
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural changes of poplar wood lignin after supercritical pretreatment using carbon dioxide and ethanol–water as co-solvents†
Xing Wang,
Yanzhu Guo *,
Jinghui Zhou* and
Guangwei Sun
*,
Jinghui Zhou* and
Guangwei Sun
Liaoning Key Laboratory of Pulp and Papermaking Engineering, Dalian Polytechnic University, Dalian, Liaoning 116034, China. E-mail: guoyz@dlpu.edu.cn; zhoujh@dlpu.edu.cn; Tel: +86 411 86323296
First published on 24th January 2017
Abstract
To delineate the structural changes of lignin during supercritical carbon dioxide pretreatment with ethanol–water as co-solvents (SCEP), enzymatic hydrolysis lignin (EHL) in original poplar chips, residual lignin in SCEP pretreated residues (SCEP-RL) and lignin dissolved in the SCEP liquors (SCEP-DL) were sequentially isolated and systematically characterized by GPC, quantitative 13C-NMR, 31P-NMR, 2D-HSQC NMR and TGA. After SCEP process, 19.2% of lignin was degraded and dissolved into SCEP liquors, while 4.8% of lignin was still present in the pretreated residues. It was also convinced that parts of the β-O-4 aryl ether linkages were cleaved and some of the stilbene, resinol (β–β) and phenylcoumaran (β-5) units were increased during SCEP process. Furthermore, the contents of free phenolic hydroxyl groups and carboxylic acids in SCEP-RL and SCEP-DL were higher than that of EHL.
1 Introduction
Lignocellulosic biomass is one of the most abundant yet underutilized bioresources on earth. It comprises large amounts of carbohydrates, such as cellulose and hemicelluloses, and is considered as a potential substrate to produce high value products, e.g., biofuels including bioethanol and biogas.1,2 Bioethanol is typically produced from biomass via three steps: pretreatment of biomass materials, enzymatic hydrolysis of pretreated residues by cellulases and fermentation of sugars by microbial organisms.3 Among these steps, the pretreatment is crucial to efficiently conversing lignocellulosic biomass to ethanol, because it can highly break down the polymeric matrix between carbohydrates and lignin in biomass, and therefore decreases the recalcitrance of biomass and enhances the accessibility of enzymes to pretreated residues during enzymatic hydrolysis.4–6 Typical methods for biomass pretreatment include auto-hydrolysis and thermochemical pretreatment using alkali,7,8 acid,9 organic solvents,10,11 or supercritical fluid,12 which have been widely studied to prepare lignocellulosic substrates from a wide range of raw materials by either removing or redistributing hemicelluloses or lignin for subsequent bioconversion.The supercritical carbon dioxide (ScCO2) is a ubiquitous, nontoxic, recyclable and low-cost reagent, which has some favorable characteristics, e.g., environment friendly, low critical temperature and pressure, gas-like mass transfer, liquid-like solvating power, low viscosity and zero surface tension. Therefore, its employment in the biomass pretreatment generally can enhance the accessibility of biomass and reduce the pretreatment temperature.13,14 Up to now, the technologies involving the application of supercritical carbon dioxide into the pretreatment have been extensively investigated with the aim of conversing lignocellulosic biomass to the bioethanol products. Commonly, a variety of organic solvents, such as ethanol–water,15 1-butanol,16 acetic acid,17 are employed as the co-solvents during the process of ScCO2 pretreatment, which can selectively isolated lignin from biomass. Ethanol is an excellent liquefaction solvent for isolating lignin from biomass due to its lower dielectric ability, higher and rapider dissolving ability to the high molecular-weight lignin fraction.15,18 Recently, the ScCO2 pretreatment with ethanol–water as co-solvents (SCEP) has been applied into the pretreatment of various biomass materials, such as corn stover,19 sugarcane bagasse,20 rice straw,21 switchgrass,22 guayule23 and rice husk.12 Due to the enhanced acidic environment caused by ScCO2, the combination of ScCO2, ethanol and water seems to be superior in the removal of lignin from biomass. It has been reported that up to 90.6% lignin was removed from biomass after SCEP, while the crystallinity and enzymatic digestibility of pretreated residues were not significantly changed.12 It is needed to notation that lignin in the pretreated residues has inhibiting property to enzyme for the subsequent enzymatic hydrolysis, because the enzyme is easily to bind on the surfaces of residues. Meanwhile, the lignin with different structures is also convinced to generate different inhibiting properties.24 Proper understanding of the structural changes of lignin during SCEP process is helpful to design suitable strategies for pretreatment and alleviates the negative effects of lignin on the enzymatic hydrolysis.
In the present study, poplar wood chips were chosen as the raw materials and employed into the SCEP process and the structural changes of lignin during SCEP process were elucidated. Three lignin samples including EHL, SCEP-DL and SCEP-DL were separately isolated from the original chips, the liquors and residual chips after SCEP. The structural features of these three preparations were comparatively investigated by quantitative 13C-NMR, 2D-HSQC NMR and 31P-NMR spectroscopy. Their molecular weights were analysed by gel permeation chromatography (GPC). Thermogravimetric analysis (TGA) was also used to elucidate the relationship between their structure features and thermal properties.
