
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient and durable platinum nanocatalysts stabilized by thiol-terminated poly(N-isopropyl acrylomide) for selective hydrogenation of halonitrobenzene to haloaniline
Wenjun Yuab,
Lan-Lan Lou*ab,
Shanshan Liab,
Tianyuan Maab,
Lezi Ouyangab,
Li Fengab and
Shuangxi Liu*abc
aInstitute of New Catalytic Materials Science and Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), School of Materials Science and Engineering, Nankai University, Tianjin 300350, China. E-mail: lllou@nankai.edu.cn; sxliu@nankai.edu.cn; Fax: +86 22 23509005; Tel: +86 22 8535 8599 Tel: +86 22 2350 9005
bNational Institute for Advanced Materials, Nankai University, Tianjin 300350, China
cCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, China
First published on 3rd January 2017
Abstract
In this paper, the selective hydrogenation of halonitrobenzenes (HNBs) to haloanilines (HANs) under mild conditions catalyzed by well-dispersed Pt nanoparticles protected by thiol-terminated poly(N-isopropyl acrylomide) (PNIPAM-SH) was firstly investigated. The polymer not only protected the Pt nanoparticles, but also inhibited the highly active Pt catalyst from producing undesired hydrodehalogenation products through anchoring the thiol groups to the surface of Pt nanoparticles. Thus high selectivities to HANs were achieved over this modified Pt catalyst for a variety of HNBs with satisfactory catalytic activities. Especially, the selectivity to HANs showed no obvious loss with the prolonging of the reaction time. Moreover, the recycling experiment showed that this Pt nanocatalyst was easier to recover and reuse based on the cononsolvency of PNIPAM-SH. Excellent stability and reusability were presented over this catalyst, and both the catalytic activity and selectivity were well maintained after fourteen runs.
1. Introduction
Haloanilines (HANs) are an important class of fine chemicals and are widely used in the synthesis of pharmaceuticals, dyes, herbicides, pesticides, and so on.1 These compounds are generally produced by selective hydrogenation of halonitrobenzenes (HNBs) catalyzed by transition metals, which is regarded as a high atom efficiency and environmentally friendly process.2 However, catalytic hydrodehalogenation of HNBs as a side reaction often occurs over most metal catalysts, which significantly limits the selectivity to HANs, especially for iodonitrobenzenes (INBs) and bromonitrobenzenes (BNBs) with a weaker carbon–halogen bond than chloronitrobenzenes (CNBs). Compared with other transition metals such as Pd,3,4 Au,5–7 Ag,8 Ir,9–11 Ru,12 Ni,13–15 and Co,16 Pt has been one of the most promising metal catalysts for selective hydrogenation of HNBs due to high catalytic activity in nitro reduction and a certain degree of selectivity to HANs.17 Several strategies have been developed to improve the selectivity of Pt catalyst, although at the expense of catalytic activity in many cases, such as alloying,18,19 using bimetallic catalysts,20,21 adjusting metal/support interaction,22–26 surface modification via ligands or modifier reagents.27–33 Among these strategies, the last one represented a widely used and powerful tool for improving the selectivity to HANs through control of the electronic properties of the catalytically active sites. Yang and Liu reported that some metal cations such as Ni2+, Fe3+ and Co2+ introduced to PVP–Pt colloidal clusters had a favorable effect on the selectivity to o-CAN in the catalytic hydrogenation of o-CNB.27 Motoyama et al. applied n-octylamine as poisoning reagent for Pt nanoparticles dispersed on nanocarbon fiber and high selectivities were obtained in the hydrogenation of HNBs.29 Recently, Lara et al. reported the Pt nanoparticles stabilized by N-heterocyclic carbine for the selective hydrogenation of different nitroaromatics under mild conditions and high levels of activity and selectivity were obtained.33 However, these modifiers usually increased the complexity of the reaction system and could not be completely avoided leaching into the products. Therefore, new surface modification of Pt nanocatalyst with excellent catalytic performance and high reusability is highly significant in the selective hydrogenation of HNBs.Poly(N-isopropylacrylamide) (PNIPAM) is the most widely studied thermosensitive polymer, and exhibits a lower critical solution temperature (LCST) of ∼32 °C, at which the single-chain polymer undergoes a reversible change from a soluble coil to an insoluble globule in water.34 This polymer has received more and more attention due to its widely applications in targeted drug delivery,35,36 catalysis,37–40 and so on. It was particularly necessary to point out that PNIPAM exhibits cononsolvency in mixed aqueous solutions, that is to say, the LCST of aqueous PNIPAM solution is remarkably decreased when being added an appropriate proportion of polar organic solvents such as methanol, ethanol and tetrahydrofuran.41,42 Thus, in this case, PNIPAM is easily precipitated from the reaction system at room temperature and below it. The phenomenon seems very interesting and surprising, because PNIPAM is well soluble in water as well as in polar solvents separately. This cononsolvency property of PNIPAM has few application in the recovery of colloid catalysts, which provides an effective and simple way to recycle the PNIPAM related colloid catalysts in polar organic solvents.
