Kato, J. Appl. Polym. Sci., 2013, 127, 530–534 CrossRef CAS.
- B. A. Noordover, V. G. van Staalduinen, R. Duchateau, C. E. Koning, R. A. van Benthem, M. Mak, A. Heise, A. E. Frissen and J. van Haveren, Biomacromolecules, 2006, 7, 3406–3416 CrossRef CAS PubMed.
- M. Majdoub, A. Loupy and G. Flèche, Eur. Polym. J., 1994, 30, 1431–1437 CrossRef CAS.
- S. Chatti, M. Bortolussi, D. Bogdal, J. Blais and A. Loupy, Eur. Polym. J., 2004, 40, 561–577 CrossRef CAS.
- R. Storbeck and M. Ballauff, J. Appl. Polym. Sci., 1996, 59, 1199–1202 CrossRef CAS.
- T. S. Kristufek, S. L. Kristufek, L. A. Link, A. C. Weems, S. Khan, S.-M. Lim, A. T. Lonnecker, J. E. Raymond, D. J. Maitland and K. L. Wooley, Polym. Chem., 2016, 7, 2639–2644 RSC.
- Y. S. Eo, H.-W. Rhee and S. Shin, J. Ind. Eng. Chem., 2016, 37, 42–46 CrossRef CAS.
- S. Chatti, G. Schwarz and H. R. Kricheldorf, Macromolecules, 2006, 39, 9064–9070 CrossRef CAS.
- M. Beldi, R. Medimagh, S. Chatti, S. Marque, D. Prim, A. Loupy and F. Delolme, Eur. Polym. J., 2007, 43, 3415–3433 CrossRef CAS.
- C. H. Lee, H. Takagi, H. Okamoto, M. Kato and A. Usuki, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6025–6031 CrossRef CAS.
- R. Marín, A. Alla, A. Martínez de Ilarduya and S. Muñoz-Guerra, J. Appl. Polym. Sci., 2012, 123, 986–994 CrossRef.
- W. J. Yoon, K. S. Oh, J. M. Koo, J. R. Kim, K. J. Lee and S. S. Im, Macromolecules, 2013, 46, 2930–2940 CrossRef CAS.
- A. B. Desai and G. L. Wilkes, J. Polym. Sci., Polym. Symp., 1974, 46, 291–319 Search PubMed.
- V. Vittoria, F. de Candia, P. Iannelli and A. Immirzi, Makromol. Chem. Rapid Comm., 1988, 9, 765–769 CrossRef CAS.
- K. Tashiro, Y. Ueno, A. Yoshioka and M. Kobayashi, Macromolecules, 2001, 34, 310–315 CrossRef CAS.
- K. Tashiro and A. Yoshioka, Macromolecules, 2002, 35, 410–414 CrossRef CAS.
- S. S. Im and H. S. Lee, J. Appl. Polym. Sci., 1989, 37, 1801–1814 CrossRef CAS.
- S.-J. Kim, J.-Y. Nam, Y.-M. Lee and S.-S. Im, Polymer, 1999, 40, 5623–5629 CrossRef CAS.
- H. Cornélis, R. G. Kander and J. P. Martin, Polymer, 1996, 37, 4573–4578 CrossRef.
- A. J. Lovinger and D. Davis, Macromolecules, 1986, 19, 1861–1867 CrossRef CAS.
- H. Harron, R. Pritchard, B. Cope and D. Goddard, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 173–180 CrossRef CAS.
- J. Mercier, G. Groeninckx and M. Lesne, J. Polym. Sci., Polym. Symp., 1967, 16(4), 2059–2067 Search PubMed.
- Z. Fan, S. Chang, Y. Yu, V. Zaporojtchenko and F. Faupel, Polym. Eng. Sci., 2006, 46, 729 CAS.
- V. Vittoria and F. Riva, Macromolecules, 1986, 19, 1975–1979 CrossRef CAS.
- V. Vittoria, Polymer, 1991, 32, 856–859 CrossRef CAS.
- J. Wang, A. DiBenedetto, J. Johnson, S. Huang and J. L. Cercena, Polymer, 1989, 30, 718–721 CrossRef CAS.
- K. R. Harris and L. A. Woolf, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 377–385 RSC.
- F. Long and L. Thompson, J. Polym. Sci., 1955, 15, 413–426 CrossRef CAS.
- J. M. Koo, S. Y. Hwang, W. J. Yoon, Y. I. Lee, S. H. Kim and S. S. Im, Polym. Chem., 2015, 6, 6973–6986 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26532d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*