2 Materials and methods
2.1. Materials
Poplar wood chips (dimension: 4 × 4 × 2 cm) were collected from Tiger Forest & Paper Co., Ltd. (Hunan, China). The moisture content in chips was 15.4 ± 0.2 wt%. Carbon dioxide (99.99% purity) was supplied by Guangming Research and Design Institute of Chemical Industry (Tianjin, China). Cellulast (80 FPU per g) and Novozyme 188 (250 FPU per g) were provided by Novozymes, China. Other reagents were of analytical or chromatographic grades and directly used without purification.2.2. Supercritical pretreatment
The supercritical carbon dioxide pretreatment of poplar chips was performed in a 1 L reactor (E1), which was connected with two 0.5 L separators in series (S1, S2) and a tank (ST1), for storage and recycle of CO2. The detail scheme for SCEP process is shown in Fig. S1.† 50 g of poplar wood chips and 500 mL of 50/50 (v/v) ethanol–water solution as co-solvent were charged into the reactor. After closed, the reactor was constantly pressurized with carbon dioxide to 20 MPa for 1 h at 120 °C. When experiments were finished, the liquid phase was collected from the separator in SCEP pretreatment systems. The pretreated poplar chips in the reactor were washed using ethanol–water solution (50/50, v/v) and dried in a cabinet oven with air circulation at 65 °C for 12 h. The dried pretreated poplar chips were kept in sealed bags for further analysis.2.3. Preparation of lignin samples
2.4. Analytical methods
The chemical compositions of original and pretreated poplar chips, including the amounts of sugars and lignin, were determined according to the National Renewable Energy Laboratory standard analytical method (NREL/TP-510-42618).25 Every experiment were repeated by two times. The contents of sugars in EHL, SCEP-RL and SCEP-DL samples were determined by ionic chromatograph (Dionex ICS-5000, USA), which was equipped with a Capillary Reagent-Free IC System and a CarboPac 20 column.The NMR determinations for all of the prepared lignin samples were performed in a Bruker AVIII 400 MHz spectrometer. For the 13C NMR analysis, 140 mg of lignin samples were dissolved in 0.5 mL of DMSO-d6. The 13C NMR spectra were acquired in the FT mode at 100.6 MHz through employing an inverse-gated decoupling pulse sequence, which was conditioned with the following parameters: 30° pulse angle, 2 s pulse delay, 1.4 s acquisition time and 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans at room temperature. The signals in 13C-NMR spectra were assigned according to the previous literature.26 The 2D-HSQC NMR determination was carried out according to recently reported method27,28 with minor modification. Briefly, 140 mg of lignin samples were dissolved in 0.5 mL DMSO-d6. The standard pulse program hsqcedetgpsisp2 was applied in the HSQC experiment. The spectral widths for the 13C and 1H dimensions were 20000 Hz and 5000 Hz, respectively. The central solvent peak was used as an internal reference (δC 39.5; δH 2.49). The application of 31P-NMR for characterization of functional groups contents in lignin moiety had been extensively explored.29 After dissolution of 20 mg of lignin samples in 500 μL of anhydrous pyridine/CDCl3 solvents (1.6:1, v/v) under magnetic stirring, 100 μL internal standard solution including 10.85 mg mL−1 cyclohexanol in pyridine/CDCl3 (1.6:1, v/v) and 100 μL relaxation reagent solution containing 5 mg mL−1 chromium(III) acetylacetonate in pyridine/CDCl3 (1.6:1, v/v) were added. Finally, 100 μL 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) was added into the above solution and the mixture was kept for 10 min. The final phosphitylated sample was transferred into a 5 mm NMR tube for 31P NMR determination.
000 scans at room temperature. The signals in 13C-NMR spectra were assigned according to the previous literature.26 The 2D-HSQC NMR determination was carried out according to recently reported method27,28 with minor modification. Briefly, 140 mg of lignin samples were dissolved in 0.5 mL DMSO-d6. The standard pulse program hsqcedetgpsisp2 was applied in the HSQC experiment. The spectral widths for the 13C and 1H dimensions were 20000 Hz and 5000 Hz, respectively. The central solvent peak was used as an internal reference (δC 39.5; δH 2.49). The application of 31P-NMR for characterization of functional groups contents in lignin moiety had been extensively explored.29 After dissolution of 20 mg of lignin samples in 500 μL of anhydrous pyridine/CDCl3 solvents (1.6:1, v/v) under magnetic stirring, 100 μL internal standard solution including 10.85 mg mL−1 cyclohexanol in pyridine/CDCl3 (1.6:1, v/v) and 100 μL relaxation reagent solution containing 5 mg mL−1 chromium(III) acetylacetonate in pyridine/CDCl3 (1.6:1, v/v) were added. Finally, 100 μL 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) was added into the above solution and the mixture was kept for 10 min. The final phosphitylated sample was transferred into a 5 mm NMR tube for 31P NMR determination.
The molecular weights of lignin preparations were analyzed by GPC, which were based on the procedures described in a previous literature.30 The analysis for thermal stability of lignin sample was carried out on a TA Q500 thermal analyzer (TA Instruments, USA), which was processed by heating the samples from 30 °C to 700 °C with a 10 °C min−1 heating rate under the protection of nitrogen.
3 Results and discussion
3.1. Composition analysis of lignin preparations, original and pretreated poplar chips