Here we would like to report the selective hydrogenation of HNBs catalyzed by colloidal Pt catalyst stabilized by thiol-terminated PNIPAM (PNIPAM-SH). The polymer here can not only stabilize the Pt nanoparticles, but also inhibit the highly active catalyst from producing hydrodehalogenation products through the strong interaction between the thiol groups and unsaturated surface of Pt nanoparticles. To the best of our knowledge, this is the first report on the utilization of thermosensitive polymer-modified Pt catalyst for selective hydrogenation of HNBs. High and steady selectivity to HANs was achieved over this Pt nanocatalyst. In addition, this catalyst could be easily recovered based on the cononsolvency of PNIPAM-SH and exhibited high durability in the selective hydrogenation reaction due to the strong interaction between PNIPAM-SH and Pt nanoparticles.
2. Experimental
2.1 General
Hexachloroplatinic acid hexahydrate (H2PtCl6·6H2O) and N-isopropylacrylamide (NIPAM) of analytical grade were supplied by Aldrich, and the monomer was recrystallized from n-hexane. 2,2-Azobisisobutyronitrile (AIBN) was purchased from Wako Pure Chemical Ind and recrystallized from pure ethanol before use. Halonitrobenzenes (HNBs) of analytical grade were obtained from J&K Chemical and used without further purification.The mean molecular weight and polydispersity of polymers were measured by gel permeation chromatography (GPC) system equipped with a Waters 1525 pump and Waters Styragel columns (MW range 3000–600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), using tetrahydrofuran as eluent at 35 °C and polystyrenes as calibration standards. Transmission electron microscopy (TEM) photographs were taken by using a Philips Tecnai G2F20 electron microscope operated at 200 kV. X-ray photoelectron spectroscopy (XPS) analysis was performed on an Axis Ultra DLD instrument, and the spectra were recorded with an Al Kα X-ray source using C 1s as a reference for binding energy. Inductively coupled plasma-atomic emission spectrometry (ICP-AES) analysis was measured with a PerkinElmer Optima 8300 spectrometer for the Pt contents in the catalysts and metal leaching into products. The products of selective hydrogenation reaction were analyzed by gas chromatography (GC) on a FL9790II gas chromatograph equipped with a flame ionization detector and a weak polarity capillary column (SE-30, 30 m × 0.53 mm × 0.50 μm). The injection and detector temperatures were set at 543 K with the column temperature of 383–413 K.
000 g mol−1), using tetrahydrofuran as eluent at 35 °C and polystyrenes as calibration standards. Transmission electron microscopy (TEM) photographs were taken by using a Philips Tecnai G2F20 electron microscope operated at 200 kV. X-ray photoelectron spectroscopy (XPS) analysis was performed on an Axis Ultra DLD instrument, and the spectra were recorded with an Al Kα X-ray source using C 1s as a reference for binding energy. Inductively coupled plasma-atomic emission spectrometry (ICP-AES) analysis was measured with a PerkinElmer Optima 8300 spectrometer for the Pt contents in the catalysts and metal leaching into products. The products of selective hydrogenation reaction were analyzed by gas chromatography (GC) on a FL9790II gas chromatograph equipped with a flame ionization detector and a weak polarity capillary column (SE-30, 30 m × 0.53 mm × 0.50 μm). The injection and detector temperatures were set at 543 K with the column temperature of 383–413 K.
2.2 Catalyst preparation
290 with a polydispersity of 1.59 as determined by GPC.
 | ||
| Scheme 1 Synthesis of PNIPAM-SH by RAFT polymerization. | ||
For comparison, PNIPAM was also prepared by polymerization of NIPAM under the same conditions in the absence of RAFT agent. The obtained PNIPAM sample possessed a mean molecular weight of 28130 with a polydispersity of 2.32 as determined by GPC.