In order to estimate the delignification efficiency during SCEP process, the chemical compositions of original and pretreated poplar chips were comparatively studied and the results are shown in Table 1. The total contents of sugars in original poplar chips was 72.7%. Among of them, the xylose and glucose were the dominant sugars, and the arabinose, galactose, and mannose were only present with low contents. After pretreatment, the total contents of sugars and glucan in chips were separately increased up to 86.31% and 71.65%. By contrast, the contents of hemicelluloses, containing xylan, arabinan, galactan and mannan, were decreased from 23.55% to 14.66%, confirming that amounts of hemicelluloses were degraded and isolated from poplar chips after SCEP. The content of acid insoluble lignin (AIL) in original poplar chips was 23.4%, while the content of acid soluble lignin (ASL) was small and only about 2.3%. After SCEP, the total content of lignin in pretreated poplar chips was 7.5%, including 6.7% AIL and 0.8% ASL. The above results indicated that about 79% of lignin was removed from original poplar chips after SCEP process. It had been convinced that the enzymatic hydrolysis efficiency of pretreated chips was higher than original chips, due to their lower contents of lignin.31 However, it was also reported that the lignin in the pretreated residues has inhibiting property to enzyme for the subsequent enzymatic hydrolysis, because the enzyme is easily to bind on the surfaces of residues.24 Therefore, it could be concluded that the correlation between lignin and enzymatic hydrolysis could not only be explained by the content of lignin. The effect of structural changes of lignin inducing by the removal of lignin on the enzymatic hydrolysis should be extensively explored. With the above aims, three lignin preparations were isolated and characterized in this study. The yields and sugar contents of EHL, SCEP-RL and SCEP-DL are shown in Table 2. The yield of EHL from original poplar chips was 25.6%, while the yield of SCEP-RL from pretreated poplar chips was 5.1%. The yield of SCEP-DL, recovered from the pretreatment liquors, was 19.8%. All of the above results were consistent with the contents of lignin shown in Table 1. The contents of total sugars in these three preparations were 1.27%, 0.79% and 0.19%, indicating that the preparations were purity enough to be characterized by NMR. The xylan was the major sugars in EHL, SCEP-DL and SCEP-RL. However, it was observed that the contents of total sugars, especially xylan, in SCEP-DL and SCEP-RL were lower than those in EHL, implying that the hemicelluloses attached to lignin were partially degraded during the SCEP process.| Samples | Yields (%) | Sugar compositions (wt%) | Lignin analysis (wt%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ara | Gal | Glu | Xyl | Man | Total | AIL | ASL | Total | ||
| a Ara, arabinose; Gal, galactose; Glu, glucose; Xyl, xylose; Man, mannose. AIL, acid insoluble lignin; ASL, acid soluble lignin. Tr: trace amount. | ||||||||||
| Original poplar | — | 0.76 | 0.87 | 49.15 | 19.62 | 2.30 | 72.7 | 23.4 | 2.3 | 25.7 |
| SCEP pretreated poplar | 71.2 | Tr | Tr | 71.65 | 12.41 | 2.25 | 86.31 | 6.7 | 0.8 | 7.5 |
| Samples | Yieldsa (%) | Sugar compositions (wt%) | |||||
|---|---|---|---|---|---|---|---|
| Ara | Gal | Glu | Xyl | Man | Total | ||
| a Yield = WL/Wmaterial × 100%, where WL is the weight of the EHL, SCEP-DL or SCEP-RL (mg), Wmaterial is the weight of the initial raw material (mg). | |||||||
| EHL | 25.6 | 0.02 | 0.05 | 0.17 | 0.96 | 0.07 | 1.27 |
| SCEP-RL | 5.1 | 0.01 | 0.02 | 0.11 | 0.62 | 0.03 | 0.79 |
| SCEP-DL | 19.8 | Tr | Tr | 0.01 | 0.18 | Tr | 0.19 |
3.2. 2D-HSQC NMR analysis
The 2D-HSQC NMR is a powerful analytic technique and can give detailed structural characterization of complex biopolymers such as lignin.5,32,33 The 2D-HSQC spectra of EHL, SCEP-RL and SCEP-DL samples are shown in Fig. 1. Based on the previous literatures,27 the main signals were assigned and the detected linkages are depicted in Fig. 1, such as the inter-unit linkages β-O-4 aryl ether (A), resinol (β–β, B) and phenylcoumaran (β-5, C). The cross-signals of Cα–Hα in β-O-4 substructure are explicitly located at δC/δH 71.8/4.86, whereas the cross-signals at δC/δH 84.3/4.30 and 86.5/4.12 are attributed to its Cβ–Hβ correlations linked to G and S units in β-O-4 substructures, respectively. Meanwhile, the resinol (β–β, B) was also detected, and the Cα–Hα, Cβ–Hβ and Cγ–Hγ correlations were observed at δC/δH 85.5/4.67, 54.1/3.08, 71.6/4.21 and 3.85, respectively. Moreover, the phenylcoumaran (β-5, C) were found at Cα–Hα (δC/δH 87.1/5.44), Cβ–Hβ (δC/δH 54.1/3.73) and Cγ–Hγ (δC/δH 62.9/3.72). In addition, the signals for the Cα-oxidized S (δC/δH 62.9/3.72). In addition, the signals for the Cα-oxidized S units (S′) was also observed at δC/δH 64.5/3.35. Furthermore, a minor C–H2,6 aromatic correlations from H units (δC/δH 127.9/7.19) could only be discovered at lower contour levels (not shown).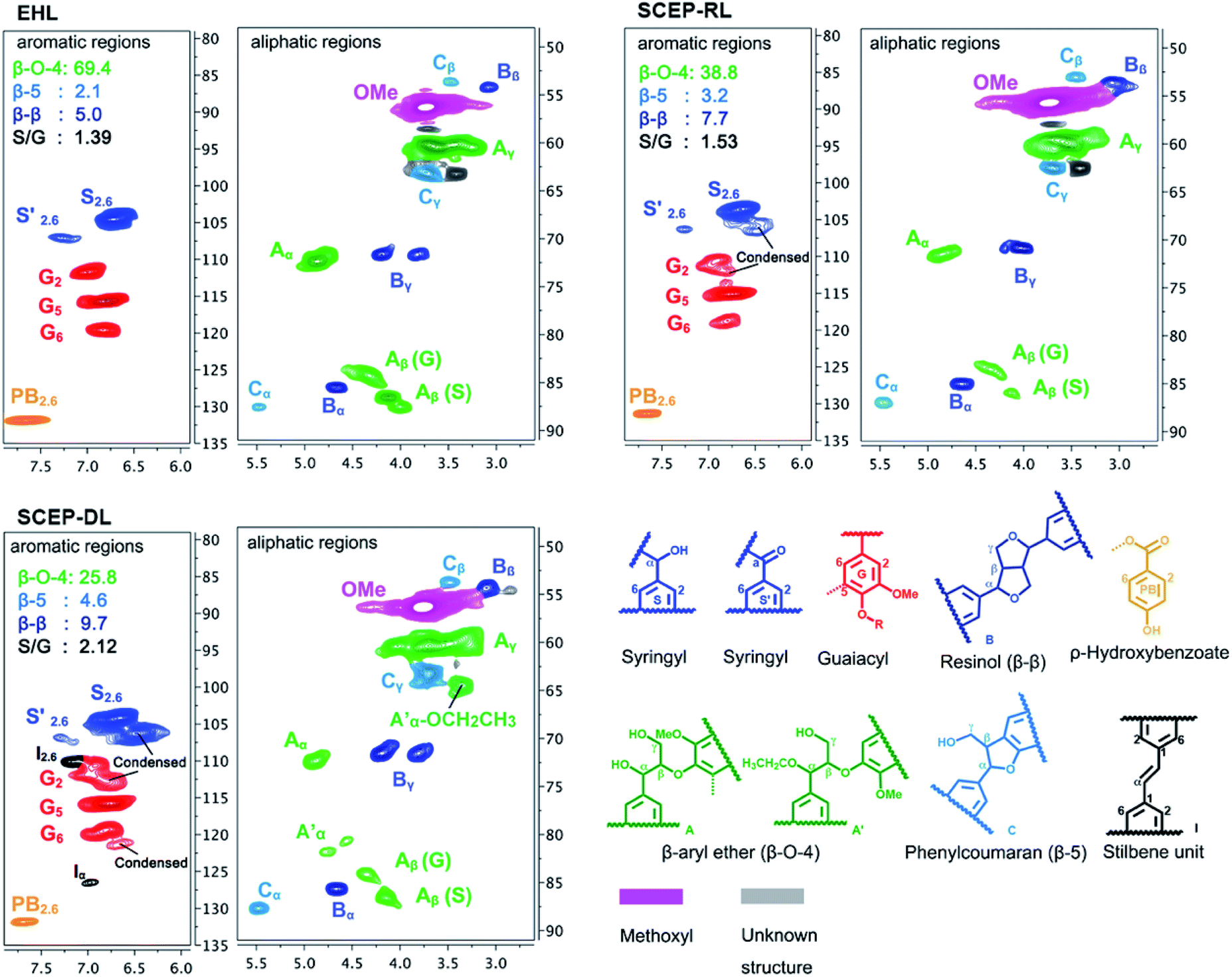 | ||
| Fig. 1 Partial 2D-HSQC NMR spectra of EHL, SCEP-RL and SCEP-DL. Volume integrals are given for the lignin side-chain structures that are color-coded to match their signal assignments in the spectrum. | ||
The structural changes of lignin during SCEP process were analyzed by comparison of the 2D-HSQC NMR spectra of EHL, SCEP-RL and SCEP-DL samples. In the aliphatic side-chain regions, two important new signals at δC/δH 64.4/3.35 and 80.6/4.55 were observed in the spectrum of SCEP-DL, which were assigned to the methylene group in α-ethoxylated β-O-4 linkages and the α-position of α-acylated β-O-4 linkages, respectively. However, these two signals were not found in the spectra of EHL and SCEP-RL preparations, indicating that the hydroxyl groups in α-position of β-O-4 linkages (A) were possibly replaced by the nucleophilic ethanol co-solvent and finally generated the α-ethoxylated β-O-4 linkages (A′).34,35
In contrast to the spectrum of EHL, some new correlations including the correlations at δC/δH 106.2/6.51 and 112.7/6.77, which were separately corresponded to the condensed S2,6 and G2 units, were observed in the spectra of SCEP-DL. More importantly, the signal at δC/δH 121.3/6.65, ascribed to the condensed G unit, was also found was also found in the spectrum of SCEP-DL. All of these results confirmed that the recondensation reaction of lignin possibly took place during the SCEP process. Moreover, two obvious correlations at δC/δH 110.0/7.16 and 126.4/6.98, which were separately assigned to the C2–H2 and Cα–Hα in the stilbene units (I), appeared in the spectrum of SCEP-DL rather than those of EHL and SCEP-RL samples. Generally, the formation of stilbene structures was mainly attributed to the degradation of β-O-4 linkages under acidic condition.31 It was previously reported that the homolytic degradation of β-O-4 linkages through an quinone methide intermediate would generate novel β-1 linkages, which were then degraded into stilbene units in acidic medium.36
The quantitative analysis of the main linkages in the lignin samples were performed on the basis of previous 2D-HSQC methods.37 As shown in Fig. 1, the content of β-O-4 linkages in EHL was 69.4/100Ar, and it decreased to 38.8/100Ar and 25.8/100Ar in SCEP-RL and SCEP-DL, respectively, suggesting that the β-O-4 linkages in SCEP-RL and SCEP-DL suffered different destruction during SCEP process. The degradation of SCEP-DL was more severely than SCEP-RL. In contrast to 5.0/100Ar β–β linkages in EHL, the contents of β–β linkages in SCEP-RL and SCEP-DL were increased to 7.7/100Ar and 9.7/100Ar, respectively. Meanwhile, the content of β-5 linkages were enhanced from 2.1/100Ar in EHL to 3.2/100Ar and 4.6/100Ar in SCEP-RL and SCEP-DL, respectively. These results confirmed that condensation probably occurred during the SCEP process and new β–β and β-5 bonds were formed.
The changes of the S/G ratio between EHL, SCEP-RL and SCEP-DL preparations were also evaluated. It can be seen that the S/G ratio in SCEP-RL (1.53) and SCEP-DL (2.12) was higher than that of original EHL (1.39). The increase of S/G ratio after SCEP process was mainly attributed to the fact that the G-type lignin was more susceptible to the condensed reaction than the S-type lignin.29
3.3. 13C-NMR analysis
The 13C-NMR spectra of three lignin preparations are depicted in Fig. S2.† The signals in the 13C-NMR spectra were carefully assigned according to previous literatures.26,27 To explore the mechanism of lignin structural transformation during SCEP process, the differences in the three spectra were profoundly investigated. It was observed that the spectra of SCEP-RL and SCEP-DL samples had a reduced signal at 152.3 ppm than EHL, which was ascribed to the C3,5 in etherified S. This result confirmed that the β-O-4 linkages were cleaved to some extent during the SCEP process. The reduced signals at 138.1 and 149.2 ppm, corresponded to the C4 of etherified S units and C3 of etherified G units, in the spectra of SCEP-RL and SCEP-DL samples, were caused by the cleavage of etherified G units after SCEP. In the spectra of SCEP-RL and SCEP-DL, the more broadened peaks at 120 and 105–110 ppm, which were assigned to the G and S units, were possibly attributed to the newly generated G and S units. This phenomenon indicated that some homologues of the common S and G units potentially formed during SCEP process.A further comparison of the integration values for the aromatic C–H (δ 123.0–103.0 ppm), aromatic C–C (δ 140.0–123.0 ppm) and aromatic C–O (δ 160.0–140.0 ppm) could give detailed information on the lignin structural features.37 As depicted in Table 3, the amounts of aromatic C–O units in SCEP-DL and SCEP-RL were reduced after SCEP, whereas their contents of aromatic C–C structures were enhanced. Considering the higher contents of β–β and β-5 linkages in SCEP-DL and SCEP-RL samples (from 2D-HSQC NMR), it was concluded that some C–C linkages were formed. This conclusion was also verified by the lower amounts of aromatic C–H in SCEP-DL (1.75/Ar) and SCEP-RL (2.11/Ar) than those in original EHL (2.20/Ar). The phenomenon was explained by the occurred condensation reactions during SCEP process, which could be resulted the replacement of C–H bonds by C–C bonds. The condensation reaction commonly was postulated in competition with the hydrolysis of β-O-4 ether linkages. It was also found that the amounts of β-O-4 linkages in SCEP-RL (0.27/Ar) and SCEP-DL (0.23/Ar) preparations were less than that in EHL sample (0.45/Ar), indicating that the β-O-4 linkages were preferentially to be hydrolyzed during the SCEP process. In addition, the contents of OCH3 in SCEP-DL and SCEP-RL samples were 1.35/Ar and 1.02/Ar and both of them were lower than those in EHL (1.39/Ar), indicating that the demethoxylation reaction of lignin probably took place during the SCEP process.