2.3 Catalytic reaction
Typically, the required amount of Pt colloid (1.9 × 10−3 mmol Pt) dispersed in 2.0 mL of ethanol and HNBs (2 mmol) were added into a glass liner tube, then transferred to a stainless steel autoclave (30 mL) and purged with pure hydrogen. After that, the reactor was conducted at 313 K with 10 bar of hydrogen and the reaction was lasted for a proper time. When the reaction was over, a certain amount of water was added to precipitate the colloid Pt catalyst from the reaction system based on the cononsolvency of polymer. The catalyst was subjected to a new run without further treatment after phase separation. The products were obtained by simple ethyl ether extraction, and the conversion of HNB and the selectivity to HAN were determined by GC-FID.3. Results and discussion
3.1 Catalyst characterization
The as-synthesized polymer protected Pt nanocatalysts Pt@PNIPAM-SH and Pt@PNIPAM were submitted to TEM characterization. As shown in Fig. 1, these two catalysts were uniformly dispersed and no aggregation of Pt nanoparticles was observed. Fig. 1b and d showed these two catalysts have sharp size distribution curves in the range of 0.6–3.0 nm, suggesting a very narrow size distribution of Pt particles. The average Pt particle diameters were about 1.7 nm and 1.8 nm for Pt@PNIPAM-SH and Pt@PNIPAM, respectively.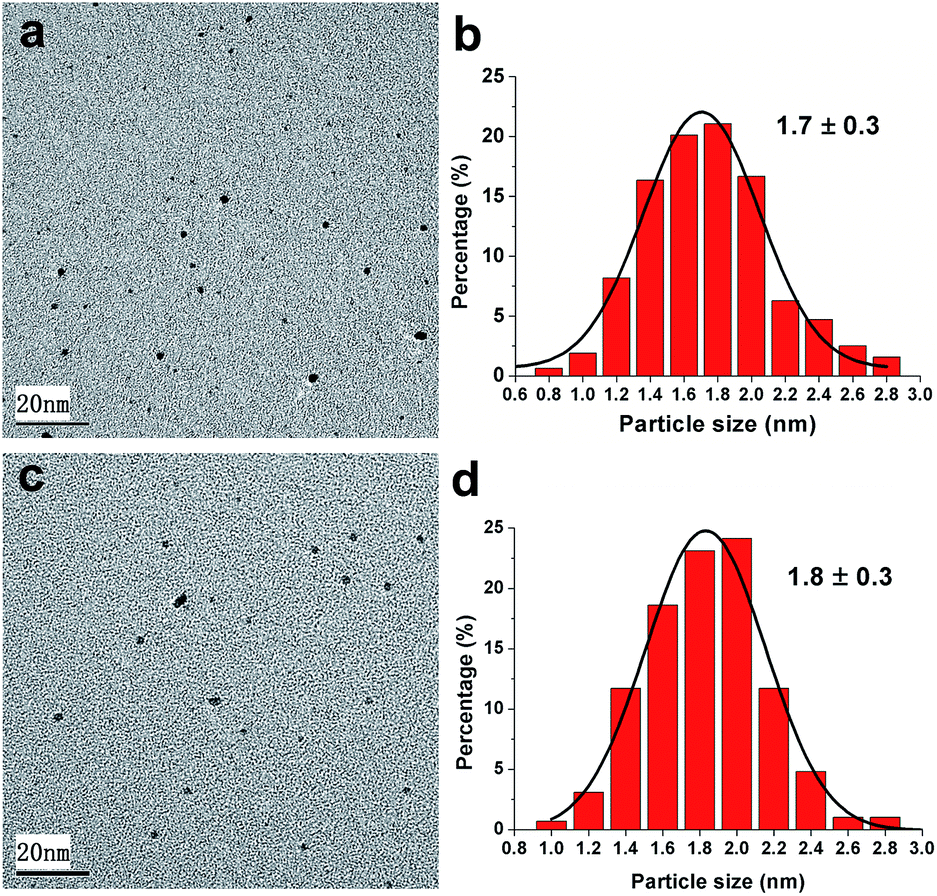 | ||
| Fig. 1 TEM micrographs and particles size distributions of Pt nanoparticles in (a and b) Pt@PNIPAM-SH and (c and d) Pt@PNIPAM. | ||
The two Pt nanocatalysts Pt@PNIPAM-SH and Pt@PNIPAM were characterized by XPS. Fig. 2 shows the high-resolution Pt 4f XPS spectra of Pt@PNIPAM-SH and Pt@PNIPAM. It could be observed that the binding energies of Pt 4f5/2 and Pt 4f7/2 in Pt@PNIPAM were 74.7 and 71.4 eV, respectively. There were no Pt 4f5/2 and Pt 4f7/2 peaks derived from Pt2+ species in the Pt 4f spectrum, which indicated that Pt2+ was entirely reduced to Pt0. Similar results were observed for Pt@PNIPAM-SH with a slightly negative shift of the Pt 4f5/2 and Pt 4f7/2 binding energies (by 0.2 and 0.1 ev, respectively).