| δ (ppm) | Assignments | Quantitative results | ||
|---|---|---|---|---|
| EHL | SCEP-RL | SCEP-DL | ||
| 160–140 | Aromatic C–O | 1.97 | 1.87 | 1.24 |
| 140–123 | Aromatic C–C | 1.87 | 2.10 | 2.18 |
| 123–103 | Aromatic C–H | 2.20 | 2.11 | 1.75 |
| 91–58 | Alkyl-O | 2.93 | 1.88 | 1.81 |
| 61.2–58 | β-O-4 | 0.45 | 0.27 | 0.23 |
| 58–54 | OCH3 | 1.39 | 1.35 | 1.02 |
According to the combined results of 2D-HSQC NMR and 13C-NMR, the breakage of the β-O-4 linkages as well as demethoxylation occurred, while the β–β and β-5 linkages were formed after SCEP. Besides, comparison of the 2.93/Ar alkyl-O groups in EHL, the contents of alkyl-O groups in SCEP-RL and SCEP-DL samples were obviously reduced to 1.88/Ar and 1.81/Ar, respectively. These results implied that the majority of alkoxy side chains had been modified during SCEP process, most likely by reactions such as dehydration, shortening of the side chain and changes of alkoxy groups into carbonyl groups such as ketones, aldehydes or carboxylic acid groups (as also evidenced by the peaks at 135.5–133.7 ppm in 31P NMR spectra).
3.4. 31P-NMR analysis
The 31P-NMR spectra of phosphytilated EHL, SCEP-RL and SCEP-DL preparations are found in Fig. S3.† Based on the previous method,34 the main peaks in 31P NMR spectra were assigned. The peaks of aliphatic hydroxyl group (OH) located at the regions of 150.0–145.4 ppm, while the peaks of carboxylic OH appeared at the regions of 135.5–133.7 ppm. The peaks present at the regions of 143.7–136.9 ppm were corresponding to the phenolic OH, including S-type OH (143.7–142.2 ppm) and G-type OH (140.2–138.4 ppm). The peaks at the regions of 141.4–142.2 ppm and 143.2–144.5 ppm were ascribed to the condensed phenolic OH. The signals at the regions of 137.1–138.2 ppm were attributed to the H-type OH in lignin. The signals of cyclohexanol at the regions of 144.9–145.3 ppm were selected as the internal standard, and the amounts of various OH in lignin were calculated. The results are depicted in Table 4.| δ (ppm) | Assignment | EHL | SCEP-RL | SCEP-DL |
|---|---|---|---|---|
| 150.0–145.4 | Aliphatic OH units | 1.95 | 1.02 | 0.70 |
| 143.7–142.2 | Syringyl OH | 0.23 | 0.47 | 0.70 |
| 142.7–141.4 | Condensed phenolic OH | 0.13 | 0.34 | 0.35 |
| 142.2–138.4 | Guaiacyl OH | 0.19 | 0.39 | 0.47 |
| 138.6–136.9 | p-Hydroxy phenyl OH | 0.02 | 0.05 | 0.09 |
| 135.5–133.7 | Carboxylic OH | 0.02 | 0.14 | 0.09 |
| Total phenol | 0.57 | 1.25 | 1.61 |
As shown in Table 4, the contents of aliphatic OH in EHL, SCEP-RL and SCEP-DL samples were 1.95, 1.02 and 0.70 mmol g−1, respectively. It was concluded that the content of aliphatic OH in lignin was obviously reduced after SCEP process. Based on the conclusion of Brandt et al.38 and Hu et al.,39 the decrease of aliphatic hydroxyl groups can be attributed to the loss of the γ-methylol group, as formaldehyde and hydroxyl groups on Cα,β, or the whole side chain in general to form β-1′ linkages, which were then converted to stilbene structures (Fig. 1 Iα structure). Moreover, the amounts of carboxylic OH in SCEP-RL (0.14 mmol g−1) and SCEP-DL (0.09 mmol g−1) preparations were slightly higher than that in EHL (0.02 mmol g−1), which was attributed to the oxidation of lignin during SCEP process. The results were in accordance with the reduced contents of aliphatic OH groups. Furthermore, the contents of condensed phenolic OH in SCEP-RL were close to those in SCEP-DL, but both of them were greatly higher than those in EHL. The amounts of G-type and S-type phenolic OH were also found to be increased after SCEP process. Meanwhile, the contents of total phenolic OH groups in lignin were obviously enhanced after pretreatment, and reached up to 1.29 mmol g−1 in SCEP-DL, which were almost two times as much as that in EHL (0.53 mmol g−1). The enhanced phenolic OH indicated the occurrence of acidic cleavage of β-O-4 linkages during SCEP process. It was also observed that enhancing rate of the G-type OH in SCEP-RL and SCEP-DL was slower than that of S-type OH. This phenomenon was possibly due to that the S unit was highly involved in the formation of β-O-4 linkages in the original lignin, and most of them could be released via the acidic cleavage reaction of β-O-4 linkages. The contents of H-type OH groups in SCEP-RL and SCEP-DL were 0.05 mmol g−1 and 0.09 mmol g−1, respectively. In contrast of 0.02 mmol g−1 in EHL, the contents of H-type OH groups after pretreatment were not significantly enhanced even if the demethoxylation reaction occurred. This result could be explained by the undetectable products of demethoxylation of G-type units in 31P-NMR spectra because they were not similar to the normal H units. The similar phenomenon was found by Wen, et al.37
3.5. Molecular weight analysis
Changes in the molecular weights of lignin could provide important insights into the depolymerization of lignin during SCEP process.40 The molar mass distribution curves of EHL, SCEP-RL and SCEP-DL samples are shown in Fig. 2, in which their weight-average (Mw) molecular weights, number-average (Mn) molecular weights and polydispersity indexes (Mw/Mn, PDI) are also summarized. The Mn of EHL was 5952 g mol−1, which was reduced by 29% to 4189 g mol−1 in SCEP-RL and further by 58% to 2494 g mol−1 in SCEP-DL. It was deduced that the lignin was greatly degraded via the depolymerization after SCEP process, which were also confirmed by the decreased amounts of β-O-4 linkages in SCEP-DL and SCEP-RL samples as determined by 2D-HSQC NMR and 13C-NMR techniques. Besides the molecular weight, the PDI was also found to be decreased from 1.78 in EHL to 1.54 in SCEP-RL and 1.70 in SCEP-DL, respectively, suggesting that the structures of SCEP-RL and SCEP-DL were more homogeneous than those of EHL. Furthermore, the SCEP-DL exhibited relatively lower molecular weights and PDI value than SCEP-RL, suggesting that the depolymerization of lignin dissolving in pretreatment liquors was more severely than those exiting in the pretreated residues. This phenomenon was also verified by the lower contents of the sensitive linkages in SCEP-DL than SCEP-RL. Due to their smaller molecular weights and higher amounts of hydroxyl groups, the SCEP-DL had excellent solubility in ethanol–water solutions. Therefore, we could get the degraded lignin products with homogeneous structures from SCEP liquors, which were ideal raw materials to be functionalized into other high-value materials.36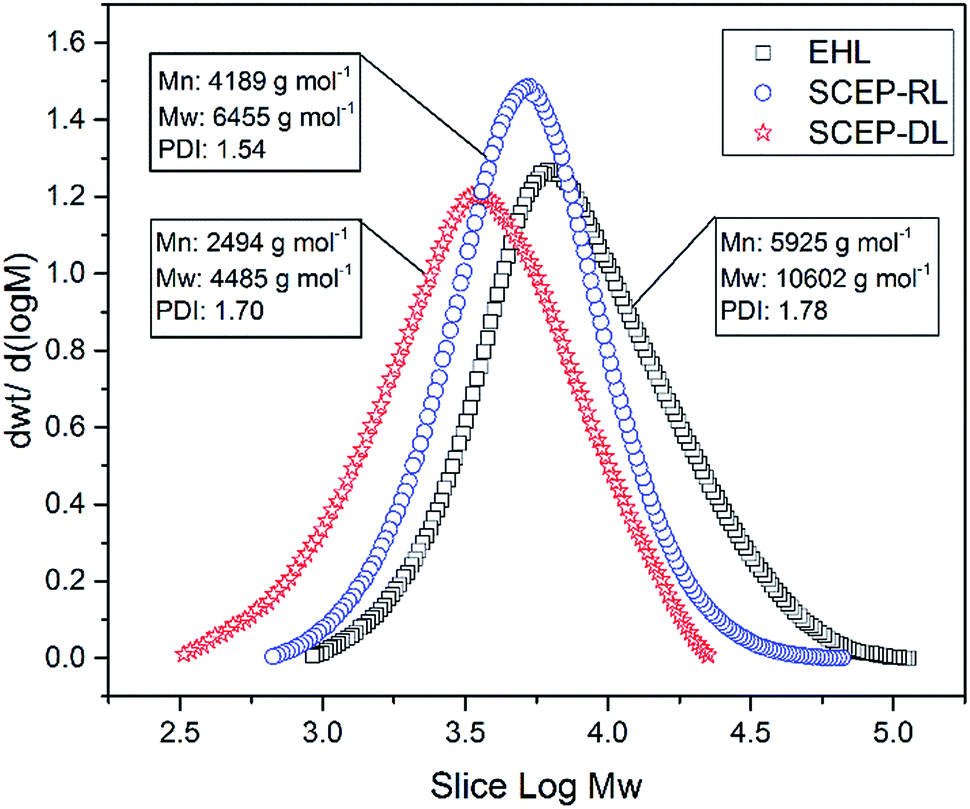 | ||
| Fig. 2 The GPC curves of EHL, SCEP-RL and SCEP-DL samples. | ||
3.6. Thermogravimetric analysis
The thermogravimetric weight loss curves (TGA) of EHL, SCEP-RL and SCEP-DL samples and their first derivative curves (DTGA) are shown in Fig. 3. It was found that the thermo-decomposition of three lignin samples took place in the temperature range of 200–600 °C. Generally, majority of aryl ether bond linkages were degraded at the temperature range of 200–350 °C. In this decomposing temperature range, the weight losses for SCEP-RL and SCEP-DL samples were separately 26.8% and 25.1%, both of which were lower than 42.7% for EHL. These results indicated that the thermal stability of SCEP-RL and SCEP-DL were higher than that of EHL at the initial decomposing phase, which were mainly attributed to their lower contents of β-O-4 linkages. Furthermore, the weight losses of SCEP-RL (13.6%) and SCEP-DL (11.0%) were higher than that of EHL (7.6%) in the decomposing temperature of 350 °C to 400 °C, the increased decomposition rate was attributed to the oxidation and degradation of side chains in lignin, i.e., carbonylation or carboxylation of aliphatic OH and dehydrogenation reaction of side chains the lignin side chain. | ||
| Fig. 3 Thermogravimetric curves and derivative thermogravimetry curves of EHL, SCEP-RL and SCEP-DL. | ||
The aromatic rings and C–C linkages (e.g., β–β and β-5 linkages) were decomposed when the temperature was above 400 °C. In the temperature of 400 °C to 600 °C, the weight losses of SCEP-RL and SCEP-DL were 14.6% and 17.5%, which were higher than that of EHL (11.9%). These results indicated that more amounts of resistant β–β and β-5 linkages were formed during SCEP process. Meanwhile, it was found that the weight values of char residues at 600 °C were 30.3% in EHL, 37.8% in SCEP-RL and 42.6% in SCEP-DL, indicating that more condensed lignin structures were generated after SCEP process, as also revealed by the aforementioned results in 13C-NMR and 2D-HSQC-NMR spectra.
In addition, the maximum decomposition temperatures (Tm) of lignin were observed to be enhanced from 289 °C in EHL to 344 °C in SCEP-RL and 349 °C in SCEP-DL, which was probably due to the generation of more stable lignin structures (e.g. condensed structures) during SCEP process.
3.7. Delignification mechanism during SCEP process
It was found that almost 79% lignin was removed from poplar chips after SCEP at 120 °C in this study. However, the delignification efficiency was only 52% after organosolv pretreatment at 165 °C, which only used the water–ethanol as pretreatment liquor without addition of ScCO2.41 It was concluded that the combined usage of ScCO2 and ethanol–water into organosolv pretreatment was beneficial to isolation of lignin from biomass, which was mainly as a result of the strongly acidic environment caused by the dissolution of ScCO2 into water. After dissolving ScCO2 into water, the carbonic acid was in situ generated and dissociated via two steps as reference with the following equations.| CO2 + H2O ↔ HCO3− + H3O+; |
| HCO3− + H2O ↔ CO32− + H3O+; |
Therefore, the hydrolysis of biomass, especially hemicelluloses and lignin, was promoted. According to the results of NMR, the mechanism of delignification during the process of SCEP was proposed and is shown in Fig. 4. The resulted included: (1) the cleavage of β-O-4 linkages would enhance the amount of phenolic OH, while the dehydration reaction could reduce the content of aliphatic OH. (2) The β-1 interlinkages were formed via the homolytic cleavage of β-O-4 with quinone methide intermediate, which then were degraded under acidic medium and generating the stilbenes structures through loss of the γ-methylol group as formaldehyde. (3) The condensation reaction commonly occurred through the couple reaction between the cationic carbons at Cα position of side chain and the free C5 or C6 position in another units of lignin another lignin unit (i.e., through the).36 (4) The demethoxylation reaction of lignin probably took place during the process of SCEP.