 | ||
| Fig. 2 High-resolution XPS spectra of Pt 4f region for Pt@PNIPAM-SH and Pt@PNIPAM. | ||
3.2 Catalytic performance
The as-synthesized catalyst Pt@PNIPAM-SH was evaluated in the selective hydrogenation of HNBs under mild conditions with hydrogen pressure of 10 bar and reaction temperature of 313 K. For comparison, Pt@PNIPAM was also investigated under the same reaction conditions.The catalytic performance of the two catalysts Pt@PNIPAM-SH and Pt@PNIPAM was compared with p-CNB as a model substrate. As shown in Fig. 3, Pt@PNIPAM showed a higher conversion of 36.4% in 15 min and a complete conversion was achieved in 90 min. Compared with Pt@PNIPAM, Pt@PNIPAM-SH exhibited a decrease reaction rate and a lower conversion of 26.7% was attained in 15 min. Thus the TOF values of Pt@PNIPAM and Pt@PNIPAM-SH were 1508 h−1 and 1106 h−1, respectively, and the inferior catalytic activity for the latter could be attributed to the partial poisoning of Pt catalytic sites by thiol groups. In term of selectivity of products, the selectivity to p-CAN over Pt@PNIPAM-SH remained stable at 99.9% within 2 h. While for Pt@PNIPAM, the selectivity was gradually decreased with reaction time and a relatively low selectivity to p-CAN of 85.7% was obtained after 1.5 h of reaction time. In summary, the modification with PNIPAM-SH could improve the selectivity of Pt nanocatalyst, although at a slight expense of reactivity, compared to PNIPAM.
 | ||
| Fig. 3 Dependence of conversion and selectivity on the reaction time in the selective hydrogenation of p-CNB. Reaction conditions: 2.0 mL EtOH; 2 mmol p-CNB, 1.9 × 10−3 mmol Pt, 10 bar H2, 40 °C. | ||
The other results of selective hydrogenation of HNBs are summarized in Table 1. It could be found that the selectivity to the undesired dechlorination product over Pt@PNIPAM for selective hydrogenation of p-CNB increased to 42.3% (Table 1, entry 2) when the reaction time was prolonged to 8 h. In addition, it is notable that Pt@PNIPAM-SH could effectively prevent the dechlorination of p-CAN and the selectivity to p-CAN was maintained 99.6% from 99.9% when prolonging the reaction time to 8 h (Table 1, entries 3 and 4). These results indicated that the catalytically active Pt sites in Pt@PNIPAM-SH, which were modified by the polymer PNIPAM-SH through the strong interaction between the thiol groups and unsaturated surface of Pt nanoparticles, were more suitable for selective hydrogenation of HNBs with a high and steady selectivity to HANs. The obtained high selectivity was mainly attributed to the moderate poisoning of Pt catalytically active sites by thiol groups in the terminal of polymer. Apart from these, the steric hindrance effect of Pt surface that was modified by PNIPAM-SH would prevent the flat adsorption of the substrate via the benzene ring,32 and consequently avoid the exposure of the C–Cl groups toward the active sites during hydrogenation of nitro groups.
|
||||||
|---|---|---|---|---|---|---|
| Entry | X | Catalyst | Time (h) | Conversionb (%) | Selectivity (%) | |
| HAN | AN | |||||
| a Reaction conditions: 2.0 mL EtOH; 2 mmol HNB, 1.9 × 10−3 mmol Pt, 10 bar H2, 40 °C.b Conversion of HNB. | ||||||
| 1 | p-Cl | Pt@PNIPAM | 1.5 | 100 | 85.7 | 10.5 |
| 2 | p-Cl | Pt@PNIPAM | 8 | 100 | 57.7 | 42.3 |
| 3 | p-Cl | Pt@PNIPAM-SH | 2 | 100 | 99.9 | 0.1 |
| 4 | p-Cl | Pt@PNIPAM-SH | 8 | 100 | 99.6 | 0.4 |
| 5 | m-Cl | Pt@PNIPAM-SH | 2 | 100 | 99.8 | 0.2 |
| 6 | m-Cl | Pt@PNIPAM | 1.5 | 100 | 92.1 | 7.9 |
| 7 | o-Cl | Pt@PNIPAM-SH | 2.5 | 100 | 99.7 | 0.3 |
| 8 | p-Br | Pt@PNIPAM-SH | 2 | 100 | 98.8 | 1.2 |
| 9 | p-Br | Pt@PNIPAM | 2 | 100 | 83.3 | 16.8 |
| 10 | m-Br | Pt@PNIPAM-SH | 2 | 100 | 98.8 | 1.2 |
| 11 | o-Br | Pt@PNIPAM-SH | 3 | 100 | 98.6 | 1.4 |
| 12 | p-I | Pt@PNIPAM-SH | 4 | 100 | 97.3 | 2.7 |
| 13 | p-I | Pt@PNIPAM | 4 | 100 | 73.3 | 26.7 |
| 14 | m-I | Pt@PNIPAM-SH | 6 | 100 | 92.9 | 7.1 |
| 15 | o-I | Pt@PNIPAM-SH | 10 | 100 | 83.7 | 16.3 |

With the same reaction conditions, we extended the scope of substrate to various HNBs to examine the generality of the reaction. In all cases, the desired HANs were obtained in satisfactory and high selectivity over Pt@PNIPAM-SH. Especially for a series of CNBs and BNBs, high selectivities of 98.5% above were obtained for CANs and BANs, respectively, along with a complete conversion achieved within 3 h (Table 1, entries 3, 5, 7, 8, 10 and 11). The selective hydrogenation of INBs (Table 1, entries 12, 14 and 15) was slightly worse than CNBs and BNBs, which could be due to the weaker C–I bond compared with C–Br and C–Cl bonds. Inferior selectivities of 83.7–93.7% to IANs were furnished with a longer reaction time of 4–10 h.