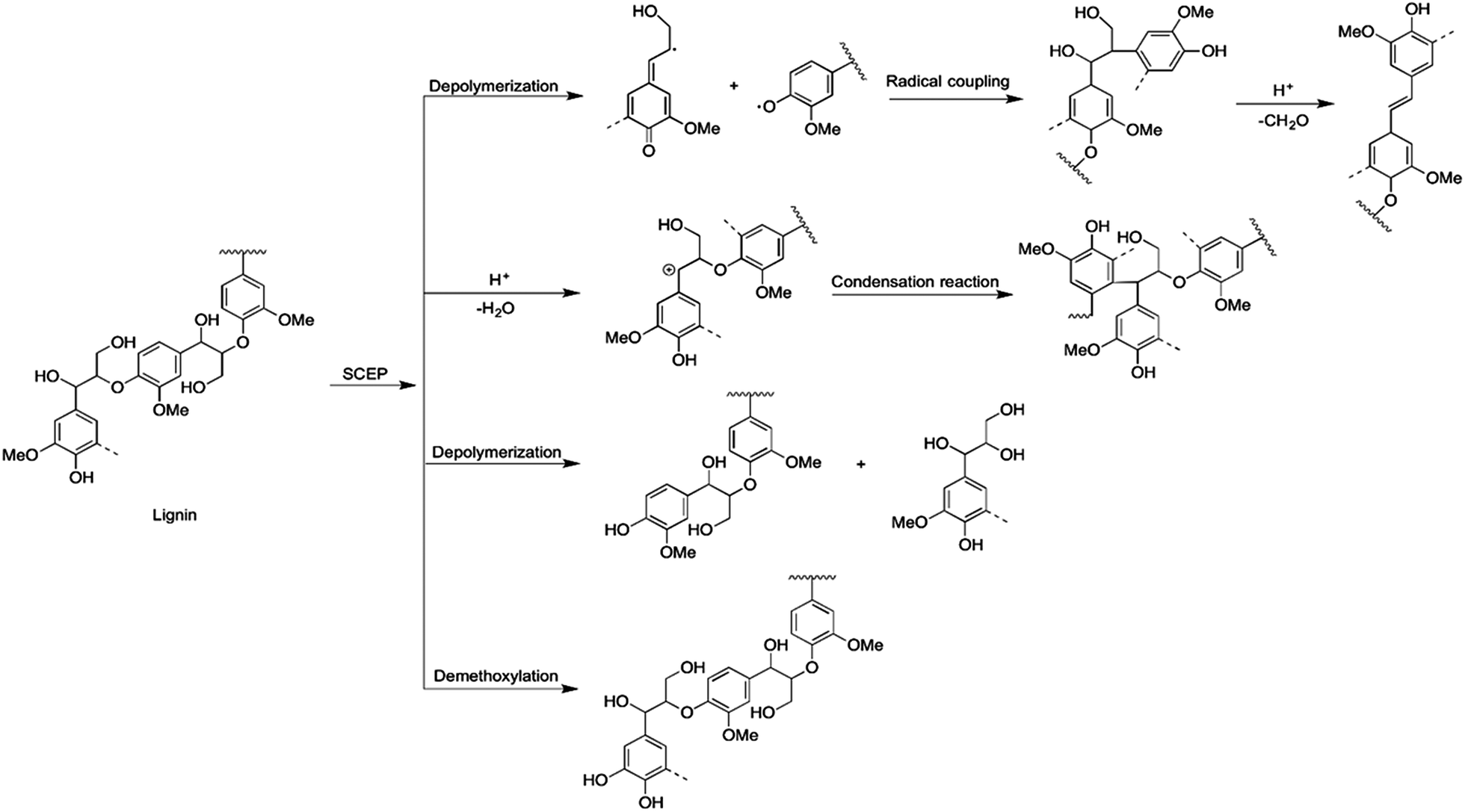 | ||
| Fig. 4 The proposed delignification reaction mechanism during SCEP process. | ||
4 Conclusions
The structural changes of lignin from poplar during SCEP process were illustrated by comparison of the chemical features of SCEP-RL and SCEP-DL samples with original EHL. After SCEP process, 79% of lignin was degraded and dissolved into the SCEP liquors. The SCEP-RL and SCEP-DL possessed higher amounts of free phenolic and carboxyl OH groups and lower molecular weights than EHL, which was mainly caused by the extensive cleavage of β-O-4 linkages, as evidenced by the quantitative results of 13C-NMR, 2D-HSQC-NMR, 31P-NMR, and GPC techniques. The founding for structural changes of lignin during SCEP process would give some highlights for optimizing, controlling the pretreatment process and obtaining the biomass residues with easily hydrolyzing property. Moreover, the well-defined and active lignin polymers isolated from biomass by SCEP could be utilized as the raw materials to develop value-added chemical products.Acknowledgements
The authors are grateful for the support by National Natural Science Foundation of China (No 31170554 and No 31470604), the Fundamental Research Funds for Dalian Polytechnic University (No 2016J001) and the 6th Session of Geping Green Action-123 Project of Liaoning Environmental Research and Education (No. CEPF2013-123-1-10).Notes and references
- R. Ravindran and A. K. Jaiswal, Bioresour. Technol., 2016, 199, 92–102 CrossRef CAS PubMed.
- C. Liu, J. Hu, H. Zhang and R. Xiao, Fuel, 2016, 182, 864–870 CrossRef CAS.
- S. K. Jang, H. Y. Kim, H. S. Jeong, J. Y. Kim, H. Yeo and I. G. Choi, Renewable Energy, 2016, 87, 599–606 CrossRef CAS.
- H. Yang, Y. Xie, X. Zheng, Y. Pu, F. Huang, X. Meng, W. Wu, A. Ragauskas and L. Yao, Bioresour. Technol., 2016, 207, 361–369 CrossRef CAS PubMed.