In addition, the position of halogeno substituent was found to have an obvious effect on the catalytic activity and product selectivity. It could be seen that the hydrogenation of o-HNBs required a longer reaction time and afforded more dehalogenation side products, compared with p-HNBs or m-HNBs. The spatial adjacence of two reducible functional groups in o-HNBs would increase the contact probability between halogeno group and catalytically active site, which resulted in dehalogenation during the hydrogenation of nitro-group. Apart from this, o-HNBs, especially o-INB, possessed comparatively larger molecule bulks, which would lead to higher steric hindrance for the reaction of o-HNBs on the surface of polymer-modified Pt nanoparticles, resulted in declined catalytic activity.
The comparison experiments showed that besides p-CNB, other substrates catalyzed by Pt@PNIPAM also gave obviously inferior selectivity to HANs compared with Pt@PNIPAM-SH (Table 1, entries 6, 9 and 13 vs. 5, 8 and 12). This further verified that the thiol groups played an irreplaceable role in inhibiting the dehalogenation side reaction.
3.3 Catalyst recycling and stability
The thermosensitive polymers PNIPAMs are well soluble in aqueous solution below the LCST at about 32 °C. Compared to water, these polymers are more easily soluble in many polar organic solvents such as alcohol, tetrahydrofuran, dioxane and acetic acid, however they show no thermosensitivity in these pure solvents. While in a mixture of water and polar organic solvent, a uniquely thermosensitive behavior was observed for PNIPAM polymers with a distinctly depressed LCST. For instance, the LCST of PNIPAM in a mixture solvent of water and methanol was reduced to −7.5 °C when the volume fraction of methanol is 0.55.41 This phenomenon has been called “cononsolvency”. In this case, PNIPAM polymers are insoluble and precipitated in mixed solvent at room temperature and below it. The catalyst Pt@PNIPAM-SH also showed cononsolvency in the mixture of water and ethanol. It could be observed that Pt@PNIPAM-SH was well soluble in ethanol (Fig. 4A), and a phase separation behavior occurred when water was added at room temperature (Fig. 4B). Thus after each hydrogenation reaction, the catalyst could be easily separated from the reaction mixture by adding deionized water at room temperature, and then well redispersed in ethanol for a new run. The cononsolvency of PNIPAM polymers provides a convenient and effective method to recycle Pt nanocatalysts in polar organ solvent. | ||
| Fig. 4 Photographs of Pt@PNIPAM-SH in ethanol (A) and in a water/ethanol mixture at room temperature (B). | ||
The recycling experiment of catalyst was carried out using p-CNB as a model substrate, in order to assess the reusability of the Pt@PNIPAM-SH, and the results are shown in Fig. 5A. It could be seen that the catalyst Pt@PNIPAM-SH could be reused for twelve runs and no decrease in both p-CNB conversion and p-CAN selectivity was observed. After twelve runs, the conversion gradually decreased, but the selectivity to p-CAN remained stable and higher than 99%. The results suggested that the Pt@PNIPAM-SH was highly stable and durable for the reaction. For the purpose of comparison, the Pt@PNIPAM catalyst was also studied in the recycling experiment. As shown in Fig. 5B, Pt@PNIPAM displayed obviously inferior catalytic stability for the selective hydrogenation of p-CNB. Both the p-CNB conversion and p-CAN selectivity decreased in the fifth run. The compared recycling experiments demonstrated that the thiol-terminated polymer can efficiently promote the stability and reusability of Pt nanocatalysts. As far as we know, the reusability achieved over Pt@PNIPAM-SH is among the best ones obtained by transition metal catalysts for selective hydrogenation of HNBs.2,9,15,21,28,30 The excellent stability and durability of Pt@PNIPAM-SH could be owing to the strong interaction between thiol group in the terminal of polymer and unsaturated surface of Pt nanoparticles as well as the good cononsolvency of thermosensitive polymer.