- C. S. Lancefield, I. Panovic, P. J. Deuss, K. Barta and N. J. Westwood, Green Chem., 2017, 19, 202–214 RSC.
- S. N. Sun, S. L. Sun, X. F. Cao and R. C. Sun, Bioresour. Technol., 2016, 199, 49–58 CrossRef CAS PubMed.
- S. McIntosh and T. Vancov, Bioresour. Technol., 2010, 101, 6718–6727 CrossRef CAS PubMed.
- M. Brahim, N. Boussetta, N. Grimi, E. Vorobiev, I. Zieger-Devin and N. Brosse, Ind. Crops Prod., 2017, 95, 643–650 CrossRef CAS.
- T. A. Lloyd and C. E. Wyman, Bioresour. Technol., 2005, 96, 1967–1977 CrossRef CAS PubMed.
- K. Zhang, Z. Pei and D. Wang, Bioresour. Technol., 2016, 199, 21–33 CrossRef CAS PubMed.
- Z. Zhang, M. D. Harrison, D. W. Rackemann, W. O. S. Doherty and I. M. O'Hara, Green Chem., 2016, 18, 360–381 RSC.
- L. V. Daza Serna, C. E. Orrego Alzate and C. A. Cardona Alzate, Bioresour. Technol., 2016, 199, 113–120 CrossRef CAS PubMed.
- M. Paljevac, M. Primožič, M. Habulin, Z. Novak and Ž. Knez, J. Supercrit. Fluids, 2007, 43, 74–80 CrossRef CAS.
- G. Simeoni, T. Bryk, F. A. Gorelli, M. Krisch, G. Ruocco, M. Santoro and T. Scopigno, Nat. Phys., 2010, 6, 503–507 CrossRef CAS.
- H. Lv, L. Yan, M. Zhang, Z. Geng, M. Ren and Y. Sun, Chem. Eng. Technol., 2013, 36, 1899–1906 CrossRef CAS.
- D. Pasquini, M. T. B. Pimenta, L. H. Ferreira and A. A. S. Curvelo, J. Supercrit. Fluids, 2005, 34, 125–131 CrossRef CAS.
- E. Kiran and H. Balkan, J. Supercrit. Fluids, 1994, 7, 75–86 CrossRef CAS.
- S. Cheng, I. D'cruz, M. Wang, M. Leitch and C. Xu, Energy Fuels, 2010, 24, 4659–4667 CrossRef CAS.
- H. Lü, M. Ren, M. Zhang and Y. Chen, Chin. J. Chem. Eng., 2013, 21, 551–557 CrossRef.
- D. Pasquini, M. T. B. Pimenta, L. H. Ferreira and A. A. D. S. Curvelo, J. Supercrit. Fluids, 2005, 36, 31–39 CrossRef CAS.
- M. Gao, F. Xu, S. Li, X. Ji, S. Chen and D. Zhang, Biosystems Engineering, 2010, 106, 470–475 CrossRef.
- N. Narayanaswamy, A. Faik, D. J. Goetz and T. Gu, Bioresour. Technol., 2011, 102, 6995–7000 CrossRef CAS PubMed.
- N. Srinivasan and L. K. Ju, Bioresour. Technol., 2010, 101, 9785–9791 CrossRef CAS PubMed.
- J. L. Rahikainen, R. Martin-Sampedro, H. Heikkinen, S. Rovio, K. Marjamaa, T. Tamminen, O. J. Rojas and K. Kruus, Bioresour. Technol., 2013, 133, 270–278 CrossRef CAS PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Laboratory Analytical Procedures, 2008, 1617 Search PubMed.
- T. Q. Yuan, S. N. Sun, F. Xu and R. C. Sun, J. Agric. Food Chem., 2011, 59, 6605–6615 CrossRef CAS PubMed.
- Y. Tobimatsu, F. Chen, J. Nakashima, L. L. Escamilla-Trevino, L. Jackson, R. A. Dixon and J. Ralph, Plant Cell, 2013, 25, 2587–2600 CrossRef CAS PubMed.
- B. L. Xue, J. L. Wen, F. Xu and R. C. Sun, J. Appl. Polym. Sci., 2013, 129, 434–442 CrossRef CAS.
- Y. Pu, S. Cao and A. J. Ragauskas, Energy Environ. Sci., 2011, 4, 3154 CAS.
- Y. X. An, N. Li, H. Wu, W. Y. Lou and M. H. Zong, ACS Sustainable Chem. Eng., 2015, 3, 2951–2958 CrossRef CAS.
- B. Wang, X. J. Shen, J. L. Wen and R. C. Sun, RSC Adv., 2016, 6, 57986–57995 RSC.
- C. G. Yoo, S. Zhang and X. Pan, RSC Adv., 2017, 7, 300–308 RSC.
- F. Zikeli, T. Ters, K. Fackler, E. Srebotnik and J. Li, Ind. Crops Prod., 2016, 85, 309–317 CrossRef CAS.
- Y. Z. Guo, J. H. Zhou, J. L. Wen, G. W. Sun and Y. J. Sun, Ind. Crops Prod., 2015, 76, 522–529 CrossRef CAS.
- J. L. Wen, B. L. Xue, S. L. Sun and R. C. Sun, J. Chem. Technol. Biotechnol., 2013, 88, 1663–1671 CrossRef CAS.
- B. B. Hallac, Y. Pu and A. J. Ragauskas, Energy Fuels, 2010, 24, 2723–2732 CrossRef CAS.
- J. L. Wen, T. Q. Yuan, S. L. Sun, F. Xu and R. C. Sun, Green Chem., 2014, 16, 181–190 RSC.
- A. Brandt, L. Chen, B. E. van Dongen, T. Welton and J. P. Hallett, Green Chem., 2015, 17, 5019–5034 RSC.
- G. Hu, C. Cateto, Y. Pu, R. Samuel and A. J. Ragauskas, Energy Fuels, 2012, 26, 740–745 CrossRef CAS.
- J. Li, G. Henriksson and G. Gellerstedt, Bioresour. Technol., 2007, 98, 3061–3068 CrossRef CAS PubMed.
- N. Kang, L. Zhong, L. F. Hui, C. L. Si, C. Li, T. Zhao and S. T. Mao, BioResources, 2011, 7, 578–592 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26122a |
| This journal is © The Royal Society of Chemistry 2017 |