 | ||
| Fig. 5 Recycle studies of Pt@PNIPAM-SH (A) and Pt@PNIPAM (B) in the selective hydrogenation of p-CNB. Reaction conditions: 2 mmol HNB, 1.9 × 10−3 mmol Pt, H2 pressure of 10 bar, reaction temperature of 40 °C, reaction time of 2 h for Pt@PNIPAM-SH and 1.5 h for Pt@PNIPAM, respectively. | ||
In order to further explore the stability of the Pt nanocatalysts during the reaction process, Pt@PNIPAM-SH after five and ten runs were characterized by TEM (Fig. 6a and b). It could be found that several particles formed small aggregates along with many monodispersed particles after the 5th run, and dozens of particles formed large aggregates along with much less monodispersed nanoparticles after the 10th run. While only a very slight increase in particle size from 1.7 ± 0.3 nm to 2.0 ± 0.4 nm was observed after ten runs (Fig. 6c). Therefore, this form of aggregation could be due to the formation of polymer aggregates and does not mean Pt nanoparticles was obviously grow larger. This suggested that the Pt nanoparticles in Pt@PNIPAM-SH were highly stable during the recycling process, which could explain the excellent reusability of this catalyst. In the case of Pt@PNIPAM, it was found that large aggregates formed only after two runs (Fig. 6d), but the Pt particle size had no obvious change. After five runs, the catalyst was seriously aggregated to form a superstructure completely including hundreds of particles, and the histogram of particle size distribution showed an increased Pt particle size of 4.3 ± 1.0 nm. These results implied that the thiol-terminated polymer could efficiently stabilize the Pt nanoparticles, and thus enhance the reusability of the catalyst Pt@PNIPAM-SH.
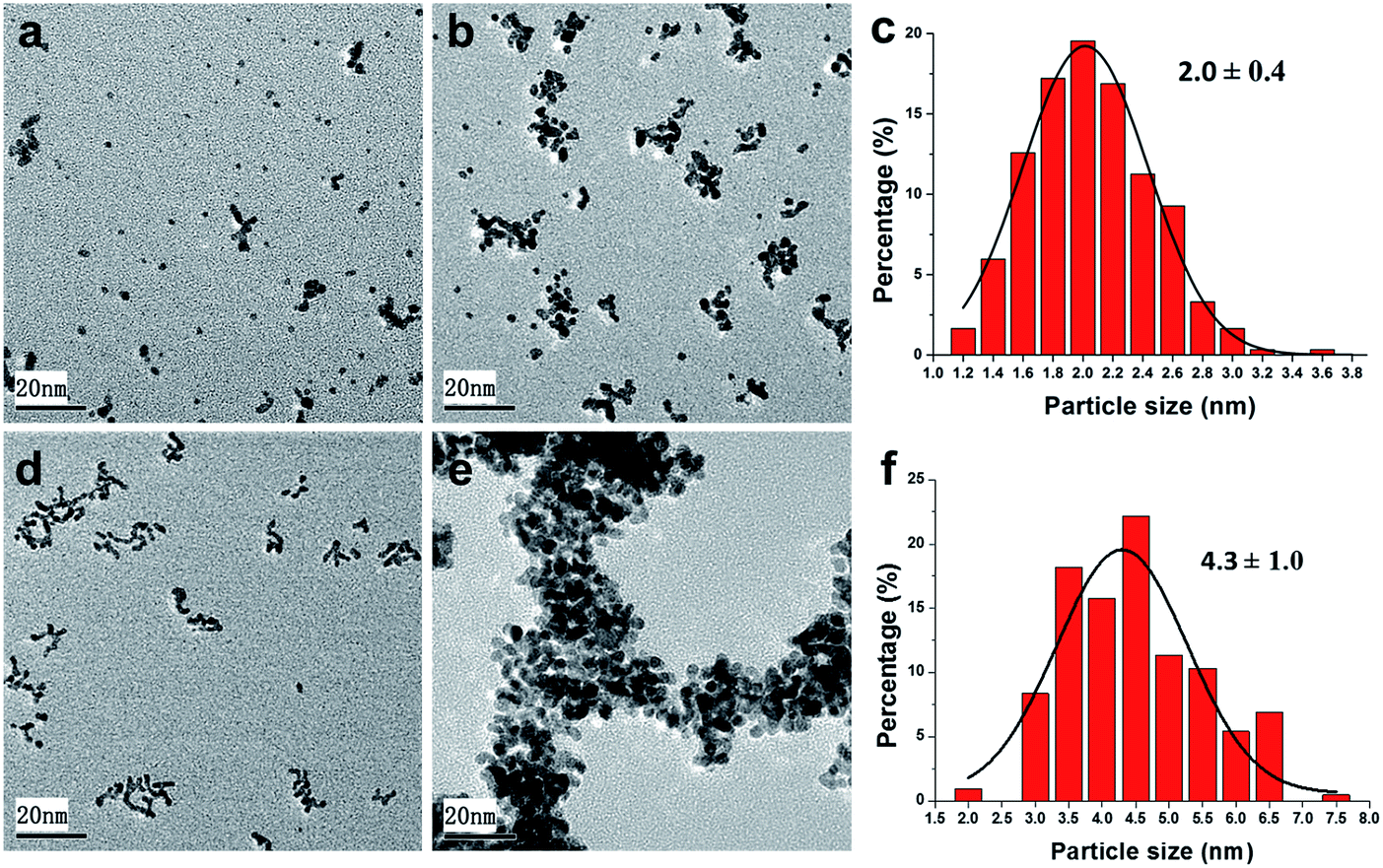 | ||
| Fig. 6 TEM images of Pt@PNIPAM-SH after five (a) and ten (b) runs, and Pt@PNIPAM after two (d) and five (e) runs; particle size distributions of Pt@PNIPAM-SH after ten runs (c) and Pt@PNIPAM after five runs (f). | ||
The contents of metal leaching are of vast importance for industrial applications, so the Pt leaching into products during the recycling experiment over Pt@NIPAM-SH was tested by ICP-AES. After each run, the liquid phase was separated from the precipitated Pt catalyst and combined together. Then the solution was vaporized to remove the solvents, and the non-volatile organics was removed by high temperature calcination. Finally, the remaining residue was dissolved in aqua regia. The result of ICP-AES showed that the total Pt leaching for fifteen runs was only 0.2%. These results demonstrated that the PNIPAM-SH protected and modified Pt nanocatalyst could be well recovered from the reaction system.
4. Conclusions
We have demonstrated that PNIPAM-SH can not only stabilize the Pt nanoparticles, but also inhibit the highly active Pt catalyst from producing hydrodehalogenation products through anchoring the thiol groups to unsaturated surface of Pt nanoparticles. The catalytic site poisoning and steric hindrance effect of Pt@PNIPAM-SH catalyst were suitable for selective hydrogenation of HNBs with high and steady selectivities to HANs, and the conclusion was further confirmed by comparing to the catalyst of Pt@PNIPAM for hydrogenation of a series of HNBs. Moreover, the recycling process of Pt@PNIPAM-SH is rather simple and practical based on the cononsolvency of PNIPAM-SH. Excellent stability and reusability were presented over Pt@PNIPAM-SH, and no decrease in catalytic activity and selectivity was observed for first twelve runs with extremely low metal leaching, which was one of the best results obtained over Pt catalysts. TEM characterization of reused catalyst revealed that the Pt nanoparticles in Pt@PNIPAM-SH were not obviously enlarged, the formation of polymer aggregates took place. These inspiring results demonstrated that the Pt nanoparticles stabilized and modified with PNIPAM-SH was an efficient strategy to improve HAN selectivity and reusability of Pt catalysts for selective hydrogenation of HNBs.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 21203102), the Tianjin Municipal Natural Science Foundation (Grant No. 14JCQNJC06000), MOE(IRT13R30) and 111 Project (B12015).References
- A. M. Tafesh and J. Weiguny, Chem. Rev., 1996, 96, 2035 CrossRef CAS PubMed.
- S. Cai, H. Duan, H. Rong, D. Wang, L. Li, W. He and Y. Li, ACS Catal., 2013, 3, 608 CrossRef CAS.
- J. Lyu, J. Wang, C. Lu, L. Ma, Q. Zhang, X. He and X. Li, J. Phys. Chem. C, 2014, 118, 2594 CAS.
- T. Chen, D. Li, H. Jiang and C. Xiong, Chem. Eng. J., 2015, 259, 161 CrossRef CAS.
- A. Serna and P. Serna, Science, 2006, 313, 332 CrossRef PubMed.
- D. He, H. Shi, Y. Wu and B.-Q. Xu, Green Chem., 2007, 9, 849 RSC.
- F. Cárdenas-Lizana, X. Wang, D. Lamey, M. Li, M. A. Keane and L. Kiwi-Minsker, Chem. Eng. J., 2014, 255, 695 CrossRef.
- Y. Chen, C. Wang, H. Liu, J. Qiu and X. Bao, Chem. Commun., 2005, 5298 RSC.
- G.-Y. Fan, L. Zhang, H.-Y. Fu, M.-L. Yuan, R.-X. Li, H. Chen and X.-J. Li, Catal. Commun., 2010, 11, 451 CrossRef CAS.
- T. Lu, H. Wei, X. Yang, J. Li, X. Wang and T. Zhang, Langmuir, 2015, 31, 90 CrossRef CAS PubMed.
- W. Lin, J. Zhao, H. Cheng, X. Li, X. Li and F. Zhao, J. Colloid Interface Sci., 2014, 432, 200 CrossRef CAS PubMed.
- M. Pietrowski, M. Zieliński and M. Wojciechowska, Catal. Lett., 2008, 128, 31 CrossRef.
- H. Li, Y. Xu, H. Yang, F. Zhang and H. Li, J. Mol. Catal. A: Chem., 2009, 307, 105 CrossRef CAS.
- X. Meng, H. Cheng, S.-i. Fujita, Y. Hao, Y. Shang, Y. Yu, S. Cai, F. Zhao and M. Arai, J. Catal., 2010, 269, 131 CrossRef CAS.
- P. Zhang, C. Yu, X. Fan, X. Wang, Z. Ling, Z. Wang and J. Qiu, Phys. Chem. Chem. Phys., 2015, 17, 145 RSC.
- Z. Wei, J. Wang, S. Mao, D. Su, H. Jin, Y. Wang, F. Xu, H. Li and Y. Wang, ACS Catal., 2015, 5, 4783 CrossRef CAS.
- P. Lara and K. Philippot, Catal. Sci. Technol., 2014, 4, 2445 CAS.
- X. Han, R. Zhou, G. Lai, B. Yue and X. Zheng, Catal. Lett., 2003, 89, 255 CrossRef CAS.
- X. Han, R. Zhou, G. Lai and X. Zheng, Catal. Today, 2004, 93–95, 433 CrossRef CAS.
- M. Liu, J. Zhang, J. Liu and W. W. Yu, J. Catal., 2011, 278, 1 CrossRef CAS.
- S. Iihama, S. Furukawa and T. Komatsu, ACS Catal., 2016, 6, 742 CrossRef CAS.
- J. Zhang, Y. Wang, H. Ji, Y. Wei, N. Wu, B. Zuo and Q. Wang, J. Catal., 2005, 229, 114 CrossRef CAS.
- M. Liang, X. Wang, H. Liu, H. Liu and Y. Wang, J. Catal., 2008, 255, 335 CrossRef CAS.
- Y. Motoyama, Y. Lee, K. Tsuji, S.-H. Yoon, I. Mochida and H. Nagashima, ChemCatChem, 2011, 3, 1578 CrossRef CAS.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed.
- V. Pandarus, R. Ciriminna, F. Béland and M. Pagliaro, Adv. Synth. Catal., 2011, 353, 1306 CrossRef CAS.
- X. Yang and H. Liu, Appl. Catal., A, 1997, 164, 197 CrossRef CAS.
- C.-X. Xiao, H.-Z. Wang, X.-D. Mu and Y. Kou, J. Catal., 2007, 250, 25 CrossRef CAS.
- M. Takasaki, Y. Motoyama, K. Higashi, S.-H. Yoon, I. Mochida and H. Nagashima, Org. Lett., 2008, 10, 1601 CrossRef CAS PubMed.
- H. Cheng, C. Xi, X. Meng, Y. Hao, Y. Yu and F. Zhao, J. Colloid Interface Sci., 2009, 336, 675 CrossRef CAS PubMed.
- M. J. Beier, J.-M. Andanson and A. Baiker, ACS Catal., 2012, 2, 2587 CrossRef CAS.
- M. Makosch, W.-I. Lin, V. Bumbálek, J. Sá, J. W. Medlin, K. Hungerbühler and J. A. van Bokhoven, ACS Catal., 2012, 2, 2079 CrossRef CAS.
- P. Lara, A. Suárez, V. Collière, K. Philippot and B. Chaudret, ChemCatChem, 2014, 6, 87 CrossRef CAS.
- J. P. Pinheiro, L. Moura, R. Fokkink and J. P. S. Farinha, Langmuir, 2012, 28, 5802 CrossRef CAS PubMed.
- Y.-Z. You, K. K. Kalebaila, S. L. Brock and D. Oupický, Chem. Mater., 2008, 20, 3354 CrossRef CAS.
- F. Mou, C. Chen, Q. Zhong, Y. Yin, H. Ma and J. Guan, ACS Appl. Mater. Interfaces, 2014, 6, 9897 CAS.
- C.-W. Chen and M. Akashi, Langmuir, 1997, 13, 6465 CrossRef CAS.
- D. E. Bergbreiter, Y.-S. Liu and P. L. Osburn, J. Am. Chem. Soc., 1998, 120, 4250 CrossRef CAS.
- R. Tan, Y. Dong, M. Peng, W. Zheng and D. Yin, Appl. Catal., A, 2013, 458, 1 CrossRef CAS.
- H. A. Zayas, A. Lu, D. Valade, F. Amir, Z. Jia, R. K. O'Reilly and M. J. Monteiro, ACS Macro Lett., 2013, 2, 327 CrossRef CAS.
- F. M. Winnik, H. Ringsdorf and J. Venzmer, Macromolecules, 1990, 23, 2415 CrossRef CAS.
- H. G. Schild, M. Muthukumar and D. A. Tirrell, Macromolecules, 1991, 24, 948 CrossRef CAS.
- W. Yu, L.-L. Lou, K. Yu, S. Li, Y. Shi and S. Liu, RSC Adv., 2016, 6, 52500 RSC.
| This journal is © The Royal Society of Chemistry 2017 |